

Abstract
Objectives
Calcium phosphate cement (CPC) is a promising material for dental, periodontal, and craniofacial repairs. However, its use requires on-site powder–liquid mixing that increases the surgical placement time and raises concerns of insufficient and inhomogeneous mixing. The objective of this study was to determine a formulation of premixed CPC (PCPC) with rapid setting, high strength, and good in vitro cell viability.
Methods
PCPCs were formulated from CPC powder + non-aqueous liquid + gelling agent + hardening accelerator. Five PCPCs were thus developed: PCPC-Tartaric, PCPC-Malonic, PCPC-Citric, PCPC-Glycolic, and PCPC-Malic. Formulations and controls were compared for setting time, diametral tensile strength, and osteoblast cell compatibility.
Results
Setting time (mean ± S.D.; n = 4) for PCPC-Tartaric was 8.2 ± 0.8 min, significantly less than the 61.7 ± 1.5 min for the Premixed Control developed previously (p < 0.001). On 7th day immersion, the diametral tensile strength of PCPC-Tartaric reached 6.5 ± 0.8 MPa, higher than 4.5 ± 0.8 MPa of Premixed Control (p = 0.036). Osteoblast cells displayed a polygonal morphology and attached to the nano-hydroxyapatite crystals in the PCPCs. All cements had similar live cell density values (p = 0.126), indicating that the new PCPCs were as cell compatible as a non-premixed CPC control known to be biocompatible. Each of the new PCPCs had a cell viability that was not significantly different (p > 0.1) from that of the non-premixed CPC control.
Significance
PCPCs will eliminate the powder–liquid mixing during surgery and may also improve the cement performance. The new PCPCs supported cell attachment and yielded a high cell density and viability. Their mechanical strengths approached the reported strengths of sintered porous hydroxyapatite implants and cancellous bone. These nano-crystalline hydroxyapatite cements may be useful in dental, periodontal, and craniofacial repairs.
Keywords: Premixed calcium phosphate, cement, Hydroxyapatite, Osteoblast cytotoxicity, Dental restorations, Craniofacial repair
1. Introduction
Hydroxyapatite is an important biomaterial because of its similarity to the apatitic mineral in natural teeth and bones [1,2]. A calcium phosphate cement (CPC) was developed with the advantage of being moldable and capable of in situ setting to form hydroxyapatite [3]. Several different cement compositions were developed [4–7]. The CPC powder consisted of tetracalcium phosphate (TTCP), Ca4(PO4)2O, and dicalcium phosphate anhydrous (DCPA), CaHPO4; when mixed with water at a powder:liquid ratio of 4:1, the paste hardened in about 30 min and formed hydroxyapatite [3,8,9]. CPC showed excellent biocompatibility and osteoconductivity, and was able to be resorbed and replaced by new bone [8,9]. It was approved in 1996 by the Food and Drug Administration for repairing craniofacial defects in humans, thus becoming the first CPC available for clinical use [9].
CPC showed promise for several dental applications, including root canal filler/sealer [10,11] and base applications [12]. CPC-containing dental composites were also developed [13]. Unlike traditional composites with glassy fillers [14–17], the CPC powder as fillers in resins resulted in the release of Ca and PO4 ions [13]. These composites showed potential for pulp capping and cavity lining applications, and remineralized in vitro the demineralized dentin [13]. Recently, CPC was also combined with a biopolymer chitosan to yield a strong and non-rigid hydroxyapatite composite envisioned for periodontal bone repair [18–20].
One disadvantage of CPC was that the clinician needed to mix the powder and liquid components thoroughly and place the paste into the defect before the paste hardens. The requirement of on-site powder–liquid mixing had drawbacks [21,22]. First, it increased the total surgical placement time. Second, it raised concerns about insufficient and inhomogeneous mixing, thus compromising the implant performance. The third issue was that all the individual components of the material and the equipment needed to be sterilized, and the mixing needed to be performed in a sterile environment. A pre-apportioned, automated mixing gun did not work well because of the CPC powder component. The powder/liquid encapsulation and automated mixing method yielded a paste of limited quantity that might be too small for many craniofacial repairs.
These difficulties prompted the development of a premixed CPC (PCPC) [21]. The CPC powder was mixed with a non-aqueous, but water-miscible liquid carrier in advance under well-controlled conditions. This water-free paste did not harden in storage in a syringe because CPC hardens only when exposed to an aqueous environment. After this paste was placed in contact with a physiological solution, exchange of the non-aqueous carrier by the aqueous solution led to cement hardening. However, the previous PCPC had a setting time >1 h and a low strength [21]. A long setting time could cause problems clinically because of the cement’s inability to maintain shape and support stresses within this time period [23]. Recently, a new PCPC was developed with the incorporation of tartaric acid and exhibited rapid setting and a high strength [22]. However, this PCPC exhibited a high level of cytotoxicity and caused cell death in vitro [22].
The objective of the present study was to develop a formulation of premixed CPC to harden rapidly, and to be mechanically strong and non-cytotoxic. The hypotheses to be tested were: (i) PCPC could be formulated via the method of “CPC powder + non-aqueous liquid + gelling agent + hardening accelerator” to possess fast-setting and high strength, with no adverse effect on the attachment and viability of osteoblast cells; and (ii) different hardening accelerators would significantly affect the cement mechanical and physical properties and osteoblast cytotoxicity.
