

Abstract
CSN5 has been implicated as a candidate oncogene in human breast cancers by genetic linkage with activation of the poor-prognosis, wound response gene expression signature. CSN5 is a subunit of the eight-protein COP9 signalosome, a signaling complex with multiple biochemical activities; the mechanism of CSN5 action in cancer development remains poorly understood. Here, we show that CSN5 isopeptidase activity is essential for breast epithelial transformation and progression. Amplification of CSN5 is required for transformation of primary human breast epithelial cells by defined oncogenes. The transforming effects of CSN5 require CSN subunits for assembly of the full COP9 signalosome and the isopeptidase activity of CSN5, which potentiates the transcriptional activity of MYC. Transgenic inhibition of CSN5 isopeptidase activity blocks breast cancer progression evoked by MYC and RAS in vivo. These results highlight CSN5 isopeptidase activity in breast cancer progression, suggesting it as a therapeutic target in aggressive human breast cancers.
Introduction
One in seven women in the United States will be diagnosed with breast cancer in their lifetime. Despite apparent similarity by conventional criteria, patients with breast cancers can show vexing heterogeneity in their disease course and response to treatment, potentially reflecting the diversity of genes that drive breast cancer progression. The long arm of human chromosome 8 is frequently amplified during the transition from ductal carcinoma in situ to invasive breast carcinoma (1), and 8q is the most common amplification in advanced human breast cancers (2). 8q is also amplified in several experimental models of mammary epithelial transformation (3), indicating that key genes controlling mammary cell growth likely reside on 8q. In addition to the well-known oncogene MYC on 8q24, additional oncogene(s) is believed to contribute to the potency of the 8q amplicon.
Based on the concept that tumor growth may recapitulate many aspects of normal wound healing, we previously identified a wound response gene expression signature that is reactivated in many types of human cancers, including breast, prostate, lung, and gastric cancers (4). The wound signature is composed of 512 genes that defined the transcriptional response of fibroblasts to serum, the soluble fraction of clotted blood. In primary breast cancer, the wound signature provides prognostic risk stratification of metastasis that is more predictive and is independent of traditional criteria, such as lymph node status, grade, and estrogen receptor status (4, 5). Using a new genetic method termed stepwise linkage analysis of microarray signatures (SLAMS), we recently identified coordinate amplification of CSN5 (also known as JAB1 or COPS5, residing on 8q13) and MYC (8q24) as the cause of wound signature activation in human breast cancers (6). Coexpression of CSN5 with MYC is sufficient to induce the wound signature and confer features of transformation to nontransformed mammary epithelial cells, including increased proliferation and matrix invasiveness. These results therefore nominate CSN5 as a candidate oncogene and potential driver of the 8q amplicon in breast cancer.
Although the role of MYC in the progression of many types of cancer has been studied extensively (7), the mechanism of CSN5 action in cancer progression is less well understood. CSN5 protein is overexpressed in many human epithelial cancers, including human breast cancers (8-10). Depletion of CSN5 can inhibit the proliferation of some pancreatic cancer cell lines in vitro (11). However, the genetic context of CSN5 action, its biochemical mode of action, and its role in cancer progression in vivo remain unclear. If we are to target CSN5 rationally in high-risk breast tumors exhibiting 8q amplification and the poor-prognosis wound signature, then a detailed understanding of the oncogenic mechanism of CSN5 is needed.
More than a dozen biochemical activities have been reported for the COP9 signalosome complex (12, 13). CSN5 may exert its oncogenic effects by at least three potential mechanisms, realized when CSN5 resides in distinct macromolecular complexes (Supplementary Fig. S1). First, CSN5 encodes the catalytic component of the COP9 signalosome, a protein complex that regulates cell proliferation, response to extracellular stimuli, cell migration, and DNA damage checkpoints (13). The COP9 signalosome, composed of eight proteins named CSN1 to CSN8, is conserved from yeast to humans and is essential for development in Drosophila and mouse (14-16). A main function of COP9 is to maintain the activity of SCF (SKP1, CUL1, and F-box) and other cullin-RING family E3 ubiquitin ligases. All cullin proteins seem to be covalently modified by the ubiquitin-like protein NEDD8 on a single lysine residue. The COP9 signalosome deneddylates CUL1; this catalytic activity resides in a metalloproteinase-like motif termed the JAMM motif in CSN5 (17). Reconstitution of the NEDD8 isopeptidase activity in vitro requires the entire COP9 signalosome (17). Deneddylation by CSN5 maintains SCF complexes in an active state (6, 18, 19), leading to the ubiquitination and proteasomal degradation of many cellular proteins. Consistent with this model, we previously found that CSN5 enhances the cotranscriptional ubiquitination of MYC that activates the transcriptional activity of MYC on a set of target genes promoting cell proliferation, invasion, and angiogenesis (6).
Second, in a small CSN complex composed of five proteins (CSN4, CSN5, CSN6, CSN7, and CSN8), CSN5 induces the cytoplasmic export and degradation of p27, the cyclin-dependent kinase (CDK) inhibitor that is targeted for destruction by both CSN5 and MYC (via transcription of CUL1; refs. 16, 20-22). Interestingly, the degradation of p27 by the proteasome is mediated by SCFSKP2 (23-25). Thus, two distinct CSN5 activities may sequentially act on p27 for nuclear export and degradation. Third, monomeric CSN5 protein can bind to and modulate the activity of multiple transcription factors and signaling proteins (8, 13). Most relevant to cancer are the interactions with hypoxia-inducible factor-1α (HIF-1α), leading to HIF-1α protein stabilization and increased angiogenic activity (26, 27), and E2F1, leading to selective activation of E2F1-mediated apoptotic activity (28). These activities seem to be independent of the isopeptidase activity because interaction of CSN5 with both HIF-1α and E2F1 is unaffected by the D151N mutation that inactivates the isopeptidase activity (27, 28). In addition, the COP9 signalosome is associated with kinase and deubiquitinase activities, which can potentially initiate oncogenic signaling (12, 29).
In the present study, we evaluate the dosage, genetic context, and specific activities of CSN5 that underlie its oncogenic role using both human and mouse models of breast epithelial transformation by defined genetic elements. Our results provide the first evidence that CSN5 isopeptidase activity is essential for breast cancer proliferation in vivo.
Materials and Methods
Antibodies, Plasmids, and Small Interfering RNAs
Antibodies for MYC (9E10), CSN5 (FL-334), p27 (F-8; Santa Cruz Biotechnology), β-actin (AC-74; Sigma), 5-bromo-2′-deoxyuridine (BrdUrd; 3D4; BD Biosciences), CSN1, CSN6 (Biomol International, LP), green fluorescent protein (GFP; Biotin), vascular endothelial growth factor (VEGF; 5C3.F8; Abcam), hemagglutinin (HA; HA.11; Covance), Ki67 (TEC-3; Dako), cleaved caspase-3 (Asp175; Cell Signaling Technology), and HIF-1α (R&D Systems) are from the indicated sources. pLXSN-CSN5 (27), pMDM2-Luc, pMDM2-Luc-Mut (30), pMIG, and pMIG-MYC (31) were previously published. pCDNA3.1-HA-CSN5 (Xin Chen, University of California, San Francisco, CA), pLZRS-GFP (Paul Khavari, Stanford University School of Medicine, Stanford, CA), pBabe-MYC (Dean Felsher, Stanford University School of Medicine), pMICD8-MYC, and pMICD8-RASV12 (Suwon Kim, University of California, and Yosef Refaeli, National Jewish Medical and Research Center, Denver, CO) were gifts of the indicated investigators. The CSN5D151N mutation was created by site-directed mutagenesis and verified by sequencing. For cloning into pMIG, HA-CSN5 and HA-CSN5D151N fragments were released by XhoI and SalI digestion and subcloned into XhoI-digested pMIG; for cloning into LZRS, HA-CSN5 and HA-CSN5D151N were cloned into pENTR and subsequently cloned into LZRS using Gateway Technology (Invitrogen). Clones were verified by sequencing and immunoblotting with HA and CSN5 antibodies. All small interfering RNAs (siRNA) were synthesized by Dharmacon. Sequences for GFP (E1; ref. 32) and CSN5 (33) are from published sequences. siMYC was an individual siGENOME duplex (sense sequence: CGACGAGACCUUCAUCAAAUU), whereas siCSN1, siCSN6, and sip27 were SMARTpool siGENOME duplexes from Dharmacon. 3′-Untranslated region (UTR)-specific siCSN5 was designed using Dharmacon siDESIGN center (sense sequence: AGAAGUACUUUACCUGAAAUU).