2. Materials and methods
2.1. CPC powder
The preparation of the TTCP and DCPA powders was described previously [3,19]. The size of the TTCP particles ranged from 1 to 80 μm with an average of 17 μm. The size of the DCPA particles ranged from 0.4 to 3 μm with an average of 1 μm. The TTCP and DCPA powders were mixed at a molar ratio of 1:1 to form the CPC powder.
2.2. Premixed CPC liquid
The liquid consisted of a non-aqueous liquid, a gelling agent, and a hardening accelerator. The non-aqueous liquid was poly(ethylene glycol) (PEG) (400, Sigma, St. Louis, MO), selected because it is non-toxic and used in biomedical applications such as drugs [24]. The gelling agent was hydroxypropyl methylcellulose (HPMC, Sigma). It is a derivative of cellulose which is one of the most common polysaccharides, and is known for its gelling ability to form viscous solutions which improved the washout resistance of CPC [21,25].
In addition to using tartaric acid as the hardening accelerator, several other organic acids were also evaluated: malic acid, malonic acid, citric acid, and glycolic acid. Table 1 lists these acids and their manufacturers. These acids were chosen for two reasons. First, they are non-toxic and are used in various foods, drinks and beauty products [26]. Second, they imparted rapid hardening to PCPC in preliminary studies. These acids can react with calcium from the dissolution of TTCP and DCPA to form calcium salts, which causes the cement to harden [27].
Table 1.
Compositions (mass fraction) of cement liquid for premixed CPC (PCPC)a
| PCPC-Tartaric | PCPC-Malonic | PCPC-Citric | PCPC-Glycolic | PCPC-Malic | |
|---|---|---|---|---|---|
| Non-aqueous liquid (% PEG) | 62.2 | 58.1 | 42.1 | 61.7 | 59.7 |
| Gelling agent (% HPMC) | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 |
| Hardening accelerator | 37.5% Tartaric acid | 41.6% Malonic acid | 57.6% Citric acid | 38.0% Glycolic acid | 40.0% Malic acid |
PCPC = CPC powder + cement liquid. Cement liquid = Nonaqueous liquid + gelling agent + hardening accelerator. PEG = poly(ethylene glycol) (400, Sigma, St. Louis, MO). HPMC = hydroxypropyl methylcellulose (Sigma). Tartaric acid (C4H6O6, Sigma). Malonic acid (C3H4O4, ACROS Organics, Morris Plain, NJ). Citric acid (C6H8O7, Fisher, Fairlawn, NJ). Glycolic acid (C2H4O3, Sigma). Malic acid (C4H6O5, Sigma).
2.3. Fabrication of PCPC specimens
Five different PCPCs were formulated by combining the same CPC powder with the five different liquids (Table 1). In preliminary studies, to achieve rapid setting, the optimum concentrations for tartaric acid, malonic acid, citric acid, glycolic acid, and malic acid were determined to be approximately 4, 7, 7, 8, and 5 mol/L, respectively. These concentrations resulted in the mass fractions listed in Table 1. The concentration 0.3% of HPMC was selected because a lower concentration did not have a sufficient gelling effect while high concentrations retarded the CPC conversion to hydroxyapatite in preliminary studies. A CPC powder:liquid mass ratio of 4:1 was used. The PCPCs were referred to as PCPC-Tartaric, PCPC-Malonic, PCPC-Citric, PCPC-Glycolic, and PCPC-Malic.
A sixth premixed CPC, developed in a previous study [21], was used as a control for physical and mechanical properties. It was designated as “Premixed Control”. Its powder consisted of the same CPC powder; its liquid consisted of 69.45% glycerol, 0.55% HPMC, and 30% Na2HPO4 [21]. The powder:liquid mass ratio was 4:1.
2.4. Hardening time
The premixed paste was placed into a mold of 6 mm diameter and 3 mm depth, and sandwiched between two porous glass slides (ACE Glass, Vineland, NJ). The assembly was immersed in a simulated physiological solution (1.15 mmol/L Ca, 1.2 mmol/L P, 133 mmol/L NaCl, 50 mmol/L HEPES, buffered to a pH of 7.4) at 37 °C [6]. The cement was considered set when a Gilmore needle with a tip diameter of 1.06 mm loaded onto the specimen under a mass of 453.5 g failed to leave a perceptible indentation [22,28]. The time from the paste being immersed to this point was used as the setting time (estimated uncertainty = ±0.5 min based on the time interval in the measurement).
2.5. Conversion to hydroxyapatite
Specimens were immersed in the physiological solution for 1, 3, and 7 days, and then milled into powder by mortar and pestle. A 6 × 3 full factorial design was used with six PCPCs and the three immersion times. A powder X-ray diffractometer (Rigaku, Danvers, MA) was used with graphite-monochromatized copper Kα radiation (λ = 0.154 nm) generated at 40 kV and 40 mA [29]. To measure the hydroxyapatite conversion, a series of samples with known amounts of hydroxyapatite (from 100% converted CPC) and known amounts of unreacted CPC powder were prepared. For example, one sample consisted of the hydroxyapatite powder mixed with the unreacted CPC powder at a hydroxyapatite:unreacted CPC mass ratio of 25:75. X-ray diffraction (XRD) patterns of these samples were obtained from the same amount of the sample packed in a sample holder. The XRD patterns were imported into a “PeakFit” software (SeaSolve Software Inc., Framingham, MA) for peaks separation. The curves were smoothed with Gauss convolution and the peaks were separated using linear baseline and Gauss area. The intensities obtained for the resolved peaks were used to construct a standard curve that described the relationship between the mass fractions of hydroxyapatite and the intensities of the (0 0 2) peak of hydroxyapatite [5,29]. Hydroxyapatite conversion for a PCPC was obtained using the standard curve and the determined (0 0 2) peak intensity (PeakFit) from the raw XRD pattern of the PCPC.