Cell Culture
PHMLEB and PHMLER cells were a kind gift of Robert Weinberg (Massachusetts Institute of Technology, Cambridge, MA). PHMLEB, PHMLER, and primary HMEC (Cambrex) were propagated in MEGM mammary epithelial cell medium (Cambrex). MCF10A cells [American Type Culture Collection (ATCC)] were propagated in DMEM/F12 (Invitrogen/Cambrex) and 5% fetal bovine serum (FBS) supplemented with epidermal growth factor (20 ng/mL), insulin (10 μg/mL), and hydrocortisone (0.5 μg/mL), and 293 cells (ATCC) were propagated in DMEM and 10% FBS. Retroviral constructs were transfected into amphotropic Phoenix cells (gift of Garry Nolan, Stanford University School of Medicine), and virus-containing supernatants were used to transduce PHMLER and MCF10A cells as described (34). Expression of transduced genes was verified by immunoblotting. siRNAs were transfected into PHMLER cells with Lipofectamine 2000 as described by the manufacturer (Invitrogen); protein knockdown was verified by immunoblotting.
Cell Proliferation, Immunofluorescence, Cell Death, Soft Agar, and Reporter Gene Assays
Cell proliferation and immunofluorescence
Cell proliferation (6) and immunofluorescence (35) were performed as described. For BrdUrd incorporation, cell proliferation was monitored 3 days after siRNA transfection by measuring incorporation of the thymidine nucleotide analogue BrdUrd (Sigma) into DNA with immunofluorescence as described (36). Percentage of BrdUrd-positive cells among >400 4′,6-diamidino-2-phenylindole (DAPI)-positive cells in multiple high-power fields was determined. For p27 immunofluorescence, the number of cells with nuclear p27 staining was determined among >160 DAPI-positive cells in multiple high-power fields.
Cell death
Terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling (TUNEL) assay for detection of apoptotic DNA fragmentation 3 or 5 days after siRNA transfection was performed per manufacturer’s instruction (Roche). Percentage of TUNEL-positive cells among >500 DAPI-positive cells in multiple high-power fields was determined.
Soft agar
Anchorage-independent growth in soft agar was performed as described with minor changes (3). A bottom layer of 0.5% low melting point agar (Invitrogen) in MEGM medium was plated in 6-cm dishes. One day after siRNA transfection, 5 × 104 cells were seeded on top in 0.34% agar. Multiple similarly dense low-power fields were quantified for colonies ≥100 μm in diameter 7 to 12 days after plating.
Reporter gene
Dual-luciferase assays (Promega) with MDM2-luciferase were performed in 293 cells as described (6).
Microarray Analysis
Array-based comparative genomic hybridization
Genomic DNA from normal male, PHMLEB, and PHMLER cells was isolated with Qiagen Blood & Cell Culture DNA Midi kit. Labeling and hybridization to Stanford cDNA microarrays containing ~23,000 genes was performed as described using normal male genomic DNA as reference (37). Array-based comparative genomic hybridization (CGH) of normal male diploid compared with normal female diploid was previously published (38) and downloaded from Stanford Microarray Database.4 Genes selected for analysis had fluorescent hybridization signal at least 1.5-fold over local background in either Cy5 or Cy3 channel. PHMLEB samples were performed in duplicate; PHMLER samples were performed in triplicate. Replicates were averaged, aligned in sequential chromosomal order as described (37), and visualized with CGH-Explorer.5
Gene expression
Total RNA was extracted with Trizol (Invitrogen) from PHMLER cells 2 days after transfection of the indicated siRNAs in duplicate; RNA was amplified using the Ambion MessageAmpII aRNA kit. Amplified total Human Universal Reference RNA (Stratagene) was used as reference. Labeling and hybridization to cDNA microarrays was performed as described (35). Genes selected for analysis had fluorescent hybridization signal at least 1.5-fold over local background in either Cy5 or Cy3 channel and had technically adequate data in at least 60% of experiments. Genes were analyzed by mean value centering within the data set. Wound signature score was calculated for each sample as described (5). Briefly, the wound signature score is the Pearson correlation coefficient of the expression pattern of genes in the wound signature in an experimental sample to the pattern observed for the same genes in serum-stimulated fibroblasts. The change in score is visualized relative to the P value of significance of coregulation as described (6).
PCR and Reverse Transcription-PCR
For quantitative reverse transcription-PCR (RT-PCR) of MYC and CSN5 (primers and Taqman probes from Applied Biosystems), Taqman One-Step RT-PCR kit (Applied Biosystems) was applied to amplified RNA from primary HMEC or PHMLER cells 48 h after siRNA transfection; all samples were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Validation of MYC and CSN5 DNA copy number was performed on genomic DNA isolated from normal male, PHMLEB, and PHMLER cells. DNA copy number was determined by quantitative microsatellite analysis as described (39) with published primers (6).
Mammary Gland Transplants
For Fig. 5B, primary mammary epithelial cells were isolated from the mammary glands (no. 2–5 glands) of 10- to 12-week-old mice, transduced with retrovirus, and transplanted into cleared no. 4 fat pads as previously described (31). For the remaining experiments, we used the following modifications to increase the efficiency of multiplexed gene transfer. After mincing of the isolated tissue, tissue chunks were treated with 0.85 to 1 mg/mL of collagenase for 1.5 h at 37°C. Differential centrifugation was used to separate blood and fibroblast cells from epithelial cells. Specifically, cells were centrifuged for 5 min at 600 × g and then washed thrice for 2 s at 450 × g. The remaining epithelial cells were resuspended in growth medium and then plated on 10-cm plates. Medium was changed every day, and then on day 4, cells were trypsinized for 5 min and the trypsinized cells were removed. The remaining cells were trypsinized for an additional 20 min and then replated for infection. Cells were infected on 2 consecutive days with the indicated retroviruses (multiplicities of infection of 2 or 3 for each virus) as described (34). Cells were grown in fresh medium for an additional day following infection, and then 65 × 104 to 100 × 104 infected cells were injected into cleared inguinal no. 4 fat pads of 3-week-old nu/nu mice. All comparisons were to the contralateral gland taken from the same mouse.
Figure 5. Catalytically inactive CSN5 inhibits breast cancer growth in vivo.
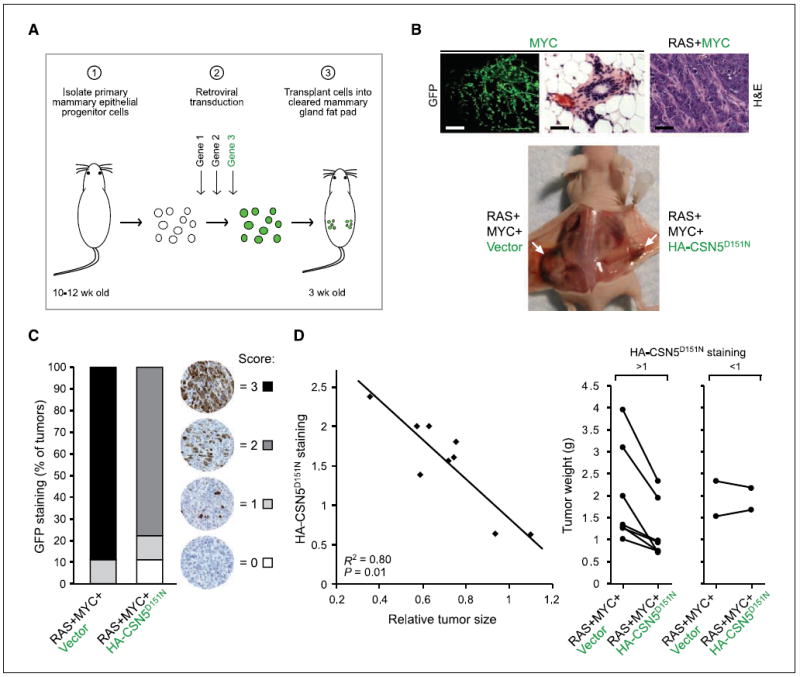
A, outline of mouse model of breast cancer. Green letters, genes linked with an internal ribosomal entry site (IRES)-GFP; green circles, cells transduced with IRES-GFP vector. B, top, mammary glands regenerated with GFP-marked MYC alone (10 wk; no tumor formation) or oncogenic RAS + GFP-marked MYC (3 wk; tumor formation). H&E staining of the tumor is shown. Bars, 1 mm (left) and 40 μm (middle and right). Bottom, tumor formation in situ of the indicated transduced genetic elements. MYC and RAS constructs are marked with IRES-CD8; vector and HA-CSN5D151N constructs are marked with IRES-GFP. White arrows, mammary tumors. C, quantification of GFP staining of tumor sections (only vector and CSN5D151N are GFP marked). D, increased HA-CSN5D151N staining correlates with decreased tumor size. Left, quantification of HA staining compared with tumor size of contralateral vector-expressing tumors. The data fit to a linear regression (R2 = 0.80; P = 0.01, one-sided t test). Right, RAS+MYC+Vector and the contralateral RAS+MYC+HA-CSN5D151N–expressing tumor sizes from each animal (tumors from the same animal are connected by a line). In tumors with HA staining >1, on average the tumor size was reduced by 40%.
Histologic and Immunohistochemical Analyses
Two to 3 weeks after mammary epithelial cell injection, mammary tumors were weighed and then fixed in 4% paraformaldehyde. Tumors were embedded in paraffin, sectioned, and stained for H&E. Immunohistochemical staining for GFP, HA, Ki67, cleaved caspase-3, HIF-1α, and VEGF protein in paraffin-embedded sections (5 μm) was performed using BioGenex immunohistochemical detection systems per the manufacturer’s instructions. A single score resenting the level of GFP staining across each tumor section was determined on a 0 to 3 scale: 0, <10% of cellular areas stained; 1, 10% to 25% of cellular areas stained; 2, 26% to 50% of cellular areas stained; 3, >50% of cellular areas stained. For HA-CSN5D151N–expressing tumors, the same 0 to 3 scoring criteria above were used to determine the level of HA staining in low-power fields (~0.2 cm2 area) across the entire tumor section; the HA staining scores were averaged and then plotted against the relative tumor size compared with the contralateral control tumor, showing a linear regression (R2 = 0.80; P = 0.01). R (correlation value) and P value (one-sided t test) were calculated with WinSTAT (R. Fitch Software). The number of Ki67-positive cells in several high-power fields was counted and averaged (minimum of 600 cells). Analysis of tumor histologic features was performed by two independent reviewers who were not aware of the tumor genotypes. Multiple medium- to high-power fields of H&E-stained sections were scored. Quantification of nuclear to cytoplasmic ratio was determined by measuring the diameter of the nucleus and cytoplasm of >25 cells for each condition. The volumes of the nucleus and cytoplasm were extrapolated [(1/6) × π × diameter3], and the ratios were averaged.
URLs
Full microarray data are available for download at Stanford Microarray Database or Gene Expression Omnibus.6
Results
Amplification of CSN5 is required for transformation of human mammary epithelial cells
Primary human mammary epithelial cells (HMEC) can be transformed into malignant breast cancer by the stepwise introduction of three genetic elements: SV40 early region [encoding large T antigen that inactivates both Rb and p53 tumor suppressors and small T antigen that inactivates protein phosphatase 2A (PP2A)], hTERT (encoding human telomerase), and oncogenic H-RASV12 (Fig. 1A; ref. 3). All four gene products are required to induce anchorage-independent growth in soft agar and tumor formation in nude mice, and these two features are completely linked in this stepwise system of transformation. In addition, spectral karyotype analysis of the resulting PHMLER cells revealed clonal amplification of endogenous chromosome 8q, which do not accompany transformation of other cell types by these genes (3). Thus, these genes targeting core pathways of growth control, plus additional gene(s) on 8q, seem to constitute a minimal genetic circuitry of human breast cancer.
Figure 1. CSN5 and MYC are coordinately amplified in PHMLEB and PHMLER cells.
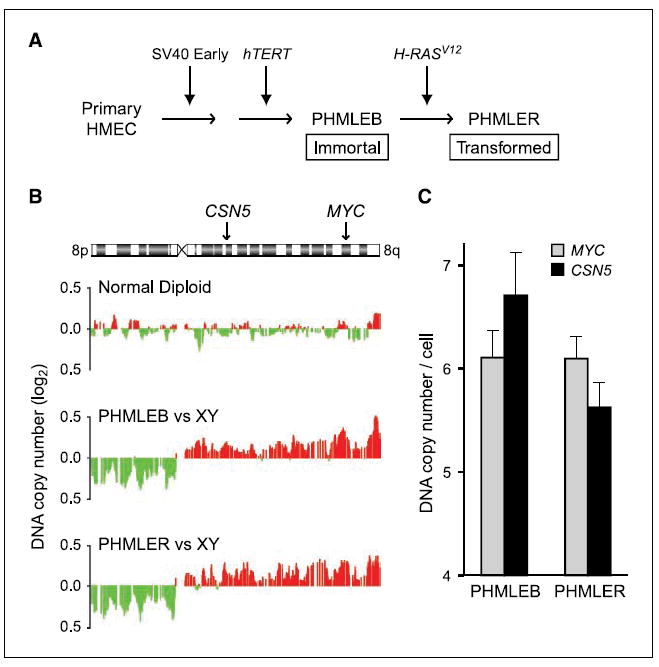
A, schematic of stepwise oncogenic transformation of primary HMEC. PHMLEB cells are immortalized; PHMLER cells are tumorigenic (3). B, array-based CGH of chromosome 8q DNA copy number in normal diploid, PHMLEB, and PHMLER cells. Red, amplification; green, deletion. Location of CSN5 and MYC on 8q is indicated. Data are shown as a moving average of the surrounding 15 genes. C, quantitative microsatellite analysis of CSN5 and MYC DNA copy number relative to normal. Columns, mean; bars, SE.