2.6. Mechanical properties
Specimens of 6 mm diameter and 3 mm thickness [22] were immersed in the physiological solution for 1, 3, and 7 days. The same 6 × 3 design as in Section 2.5 was used. Diametral tensile strength (DTS) (estimated standard uncertainty was 3% based on the accuracy in the specimen dimension measurement) of the cement specimens was measured on a computer-controlled Universal Testing Machine (5500R, MTS, Cary, NC) at a crosshead speed of 1 mm/min [22]. A sheet of filter paper (Whatman Type #1, Whatman International, Spring-field Mill, Maidstone, Kent, England) was placed underneath, and another sheet was placed on the top of the cement specimen during the loading.
2.7. Cell attachment and live/dead staining
MC3T3-E1 mouse osteoblast cells (Riken, Hirosaka, Japan) were cultured at 100% humidity in α-modified Eagle’s minimum essential medium (Biowhittaker, Walkersville, MD) [30–33]. Seven materials were tested: the five new PCPCs, the non-premixed Conventional CPC Control, and the tissue culture polystyrene (TCPS) wells without CPC serving as TCPS Control. Conventional CPC, with water as the liquid at a powder:liquid ratio 4:1, was used as a control for cell culture because of its known biocompatibility [9]. Cement specimens with dimensions of 3 mm × 4 mm × 12 mm, similar to those in a previous study [22], were set in the simulated physiologic solution as in Section 2.4 for 3 days. The set specimens were sterilized by autoclaving at 121 °C for 20 min [33].
Fifty thousand cells diluted into 2 mL of media were added to each well (24-well Falcon plate, circular wells, BD Biosciences, San Jose, CA) containing a specimen and incubated for 1 day [32,33]. Each well was approximately 17 mm deep and had a diameter of 16 mm. Thus, cells were seeded both on top and around the cement specimens. It was not necessary to restrict the cell attachment to the cement specimen only; cells were able to attach to the specimen as well as to the exposed TCPS areas around the specimen. Staining was done with 1 mL of cell media containing 2 μmol/L calcein-AM and 2 μmol/L ethidium homodimer-1 (Molecular Probes, Eugene, OR). The live cells were stained green and the dead cells were stained red [32,33]. The stained cells were examined using epifluorescence microscopy (Eclipse TE300, Nikon, Melville, NY). Calcein-AM is a non-fluorescent, cell-permeant fluorescein derivative that is converted by cellular enzymes into cell-impermeant and highly fluorescent calcein. Calcein accumulates inside live cells having intact membranes causing them to fluoresce green. Ethidium-homodimer-1 enters dead cells with damaged membranes and undergoes a 40-fold enhancement of fluorescence upon binding to their DNA causing the nuclei of dead cells to fluoresce red. To estimate the live cell density, two randomly chosen fields of view were photographed for each specimen through a green filter and a red filter using a 10× objective (100× magnification), yielding four pictures from each specimen. Each image had an area of 1.8 mm2 and each specimen had a top surface area of approximately 48 mm2. Thus, approximately 8% of the top surface of each specimen was imaged. Five specimens (n = 5) for each of the six cements yielded a total of 120 images. Each image was printed and the live (green) and dead (red) cells were counted. The percent of live cells = the number of live cells/(the number of live cells + the number of dead cells) [22].
2.8. Quantitative cell viability
The same cells as in Section 2.7 were seeded with 10,000 cells per well in 2 mL of media into a 24-well plate. Cell attachment was observed to be uniform and even in all cases. In a separate 24-well plate, each cement specimen was immersed in a well with 2 mL of fresh medium without cells and extracted overnight in the incubator to accumulate any possible harmful leachout in the medium [22]. Each cement specimen had dimensions of approximately 3 mm × 4 mm × 12 mm (volume = 0.144 mm3) similar to that of a previous extraction study [22], yielding: specimen volume/culture medium volume = 7.2%. After 24 h, the medium from each well containing the cells was removed and replaced with 2 mL of extraction medium containing any harmful leachout from the cement. Note that cells for this assay were seeded directly onto the bottom of the tissue culture polystyrene wells and were never in direct contact with the cement specimens. Cells only had contact with extracts from the cement specimens.