We obtained this series of cells representing stepwise mammary transformation to investigate the role of CSN5 in 8q-amplified breast cancers. Using array-based CGH, we found that clonal 8q amplification arose before the introduction of RAS in PHMLEB cells, and the amplification encompassed both CSN5 and MYC (Fig. 1B). Quantitative microsatellite analysis, which measures loci-specific DNA copy number relative to the genome (39), confirmed that the copy number of CSN5 and MYC loci increased to six to seven copies per cell in PHMLEB and PHMLER cells (Fig. 1C), comparable with levels seen in human breast tumors exhibiting the wound signature (6). CSN5 and MYC protein levels are correspondingly increased in PHMLER cells compared with HMEC (Fig. 2A); no difference in protein levels was observed between PHMLER and PHMLEB cells (data not shown). These results suggest that 8q amplification likely provides an early growth advantage during transformation and contributed to all of the previously documented phenotypes of transformation in PHMLER cells.
Figure 2. CSN5 and MYC are required for transformed phenotypes of PHMLER cells.
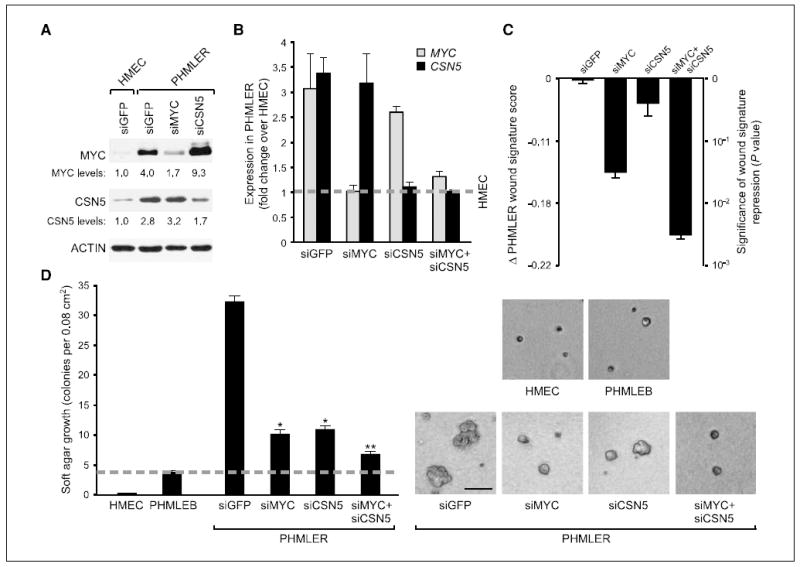
A, immunoblot 3 d after siRNA transfection in the indicated cells. Relative levels of MYC and CSN5 protein were quantified with ImageJ. B, fold change of MYC and CSN5 mRNA in PHMLER cells over primary HMEC (dashed line) as determined by quantitative RT-PCR. Columns, mean; bars, SE. C, normalization of CSN5 and MYC synergistically inhibits wound signature expression. Change in wound signature score (see Materials and Methods) of each sample (left) plotted on a scale of the significance of the change in score (right). Columns, mean; bars, SE. The significance scale reflects the exponential increase in the risk of metastasis and death with linear increase of the wound score (6). D, normalization of CSN5 and MYC synergistically inhibits anchorage-independent growth. Left, quantification of soft agar colonies ≥100 μm in diameter 8 d after plating. Columns, mean; bars, SE. Dashed line, background colony growth of the nontumorigenic PHMLEB cells. *, P < 0.002, Student’s t test, compared with siGFP; **, P < 0.002, Student’s t test, compared with siMYC and siCSN5. Right, images of soft agar colonies. Bar, 100 μm.
We next determined the requirement of CSN5 and MYC overexpression for features of transformation in PHMLER cells. We chose siRNAs that reduced CSN5 or MYC protein levels in PHMLER cells to those equivalent to or slightly above primary HMEC (Fig. 2A and B), and we used a siRNA against GFP as a control. Reduction of endogenous CSN5 protein stabilized MYC protein level as previously observed in 293 HEK cells (Fig. 2A; refs. 6, 19). Consistent with previous linkage analysis prediction (6), normalization of CSN5 and MYC level in PHMLER cells synergistically and strongly inhibited expression of the wound signature as determined by microarray analysis (Fig. 2C). Normalization of either CSN5 or MYC protein level in PHMLER cells also greatly inhibited the number and size of anchorage-independent colonies in soft agar, a stringent assay of transformation in these cells (3); a further decrease was observed on depletion of both proteins (Fig. 2D). Normalization of CSN5 level seemed to have a stronger effect on soft agar growth than wound signature expression, implying that transformation and wound signature can be unlinked in these cells. Normalization of CSN5 and MYC significantly decreased the rate of PHMLER cell accumulation mainly due to decreased cell proliferation but not increased apoptosis (Supplementary Fig. S2). In contrast, whereas reduction of MYC slowed the proliferation of HMEC, reduction of CSN5 had little effect on the normal rate of HMEC proliferation (Supplementary Fig. S3). These results indicate that, despite the ongoing dysregulation of RAS, p53, Rb, PP2A, and TERT in PHMLER cells, overproduction of CSN5 and MYC proteins is individually and continually required to maintain the transformed phenotype of PHMLER cells.
The isopeptidase activity of CSN5 is required for mammary epithelial transformation
Because CSN5 is found in several distinct macromolecular complexes that have distinct modes of action, we reasoned that an efficient strategy to determine the mechanism of CSN5 action is to dissect the CSN subunit requirement (Supplementary Fig. S1). We chose to analyze CSN1 and CSN6 because CSN1 is only found in the full COP9 complex, whereas CSN6 can be either in the full or small CSN complexes (22). We found that depletion of either CSN1 or CSN6 strongly decreased the proliferation rate and anchorage-independent growth of PHMLER cells (Fig. 3A–C). The stronger inhibitory effect of CSN1 or CSN6 depletion on cell proliferation is likely explained by more complete protein depletion compared with CSN5 knockdown. Because depletion of either CSN1 or CSN6 led to similar phenotypes as CSN5 normalization, these data suggest that the full COP9 signalosome is required for the transformed phenotypes of PHMLER cells. It is formally possible that CSN1 and CSN6 may have roles independent of CSN5, although biochemical studies indicate that CSN1 and CSN6 are quantitatively associated with CSN5 (22). Consistent with the idea that additional CSN subunits participate in CSN5-amplified breast cancers, analysis of array-based CGH data revealed that genes encoding several CSN subunits, particularly CSN1, CSN6, and CSN7A, are also amplified in a subset of primary human breast cancers (Supplementary Fig. S4).
Figure 3. Transformed phenotypes of PHMLER cells require additional CSN subunits.
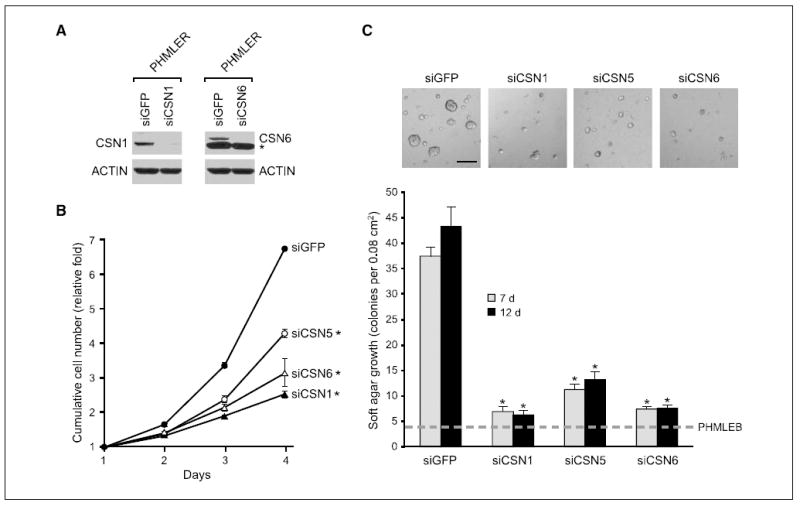
A, immunoblot 3 d after siRNA transfection in PHMLER cells. *, nonspecific band. B, cell proliferation by daily counts of transfected PHMLER cells. Points, mean; bars, SE. *, P < 0.001, Student’s t test, compared with siGFP at days 3 and 4. C, silencing of CSN1 and CSN6 inhibits anchorage-independent growth. Top, images of soft agar colonies 7 d after plating. Bar, 200 μm. Bottom, quantification of soft agar colonies 7 or 12 d after plating. Columns, mean; bars, SE. Dashed line, colony growth of PHMLEB cells. *, P < 0.01, Student’s t test, compared with siGFP.
We next determined whether the NEDD8 isopeptidase activity of CSN5 is required for PHMLER cell transformation by use of the isopeptidase mutant CSN5D151N. D151N alters one of the key conserved residues coordinating the active site zinc without affecting COP9 complex assembly (40); the defective CSN5D151N subunit poisons the entire COP9 complex and exerts a dominant-negative effect (17). Importantly, the D151N mutation does not affect the function of the small CSN complex nor monomeric CSN5 interaction and regulation of target proteins, such as HIF-1α or E2F1 (22, 27, 28). Because the full COP9 complex is required to reconstitute CSN catalytic activity (17), involvement of CSN5 isopeptidase activity would provide further evidence that CSN5 is acting within the full COP9 signalosome.
We first analyzed the ability of CSN5D151N to regulate MYC protein stability and MYC target gene activation. In stably transduced MCF10A cells, a nontransformed breast epithelial cell line, wild-type CSN5 decreased MYC protein accumulation [as a result of stimulating MYC turnover (6)], but CSN5D151N had the opposite effect and increased MYC protein accumulation (Fig. 4A, lane 3 versus lane 5 and lane 4 versus lane 6). MYC mRNA level was equivalent in each pair of CSN5 versus CSN5D151N comparisons (data not shown). Based on the known cotranscriptional ubiquitination and turnover of MYC with target gene activation (41), we hypothesized that the isopeptidase-null CSN5D151N will inhibit MYC target gene activation. We previously identified the oncogene MDM2 as a MYC target gene that required CSN5 activity for full activation (6), and we recapitulated the functional collaboration of MYC and CSN5 using an MDM2-luciferase reporter gene assay in 293 HEK cells (Fig. 4B). CSN5 and MYC individually activated the MDM2 promoter and superactivated the promoter in combination. Importantly, a mutation of the E-box binding site for MYC in the MDM2 promoter completely abrogated transcriptional activation by both CSN5 and MYC, indicating that CSN5 likely activates the MDM2 promoter via endogenous MYC (Fig. 4B, lanes 7 and 8). The isopeptidase mutant CSN5D151N failed to stimulate the MDM2 reporter gene and, importantly, completely inhibited the ability of MYC to activate the MDM2 reporter (Fig. 4B, lanes 5 and 6). These results indicate that CSN5 isopeptidase activity is required for MYC protein turnover and activation of certain target genes and further suggest that CSN5D151N acts as a dominant negative with respect to MYC activation.
Figure 4. Catalytic activity of CSN5 is required for MYC activation and transformation of PHMLER cells.
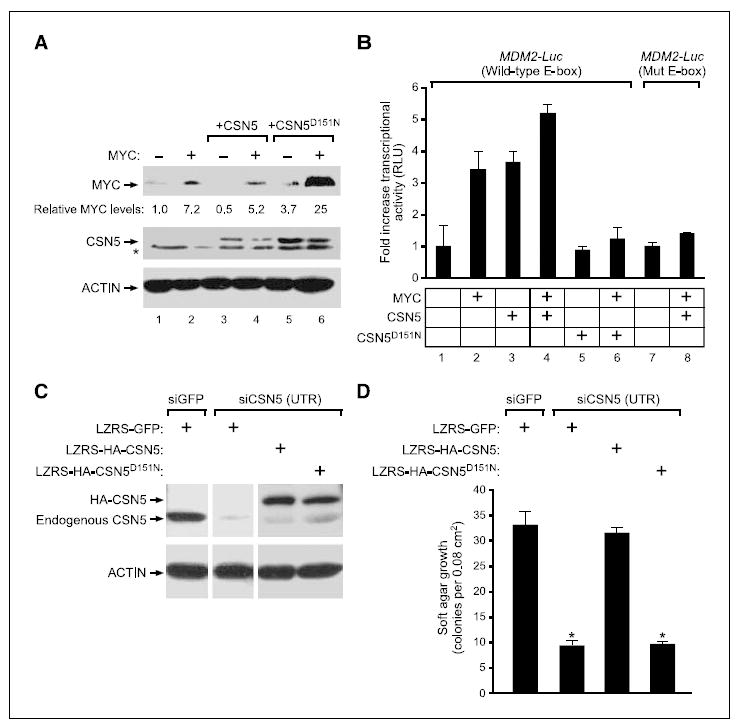
A, immunoblot of MYC and CSN5 or CSN5D151N protein in transduced MCF10A cells. For the CSN5 blot, arrow indicates transduced CSN5, which contains a MYC epitope tag. *, nonspecific band. Relative levels of MYC protein are provided. B, CSN5D151N inhibits activation of MDM2 promoter by MYC. Reporter gene activity in the presence of the indicated gene combinations in transfected 293 cells, normalized to input MYC levels. Columns, mean relative luciferase units (RLU); bars, SE. Mut E-box, MDM2 promoter with mutated E-box element. C, immunoblot of transduced PHMLER cells 3 d after siRNA transfection. UTR, siRNA targeting 3′-UTR of endogenous CSN5. D, catalytic activity of CSN5 is required for anchorage-independent growth. Quantification of soft agar colonies 7 d after plating. Columns, mean; bars, SE. *, P < 10−4, Student’s t test, compared with siGFP or siCSN5+LZRS-HA-CSN5.
To analyze the effect of CSN5D151N in PHMLER cells that carry endogenous amplification of CSN5 and MYC, we used an orthogonal expression strategy to, in effect, “knock in” CSN5D151N. We normalized endogenous CSN5 levels by use of a siRNA that targets the 3′-UTR of CSN5 transcript and then reconstituted the cells with cDNAs encoding HA-tagged wild-type or mutant CSN5 fused to a heterologous 3′-UTR sequence to levels similar to endogenous CSN5 (Fig. 4C). As expected, wild-type CSN5 fully rescued the defect in proliferation and anchorage-independent growth induced by depletion of endogenous CSN5, but CSN5D151N was unable to rescue growth (Fig. 4D; data not shown). These results indicate that CSN5 isopeptidase activity is required for phenotypes of transformation in PHMLER cells.