Seven materials were tested: the five new PCPCs, and the Conventional CPC and the TCPS controls. The cells were incubated in the extracts for 3 days. Digital photography with a phase contrast microscope (Nikon TE300, Melville, NY) was used to examine the cells (10× objective, 100× magnification). Cell viability was measured by using the Wst-1 assay [32,33]. This is a colorimetric assay of mitochondrial dehydrogenase activity where the absorbance at 450 nm is proportional to the amount of dehydrogenase activity in the cell. Specimens with cells were transferred to a new 24-well plate. One milliliter of Tyrode’s HEPES buffer (140 mmol/L NaCl, 0.34 mmol/L Na2HPO4, 2.9 mmol/L KCl, 10 mmol/L Hepes, 12 mmol/L NaHCO3, 5 mmol/L glucose, pH 7.4) and 0.1 mL of Wst-1 solution were then added to each well. After a 2-h incubation, a 0.2 mL aliquot from each well was transferred to a 96-well plate and the absorbance at 450 nm was measured with a platereader (Perkin-Elmer, Gaithersburg, MD) [32,33].
A scanning electron microscope (SEM, JEOL 5300, Peabody, MA) was used to examine the specimens. Cells cultured for 1 day while anchored onto the cement specimens were rinsed with saline, fixed with 1% volume fraction of glutaraldehyde, subjected to graded alcohol dehydrations, rinsed with hexamethyldisilazane, and sputter coated with gold.
Two-way and one-way ANOVA were performed to detect significant effects of the variables. Tukey’s multiple comparison test was used with p ≤ 0.05 to compare the formulations.
3. Results
The cement setting time (mean ± S.D.; n = 4) was measured to be 8.2 ± 0.8, 17.0 ± 0.8, 40.3 ± 3.1, 23.3 ± 1.0, 1.0 ± 0.5, and 61.7 ± 1.6, for PCPC-Tartaric, PCPC-Malonic, PCPC-Citric, PCPC-Malic, PCPC-Glycolic, and Premixed Control, respectively. These values are significantly different from each other (p < 0.05). All the new PCPCs with various organic acids as hardening accelerators hardened much faster than the Premixed Control.
The diametral tensile strength (mean ± S.D.; n = 6) results are plotted in Fig. 1. Most cements showed moderate strength increases upon increasing the immersion time (p < 0.001). Different types of materials also had a significant effect on the strength values (p < 0.001). Between different materials, on 7th day, the strength of PCPC-Tartaric was 6.5 ± 0.8 MPa, not significantly different from 4.7 ± 1.0 MPa of PCPC-Malonic (p = 0.135) and 5.3 ± 2.0 MPa of PCPC-Malic (p = 0.579). The strength of PCPC-Tartaric was significantly higher than 3.5 ± 0.6 MPa of PCPC-Citric (p < 0.001), 4.0 ± 1.3 MPa of PCPC-Glycolic (p < 0.001), and 4.5 ± 0.8 MPa of Premixed Control (p = 0.036).
Fig. 1.
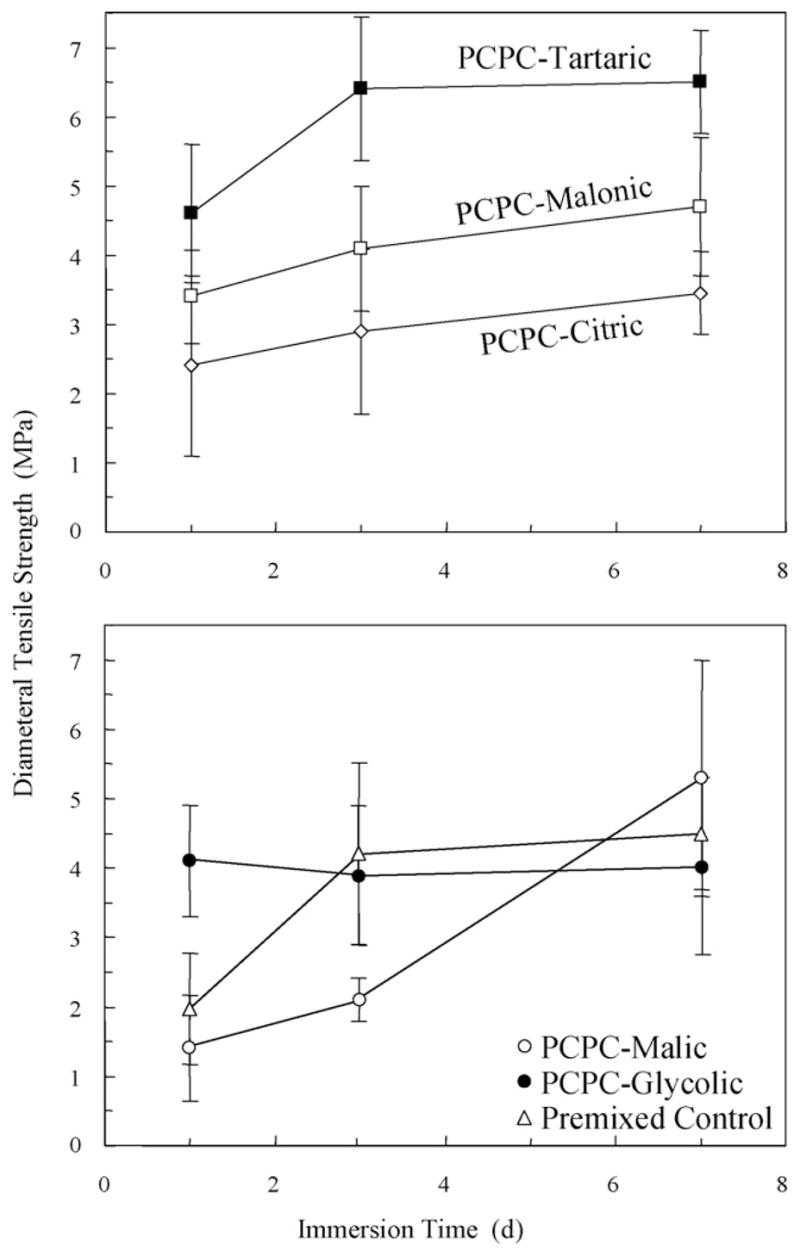
Diametral tensile strength. Each value is the mean of six measurements with the error bar showing one standard deviation (mean ± S.D.; n = 6). Both material type and immersion time had significant effects on the strength values (p < 0.001).