The central role of the CDK inhibitor p27 in limiting cell cycle progression and its conserved regulation by CSN5 isopeptidase activity (13) suggested p27 as a possible target of the oncogenic effects of CSN5 (9, 10). Previous studies revealed that CSN5 is required in mammalian cells for the nuclear export and proteolytic degradation of p27 (20). We found that depletion of CSN5 alone or in combination with MYC increased nuclear p27 protein levels in PHMLER cells (Supplementary Fig. S5). Cell cycle arrest and loss of anchorage-independent growth induced by CSN5 depletion were specifically reversed by concomitant depletion of p27 (Supplementary Fig. S5), suggesting that a key role of CSN5 activation in breast epithelial transformation is the removal of CDK inhibitor p27 to enable unfettered cell proliferation.
CSN5 isopeptidase activity is required for de novo mammary cancer growth evoked by MYC and RAS in vivo
To dissect mechanisms of cancer progression in vivo in greater detail, we transduced genes of interest into primary mammary epithelial progenitor cells by retrovirus-mediated gene transfer followed by transplantation of transduced cells into cleared mammary fat pads to regenerate transgenic mammary glands and breast cancer development (Fig. 5A) as described previously (31). To control for hormonal and other environmental variations, we transplanted mammary epithelial cells transduced with different genes on opposite sides of the same host animal. Expression of MYC and oncogenic RASV12 in primary mammary progenitor cells gave rise to highly proliferative mammary adenocarcinomas in 100% of mice within 3 weeks (n = 15; Fig. 5B). In contrast, MYC overexpression alone induced ductal hyperplasia without invasion of epithelial cells beyond basement membrane, even after 3 months (n = 7; Fig. 5B), as previously observed (31).
If the collaboration of MYC and RAS in vivo leads to increased requirement of CSN5 isopeptidase activity, as in HMECs, then this rapidly invasive tumor model should be sensitive to inhibition by enforced expression of CSN5D151N. We compared coexpression of MYC and RAS with either vector or HA-tagged CSN5D151N. We found a strong tumor-suppressive effect of CSN5D151N as evidenced by both tumor size reduction and transgene-negative selection. Coexpression of CSN5D151N decreased the tumor size relative to the contralateral vector-expressing lesion (Fig. 5B). Analysis of GFP expression, which marks expression of HA-CSN5D151N or control vector, showed that whereas the vector remained strongly expressed in 90% of the control tumors, all tumors arising from cells transduced with CSN5D151N had greatly diminished or even extinguished expression of this transgene (Fig. 5C). This suggests that CSN5D151N expression was actively selected against, most likely due to the requirement of CSN5 catalytic activity for efficient cell proliferation in mammary cells (Fig. 4D). Further, there was a linear association between the level of HA-CSN5D151N expressed and the size of the resulting tumor: increasing HA-CSN5D151N expression was significantly correlated with reduced tumor size relative to the contralateral vector-expressing tumors (P = 0.01; Fig. 5D). Despite the incomplete CSN5D151N expression throughout the tumors, on average we still observed a 40% decrease in the size of RAS+MYC tumors upon expression of catalytically inactive CSN5 in tumors with HA-CSN5D151N expression (Fig. 5D).
As in PHMLER cells, we found that in vivo expression of CSN5D151N led to decreased proliferation but not increased apoptosis. Ki67 staining showed substantially decreased number of cycling cells in regions with CSN5D151N expression but not in regions where CSN5D151N was lost (Fig. 6A and B). Staining for cleaved caspase-3, a marker of apoptosis, showed no change in apoptosis on CSN5D151N expression (Fig. 6A). Histologic analysis further showed that whereas the RAS+MYC tumors were pleomorphic with areas of necrosis, and showed high nuclear to cytoplasmic ratio, CSN5D151N-expressing tumors were less necrotic and had significantly lower nuclear to cytoplasmic ratio (Fig. 6A and C; data not shown). Our previous microarray analysis identified the hypoxia-inducible transcription factor HIF-1α as a MYC target gene that was coactivated by CSN5 in human breast cells (6). Accordingly, expression of CSN5D151N in vivo decreased protein expression of HIF-1α and its target gene VEGF, a key factor in tumor angiogenesis (Supplementary Fig. S6; ref. 27). Taken together, these results suggest that the isopeptidase activity of CSN5 is essential for MYC-driven tumor progression in vivo.
Figure 6. CSN5D151N inhibits tumor cell proliferation and lowers tumor grade.
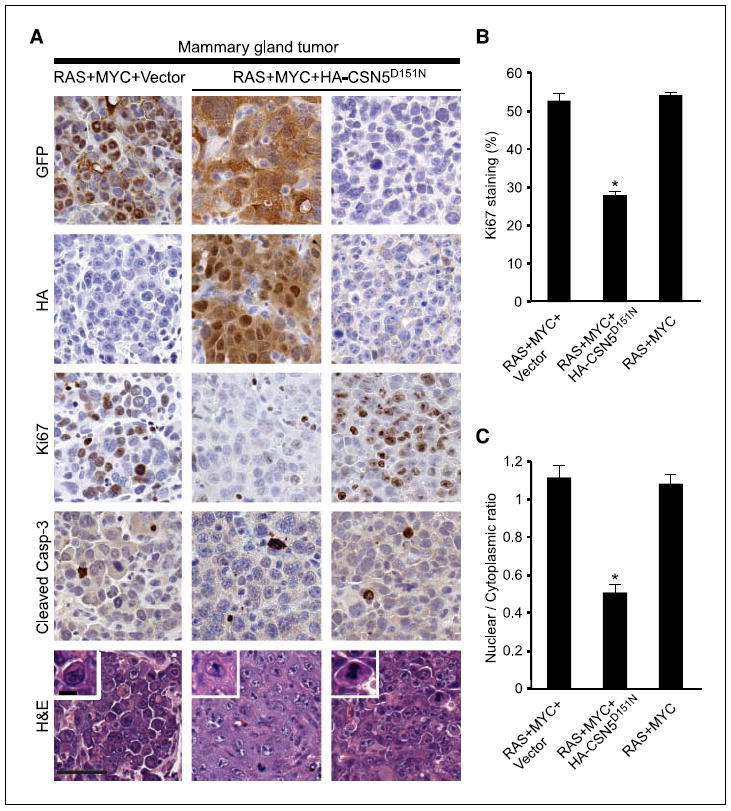
A, H&E staining and immunohistochemistry of the indicated proteins in RAS+MYC+Vector, RAS+MYC+HA-CSN5D151N, and RAS+MYC (CSN5D151N with no GFP or HA staining) tumors. Bar, 50 μm. Inset of H&E shows single cells magnified, highlighting the difference in nuclear to cytoplasmic ratios. Bar, 10 μm. B, quantification of Ki67-positive cells. Columns, mean; bars, SE. *, P < 10−6, Student’s t test, compared with all other samples. C, quantification of nuclear to cytoplasmic ratio. Columns, mean; bars, SE. *, P < 10−8, Student’s t test, compared with all other samples.
Discussion
In this report, we provide the first evidence that CSN5 isopeptidase activity is an essential component for human and mouse mammary epithelial transformation by defined genetic elements. Much like authentic human breast cancers, increased copy number of chromosome 8q encompassing both CSN5 and MYC arose early during the transformation process in HMEC cells. Attenuation of either CSN5 or MYC overexpression in PHMLER cells to the level found in HMEC greatly inhibited all features of transformation tested. These observations are congruent with our previous finding that overexpression of CSN5 and MYC in nontransformed breast epithelial cells collaborates to induce features of metastatic cancers (6). The loss-of-function analyses presented here, in conjunction with gain-of-function studies by us and other investigators (11, 20, 27), suggest that CSN5 activation is likely a key driver of epithelial transformation.
Our findings provide two new insights that assist the molecular dissection of cancer genomes. First, the functional interaction between CSN5 and MYC was initially predicted by genetic analysis of gene copy number aberrations linked with the poor-prognosis wound signature (6). The validation of a functional oncogenic and biochemical pathway connecting CSN5 to MYC ubiquitination, target gene activation, and MYC-dependent tumor growth suggests that integrated oncogenomic approaches (such as the SLAMS method) may be useful in deciphering mechanisms of cancer development. Second, the minimal genetic circuitry of human breast epithelial transformation is more complex than previously thought. Increased copy number of CSN5 and MYC is required to maintain the transformed phenotype of PHMLER cells in the face of dysregulated hTERT, Rb, p53, PP2A, and RAS. Detailed genomic studies of other “genetically defined” cancer models (42-45) may uncover key endogenous mutations that complement the transduced genes to achieve transformation.
Because of its frequent overexpression in human cancers, CSN5 has been suggested as a potential drug target (46). 8q amplification encompassing both CSN5 and MYC has been observed in additional tumor types, including prostate, pancreatic, and liver cancer (38, 47, 48), and in vitro studies suggested a role for CSN5 to regulate pancreatic cancer cell proliferation (11). Thus, CSN5 regulation of MYC and cell proliferation may be a common feature of many human cancers. Due to the large number of biochemical activities associated with CSN5 and the COP9 signalosome, it was previously unclear which CSN5 activity should be targeted. Our data suggest that CSN5 isopeptidase activity is a relevant target in breast cancer and potentially other types of human cancers. The isopeptidase activity of CSN5 resides in a zinc metalloproteinase-like domain termed the JAMM motif. The JAMM motif of CSN5 is an attractive therapeutic target because there is much experience in pharmacologic inhibition of metalloproteinases with small-molecule drugs. Indeed, >10 drugs have been approved for human use that target another zinc metalloproteinase, angiotensin-converting enzyme (including popular antihypertensive drugs such as captopril and benazepril; ref. 46). In contrast, inhibition of transcription factors or signaling proteins working via protein-protein interaction is challenging, and to date, no drug that specifically inhibits MYC has been developed. An intriguing possibility is that MYC-driven tumor progression may be blocked by inhibition of CSN5, a far more tractable drug target. The degree, duration, and cell specificity required for CSN5 blockade to attenuate cancer growth should be addressed in future studies.
Supplementary Material
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Acknowledgments
Grant support: National Cancer Institute grants CA118750 (H.Y. Chang) and CA072006 and CA057621 (Z. Werb), California Breast Cancer Research Program (H.Y. Chang and A.S. Adler), Dermatology Foundation Research Career Development Award (D. J. Wong), American Cancer Society (L.E. Littlepage and H.Y. Chang), and Ruth L. Kirschstein National Research Service Award CA103534-03 (L.E. Littlepage). H.Y. Chang is the Kenneth G. and Elaine A. Langone Scholar of the Damon Runyon Cancer Research Foundation.
We thank Robert Weinberg, Paul Khavari, Xin Chen, Dean Felsher, Jason Shohet, Suwon Kim, Yosef Refaeli, Alex Swarbrick, Alana Welm, and Garry Nolan for reagents and Anthony Oro for comments on the manuscript.
Footnotes
Gene Expression Omnibus series accession number GSE9206, http://www.ncbi.nlm.nih.gov/geo/.
References
- 1.Robanus-Maandag EC, Bosch CA, Kristel PM, et al. Association of C-MYC amplification with progression from the in situ to the invasive stage in C-MYC-amplified breast carcinomas. J Pathol. 2003;201:75–82. doi: 10.1002/path.1385. [DOI] [PubMed] [Google Scholar]
- 2.Tirkkonen M, Tanner M, Karhu R, Kallioniemi A, Isola J, Kallioniemi OP. Molecular cytogenetics of primary breast cancer by CGH. Genes Chromosomes Cancer. 1998;21:177–84. [PubMed] [Google Scholar]
- 3.Elenbaas B, Spirio L, Koerner F, et al. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 2001;15:50–65. doi: 10.1101/gad.828901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang HY, Sneddon JB, Alizadeh AA, et al. Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol. 2004;2:E7. doi: 10.1371/journal.pbio.0020007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang HY, Nuyten DS, Sneddon JB, et al. Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proc Natl Acad Sci U S A. 2005;102:3738–43. doi: 10.1073/pnas.0409462102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adler AS, Lin M, Horlings H, Nuyten DS, van de Vijver MJ, Chang HY. Genetic regulators of large-scale transcriptional signatures in cancer. Nat Genet. 2006;38:421–30. doi: 10.1038/ng1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pelengaris S, Khan M, Evan G. c-MYC: more than just a matter of life and death. Nat Rev Cancer. 2002;2:764–76. doi: 10.1038/nrc904. [DOI] [PubMed] [Google Scholar]
- 8.Richardson KS, Zundel W. The emerging role of the COP9 signalosome in cancer. Mol Cancer Res. 2005;3:645–53. doi: 10.1158/1541-7786.MCR-05-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esteva FJ, Sahin AA, Rassidakis GZ, et al. Jun activation domain binding protein 1 expression is associated with low p27(Kip1)levels in node-negative breast cancer. Clin Cancer Res. 2003;9:5652–9. [PubMed] [Google Scholar]
- 10.Kouvaraki MA, Rassidakis GZ, Tian L, Kumar R, Kittas C, Claret FX. Jun activation domain-binding protein 1 expression in breast cancer inversely correlates with the cell cycle inhibitor p27(Kip1) Cancer Res. 2003;63:2977–81. [PubMed] [Google Scholar]
- 11.Fukumoto A, Tomoda K, Yoneda-Kato N, Nakajima Y, Kato JY. Depletion of Jab1 inhibits proliferation of pancreatic cancer cell lines. FEBS Lett. 2006;580:5836–44. doi: 10.1016/j.febslet.2006.09.042. [DOI] [PubMed] [Google Scholar]
- 12.Bech-Otschir D, Seeger M, Dubiel W. The COP9 signalosome: at the interface between signal transduction and ubiquitin-dependent proteolysis. J Cell Sci. 2002;115:467–73. doi: 10.1242/jcs.115.3.467. [DOI] [PubMed] [Google Scholar]
- 13.Cope GA, Deshaies RJ. COP9 signalosome: a multifunctional regulator of SCF and other cullin-based ubiquitin ligases. Cell. 2003;114:663–71. doi: 10.1016/s0092-8674(03)00722-0. [DOI] [PubMed] [Google Scholar]
- 14.Doronkin S, Djagaeva I, Beckendorf SK. CSN5/Jab1 mutations affect axis formation in the Drosophila oocyte by activating a meiotic checkpoint. Development. 2002;129:5053–64. doi: 10.1242/dev.129.21.5053. [DOI] [PubMed] [Google Scholar]
- 15.Oron E, Mannervik M, Rencus S, et al. COP9 signalosome subunits 4 and 5 regulate multiple pleiotropic pathways in Drosophila melanogaster. Development. 2002;129:4399–409. doi: 10.1242/dev.129.19.4399. [DOI] [PubMed] [Google Scholar]