Fig. 2 plots the mass percentages of CPC converted to hydroxyapatite. The six cements had initial (1 day) hydroxyapatite conversions of about 20–30%. With further increase in the immersion time, some materials had more substantial increases in the hydroxyapatite conversion than others. On 7th day, the hydroxyapatite conversion (mean ± S.D.; n = 3) was 78.5 ± 3.5%, 70.3 ± 2.5%, 66.0 ± 1.4%, 50.6 ± 1.5%, 45.1 ± 0.8%, and 33.4 ± 1.4%, for Premixed Control, PCPC-Glycolic, PCPC-Malic, PCPC-Tartaric, PCPC-Malonic, and PCPC-Citric, respectively. They are all significantly different from each other (p < 0.05), except between PCPC-Glycolic and PCPC-Malic (p = 0.067).
Fig. 2.
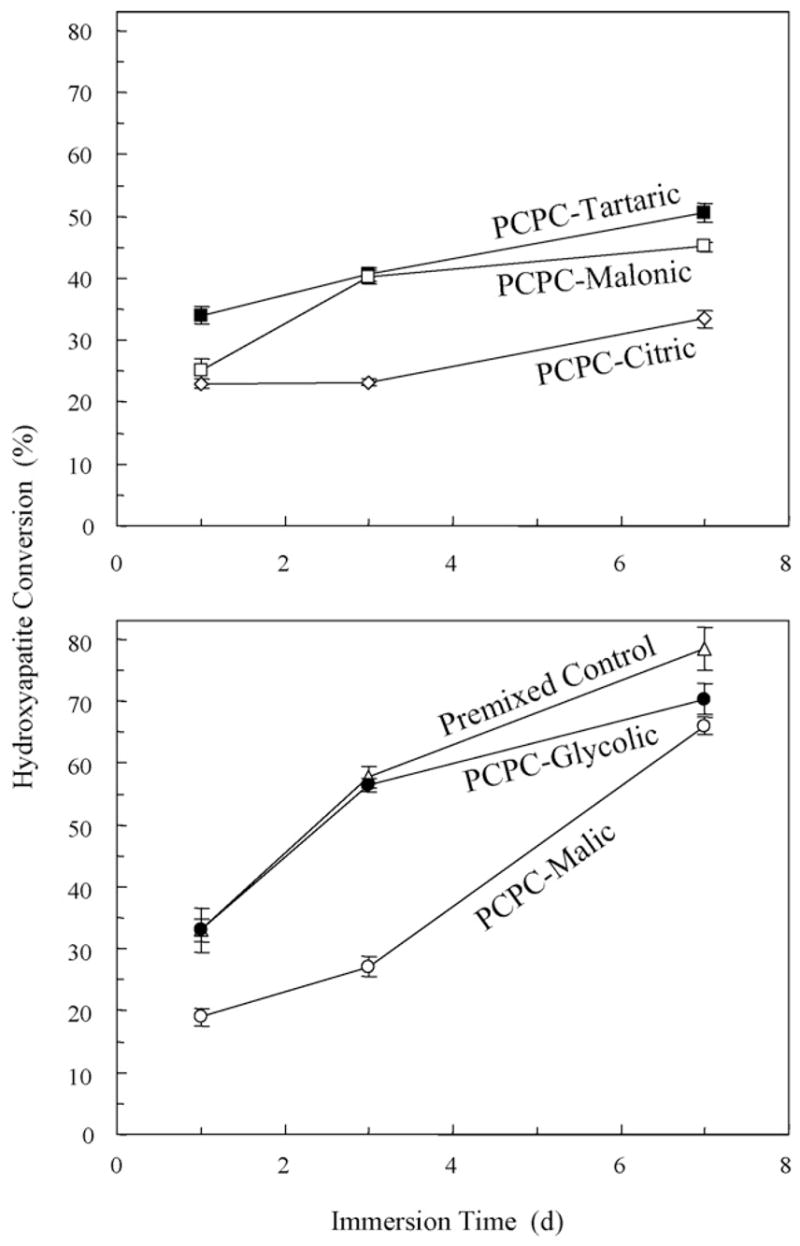
The mass percentages of CPC conversion to hydroxyapatite. Each value is the mean of six measurements with the error bar showing one standard deviation (mean ± S.D.; n = 3).
Cells were seeded onto the cement specimens, incubated for 1 day, and observed with fluorescence microscopy. Cell attachment to the specimens was even and uniform in most cases. In Fig. 3, cells cultured on Conventional CPC Control and PCPC-Tartaric are shown as examples. Epifluorescence microscopy showed that the live cells, stained green, appeared to have attained a polygonal morphology on all six cements. Dead cells, stained red, were few on all cements. The live cell density is plotted in (E) for the six cements and the TCPS Control. The horizontal line in (E) shows that these values are not significantly different (p = 0.126), indicating that the new PCPCs were as non-cytotoxic as the TCPS and the Conventional CPC controls.
Fig. 3.

Cells seeded on the cements and cultured for 1 day. Examples are shown for Conventional CPC Control (known to be biocompatible) and PCPC-Tartaric. Other PCPCs and TCPS Control had similar features and are not shown here. Live cells were stained green and had developed a normal polygonal morphology. Dead cells were stained red and were very few on all the materials. In (E), the six cements and the tissue culture polystyrene (TCPS) Control had similar live cell density values (p = 0.126), as indicated by the horizontal line.
Fig. 4 shows the SEM micrographs of cells cultured for 1 day. The body of the osteoblast “O” had a size of approximately 20 μm, but it had developed cytoplasmic extensions “E” with lengths up to 50 μm. These protruding extensions, also termed lamellipodia, are regions of the plasma membrane that contain a meshwork or bundles of actin-containing microfilaments, which permit the migration of cells along a substratum [34]. Cell–cell junctions (“J” in C) were also formed. In (B) and (D), secondary extensions with a diameter of 100–300 nm were sprouted near the tips of “E” (arrows). They appeared to have firmly anchored on the cement surfaces. Similar cell spreading and attachment were observed on all six cements.
Fig. 4.

SEM of cells seeded on the cements and cultured for 1 day. Examples are shown for Conventional CPC Control (A and B) and PCPC-Tartaric (C and D). Other cements had similar features and are not shown here. O, osteoblast cell; E, cytoplasmic extension or lamellipodium; J, cell–cell junction. Arrows in (B and D) indicate secondary extensions anchored on the cements.
Fig. 5 shows an example of cells at a higher magnification attaching to a PCPC-Tartaric specimen. Similar features were observed for the other cements. The cytoplasmic extensions were anchored on the nano-sized hydroxyapatite crystals (arrows). These were likely to be hydroxyapatite because these nano-sized crystals were not present in the starting CPC powder (which had TTCP particles of about 17 μm and DCPA particles of 1 μm), and XRD detected the formation of hydroxyapatite. While the sizes of the crystals differed in different cements, they generally had elongated shapes with diameters ranging from about 50 to 300 nm. The sizes and shapes of the crystals were similar to the nano-hydroxyapatite crystals from CPC conversion observed in previous studies [29].
Fig. 5.
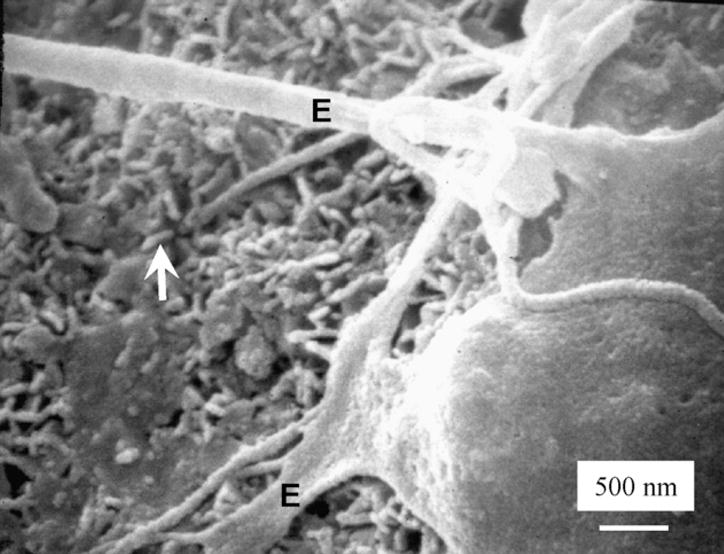
SEM at a higher magnification showing extensions of the cells anchoring on the nano-sized hydroxyapatite crystals (arrows). This example is shown for PCPC-Tartaric. Similar features were observed for other PCPCs.
Fig. 6 plots the quantitative cell viability results measured using the Wst-1 assay which is a colorimetric assay of dehydrogenase activity quantified by the absorbance at 450 nm. Dissimilar letters at the upper right corner of each bar indicate values that are significantly different (p < 0.05). The TCPS Control had a moderately higher value than most of the cements. Each of the five new PCPCs had a cell viability that was not significantly different (p > 0.1) from the cell viability of the FDA approved, non-premixed Conventional CPC Control.
Fig. 6.

Quantitative cell viability was assessed using a Wst-1 assay by measuring the mitochondrial dehydrogenase. The absorbance at 450 nm showed that each of the five new PCPCs had cell viability not significantly different (p > 0.1) from that of the non-premixed Conventional CPC Control. Dissimilar letters at the upper right corner of each bar indicate values that are significantly different (p < 0.05). Each value is mean ± S.D.; n = 5.