- 16.Tomoda K, Yoneda-Kato N, Fukumoto A, Yamanaka S, Kato JY. Multiple functions of Jab1 are required for early embryonic development and growth potential in mice. J Biol Chem. 2004;279:43013–8. doi: 10.1074/jbc.M406559200. [DOI] [PubMed] [Google Scholar]
- 17.Cope GA, Suh GS, Aravind L, et al. Role of predicted metalloprotease motif of Jab1/Csn5 in cleavage of Nedd8 from Cul1. Science. 2002;298:608–11. doi: 10.1126/science.1075901. [DOI] [PubMed] [Google Scholar]
- 18.Wee S, Geyer RK, Toda T, Wolf DA. CSN facilitates Cullin-RING ubiquitin ligase function by counteracting autocatalytic adapter instability. Nat Cell Biol. 2005;7:387–91. doi: 10.1038/ncb1241. [DOI] [PubMed] [Google Scholar]
- 19.Cope GA, Deshaies RJ. Targeted silencing of Jab1/Csn5 in human cells downregulates SCF activity through reduction of F-box protein levels. BMC Biochem. 2006;7:1. doi: 10.1186/1471-2091-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tomoda K, Kubota Y, Kato J. Degradation of the cyclin-dependent-kinase inhibitor p27Kip1 is instigated by Jab1. Nature. 1999;398:160–5. doi: 10.1038/18230. [DOI] [PubMed] [Google Scholar]
- 21.O’Hagan RC, Ohh M, David G, et al. Myc-enhanced expression of Cul1 promotes ubiquitin-dependent proteolysis and cell cycle progression. Genes Dev. 2000;14:2185–91. doi: 10.1101/gad.827200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tomoda K, Kubota Y, Arata Y, et al. The cytoplasmic shuttling and subsequent degradation of p27Kip1 mediated by Jab1/CSN5 and the COP9 signalosome complex. J Biol Chem. 2002;277:2302–10. doi: 10.1074/jbc.M104431200. [DOI] [PubMed] [Google Scholar]
- 23.Sutterluty H, Chatelain E, Marti A, et al. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat Cell Biol. 1999;1:207–14. doi: 10.1038/12027. [DOI] [PubMed] [Google Scholar]
- 24.Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999;1:193–9. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- 25.Tsvetkov LM, Yeh KH, Lee SJ, Sun H, Zhang H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr Biol. 1999;9:661–4. doi: 10.1016/s0960-9822(99)80290-5. [DOI] [PubMed] [Google Scholar]
- 26.Bae MK, Ahn MY, Jeong JW, et al. Jab1 interacts directly with HIF-1α and regulates its stability. J Biol Chem. 2002;277:9–12. doi: 10.1074/jbc.C100442200. [DOI] [PubMed] [Google Scholar]
- 27.Bemis L, Chan DA, Finkielstein CV, et al. Distinct aerobic and hypoxic mechanisms of HIF-α regulation by CSN5. Genes Dev. 2004;18:739–44. doi: 10.1101/gad.1180104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hallstrom TC, Nevins JR. Jab1 is a specificity factor for E2F1-induced apoptosis. Genes Dev. 2006;20:613–23. doi: 10.1101/gad.1345006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schweitzer K, Bozko PM, Dubiel W, Naumann M. CSN controls NF-κB by deubiquitinylation of IκBα. EMBO J. 2007;26:1532–41. doi: 10.1038/sj.emboj.7601600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Slack A, Chen Z, Tonelli R, et al. The p53 regulatory gene MDM2 is a direct transcriptional target of MYCN in neuroblastoma. Proc Natl Acad Sci U S A. 2005;102:731–6. doi: 10.1073/pnas.0405495102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Welm AL, Kim S, Welm BE, Bishop JM. MET and MYC cooperate in mammary tumorigenesis. Proc Natl Acad Sci U S A. 2005;102:4324–9. doi: 10.1073/pnas.0500470102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chi JT, Chang HY, Wang NN, Chang DS, Dunphy N, Brown PO. Genomewide view of gene silencing by small interfering RNAs. Proc Natl Acad Sci U S A. 2003;100:6343–6. doi: 10.1073/pnas.1037853100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim BC, Lee HJ, Park SH, et al. Jab1/CSN5, a component of the COP9 signalosome, regulates transforming growth factor β signaling by binding to Smad7 and promoting its degradation. Mol Cell Biol. 2004;24:2251–62. doi: 10.1128/MCB.24.6.2251-2262.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pear WS, Nolan GP, Scott ML, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci U S A. 1993;90:8392–6. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang HY, Chi JT, Dudoit S, et al. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci U S A. 2002;99:12877–82. doi: 10.1073/pnas.162488599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sage J, Miller AL, Perez-Mancera PA, Wysocki JM, Jacks T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature. 2003;424:223–8. doi: 10.1038/nature01764. [DOI] [PubMed] [Google Scholar]
- 37.Pollack JR, Perou CM, Alizadeh AA, et al. Genomewide analysis of DNA copy-number changes using cDNA microarrays. Nat Genet. 1999;23:41–6. doi: 10.1038/12640. [DOI] [PubMed] [Google Scholar]
- 38.Bashyam MD, Bair R, Kim YH, et al. Array-based comparative genomic hybridization identifies localized DNA amplifications and homozygous deletions in pancreatic cancer. Neoplasia. 2005;7:556–62. doi: 10.1593/neo.04586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ginzinger DG, Godfrey TE, Nigro J, et al. Measurement of DNA copy number at microsatellite loci using quantitative PCR analysis. Cancer Res. 2000;60:5405–9. [PubMed] [Google Scholar]
- 40.Ambroggio XI, Rees DC, Deshaies RJ. JAMM: a metalloprotease-like zinc site in the proteasome and signalosome. PLoS Biol. 2004;2:E2. doi: 10.1371/journal.pbio.0020002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim SY, Herbst A, Tworkowski KA, Salghetti SE, Tansey WP. Skp2 regulates Myc protein stability and activity. Mol Cell. 2003;11:1177–88. doi: 10.1016/s1097-2765(03)00173-4. [DOI] [PubMed] [Google Scholar]
- 42.Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–8. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 43.Rich JN, Guo C, McLendon RE, Bigner DD, Wang XF, Counter CM. A genetically tractable model of human glioma formation. Cancer Res. 2001;61:3556–60. [PubMed] [Google Scholar]
- 44.Lundberg AS, Randell SH, Stewart SA, et al. Immortalization and transformation of primary human airway epithelial cells by gene transfer. Oncogene. 2002;21:4577–86. doi: 10.1038/sj.onc.1205550. [DOI] [PubMed] [Google Scholar]
- 45.Gupta PB, Kuperwasser C, Brunet JP, et al. The melanocyte differentiation program predisposes to metastasis after neoplastic transformation. Nat Genet. 2005;37:1047–54. doi: 10.1038/ng1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nalepa G, Wade Harper J. Therapeutic anti-cancer targets upstream of the proteasome. Cancer Treat Rev. 2003;29(Suppl 1):49–57. doi: 10.1016/s0305-7372(03)00083-5. [DOI] [PubMed] [Google Scholar]
- 47.Lapointe J, Li C, Giacomini CP, et al. Genomic profiling reveals alternative genetic pathways of prostate tumorigenesis. Cancer Res. 2007;67:8504–10. doi: 10.1158/0008-5472.CAN-07-0673. [DOI] [PubMed] [Google Scholar]
- 48.Patil MA, Gutgemann I, Zhang J, et al. Array-based comparative genomic hybridization reveals recurrent chromosomal aberrations and Jab1 as a potential target for 8q gain in hepatocellular carcinoma. Carcinogenesis. 2005;26:2050–7. doi: 10.1093/carcin/bgi178. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).