4. Discussion
Premixed CPC was developed to shorten the surgical time by avoiding the on-site powder–liquid mixing, and to improve the cement properties by mixing in advance under well-controlled conditions. The new PCPCs had live cell density values not significantly different from that of a FDA-approved non-premixed CPC and that of the TCPS Control. The non-premixed CPC control showed excellent biocompatibility and osteoconductivity, and was able to be resorbed and replaced by new bone as demonstrated in previous studies [8,9]. While the TCPS control had a moderately higher cell viability, all the new PCPCs had cell viability values matching that of the non-premixed CPC control. The cells developed cytoplasmic extensions anchoring on the nano-sized hydroxyapatite crystals in the PCPCs. A biomimetic advantage of the PCPCs was that they consisted of nano-hydroxyapatite crystals with sizes similar to those found in natural bones and teeth. Enamel rods consisted of apatite crystallites about 100 nm in diameter [35]. Dentin and bone had smaller apatite crystals, with dimensions of approximately 5 nm × 30 nm × 100 nm [36]. In biomimetic fabrication of biomaterials, bone was considered a nano-composite of apatite minerals and proteins [2]. Nanostructured engineering of biomaterial surfaces enhanced the cell adhesion [37]. Hence the nano-hydroxyapatite crystals in the PCPCs likely enhanced the cell attachment due to their sizes being similar to the apatite crystallites in natural hard tissues. Another advantage of the PCPCs was that the nano-hydroxyapatite was formed in an aqueous environment at body temperature. It was shown that hydroxyapatite in CPC was more similar to the biological apatites than the sintered hydroxyapatite formed at high temperatures [3,8,9].
Sintered porous hydroxyapatite implants had flexural strengths of 2–11 MPa [38]. Cancellous bone had a tensile strength of about 3.5 MPa [39]. Although the measurements are not identical and direct comparison cannot be made, these data do suggest that the strengths of the PCPCs (Fig. 1) approached those of sintered porous hydroxyapatite implants and cancellous bone. Among the PCPCs, Premixed Control had an undesirably long setting time of 62 min. PCPC-Citric had a low strength and a long setting time of 40 min. Faster setting was desirable because it would enable the graft to attain significant strength and geometric integrity within a short period of time postoperatively. PCPC-Tartaric, PCPC-Malic and PCPC-Malonic appeared to have the best combination of relatively high strength, fast-setting, and several minutes of working time for the clinician to mold the cement before it hardens. It should be noted that the PCPC paste does not harden while in storage or in a syringe. The paste hardens only after being placed into contact with physiological fluids. Certain craniofacial repairs may prefer a working time of about 20 min such as PCPC-Malic and PCPC-Malonic. Root canal filler/sealer and pulp capping/cavity lining uses may prefer a setting time of a few minutes such as PCPC-Tartaric. Further studies are needed to examine the use of these PCPCs in vivo.
After 7 days immersion, Premixed Control had a hydroxyapatite conversion of nearly 80%. PCPC-Tartaric, the mechanically strongest cement, had a hydroxyapatite conversion of about 50%. The present study focused on developing new PCPC with high strength, workable setting time, and biocompatibility; further study is needed to investigate the mechanisms by which the different PCPCs convert to hydroxyapatite. Furthermore, because TTCP and DCPA react to form hydroxyapatite via 2Ca4(PO4)2O + 2CaHPO4 → Ca10(PO4)6(OH)2, a lower percentage of hydroxyapatite indicates the presence of unreacted TTCP and DCPA. Both TTCP and DCPA have higher solubility values than hydroxyapatite [3]. In addition, TTCP is more basic and more soluble than hydroxyapatite, especially at acidic pH [3], such as that produced by osteoclasts in vivo [40]. Therefore, PCPC-Tartaric with a low hydroxyapatite conversion may be more rapidly resorbed in vivo than the Premixed Control with a high hydroxyapatite conversion. Further studies are needed to investigate the resorption rates of PCPCs in animal models.
Acknowledgments
We thank Drs. F.C. Eichmiller and S.H. Dickens for discussions, and A.A. Giuseppetti for experimental assistance. This study was supported by USPHS grants DE14190, DE11789, Y1-DE-1021, NIST, and the ADAF.
References
- 1.LeGeros RZ. Biodegradation and bioresorption of calcium phosphate ceramics. Clin Mater. 1993;14:65–88. doi: 10.1016/0267-6605(93)90049-d. [DOI] [PubMed] [Google Scholar]
- 2.Chang MC, Ko CC, Douglas WH. Preparation of hydroxyapatite-gelatin nanocomposites. Biomaterials. 2003;24:2853–62. doi: 10.1016/s0142-9612(03)00115-7. [DOI] [PubMed] [Google Scholar]
- 3.Brown WE, Chow LC. A new calcium phosphate water setting cement. In: Brown PW, editor. Cements research progress. Westerville, OH: Am. Ceram. Soc.; 1986. pp. 352–79. [Google Scholar]
- 4.Ginebra MP, Fernandez E, De Maeyer EAP, Verbeeck RMH, Boltong MG, Ginebra J, et al. Setting reaction and hardening of an apatite calcium phosphate cement. J Dent Res. 1997;76:905–12. doi: 10.1177/00220345970760041201. [DOI] [PubMed] [Google Scholar]
- 5.Miyamoto Y, Ishikawa K, Takechi M, Toh T, Yuasa T, Nagayama M, et al. Histological and compositional evaluations of three types of calcium phosphate cements when implanted in subcutaneous tissue immediately after mixing. J Biomed Mater Res. 1999;48B:36–42. doi: 10.1002/(sici)1097-4636(1999)48:1<36::aid-jbm8>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 6.Xu HHK, Quinn JB, Takagi S, Chow LC. Processing and properties of strong and non-rigid calcium phosphate cement. J Dent Res. 2002;81:219–24. [PubMed] [Google Scholar]
- 7.Gbureck U, Barralet JE, Hofmann MP, Thull R. Nanocrysttaline tetracalcium phosphate cement. J Dent Res. 2004;83:425–8. doi: 10.1177/154405910408300514. [DOI] [PubMed] [Google Scholar]
- 8.Shindo ML, Contantino PD, Friedman CD, Chow LC. Facial skeletal augmentation using hydroxyapatite cement. Arch Otolaryngol Head Neck Surg. 1993;119:185–90. doi: 10.1001/archotol.1993.01880140069012. [DOI] [PubMed] [Google Scholar]
- 9.Friedman CD, Costantino PD, Takagi S, Chow LC. BoneSource™ hydroxyapatite cement: a novel biomaterial for craniofacial skeletal tissue engineering and reconstruction. J Biomed Mater Res. 1998;43B:428–32. doi: 10.1002/(sici)1097-4636(199824)43:4<428::aid-jbm10>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 10.Sugawara A, Kusama K, Nishimura S, Nishiyama M, Moro I, Kudo I, et al. Histopathological reaction of a calcium phosphate cement root canal filler. J Hard Tissue Biol. 1995;4:1–7. [Google Scholar]
- 11.Cherng AM, Chow LC, Takagi S. In vitro evaluation of a calcium phosphate cement root canal filler/sealer. J Endodontics. 2001;27:613–5. doi: 10.1097/00004770-200110000-00003. [DOI] [PubMed] [Google Scholar]
- 12.Dickens-Venz SH, Takagi S, Chow LC, Bowen RL, Johnston AD, Dickens B. Physical and chemical properties of resin-reinforced calcium phosphate cements. Dent Mater. 1994;10:100–6. doi: 10.1016/0109-5641(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 13.Dickens SH, Flaim GM, Takagi S. Mechanical properties and biochemical activity of remineralizing resin-based Ca-PO4 cements. Dent Mater. 2003;19:558–66. doi: 10.1016/s0109-5641(02)00105-7. [DOI] [PubMed] [Google Scholar]
- 14.Bayne SC, Thompson JY, Swift EJ, Jr, Stamatiades P, Wilkerson M. A characterization of first-generation flowable composites. J Am Dent Assoc. 1998;129:567–77. doi: 10.14219/jada.archive.1998.0274. [DOI] [PubMed] [Google Scholar]
- 15.Ferracane JL, Berge HX, Condon JR. In vitro aging of dental composites in water—effect of degree of conversion, filler volume, and filler/matrix coupling. J Biomed Mater Res. 1998;42:465–72. doi: 10.1002/(sici)1097-4636(19981205)42:3<465::aid-jbm17>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 16.Watts DC, Hindi AA. Intrinsic “soft-start” polymerization shrinkage-kinetics in an acrylate-based resin composite. Dent Mater. 1999;15:39–45. doi: 10.1016/s0109-5641(99)00012-3. [DOI] [PubMed] [Google Scholar]
- 17.Sakaguchi RL. Review of the current status and challenges for dental posterior restorative composites: clinical, chemistry, and physical behavior considerations. Dent Mater. 2005;21:3–6. doi: 10.1016/j.dental.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 18.Takagi S, Chow LC, Hirayama S, Eichmiller FC. Properties of novel resorbable chitosan-calcium phosphate composites. Dent Mater. 2003;19:797–804. doi: 10.1016/s0109-5641(03)00028-9. [DOI] [PubMed] [Google Scholar]
- 19.Xu HHK, Quinn JB, Takagi S, Chow LC. Synergistic reinforcement of in situ hardening calcium phosphate composite scaffold for bone tissue engineering. Biomaterials. 2004;25:1029–37. doi: 10.1016/s0142-9612(03)00608-2. [DOI] [PubMed] [Google Scholar]
- 20.Xu HHK, Simon CG., Jr Fast setting calcium phosphate-chitosan scaffold: mechanical properties and biocompatibility. Biomaterials. 2005;26:1337–48. doi: 10.1016/j.biomaterials.2004.04.043. [DOI] [PubMed] [Google Scholar]
- 21.Takagi S, Chow LC, Hirayama S, Sugawara A. Premixed calcium-phosphate cement pastes. J Biomed Mater Res. 2003;67B:689–96. doi: 10.1002/jbm.b.10065. [DOI] [PubMed] [Google Scholar]
- 22.Carey LE, Xu HHK, Simon CG, Jr, Takagi S, Chow LC. Premixed rapid-setting calcium phosphate composites for bone repair. Biomaterials. 2005;26:5002–14. doi: 10.1016/j.biomaterials.2005.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ishikawa K, Miyamoto Y, Takechi M, Toh T, Kon M, Nagayama M, et al. Non-decay type fast-setting calcium phosphate cement: hydroxyapatite putty containing an increased amount of sodium alginate. J Biomed Mater Res. 1997;36A:393–9. doi: 10.1002/(sici)1097-4636(19970905)36:3<393::aid-jbm14>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 24.For example, www.drugdigest.org.
- 25.Xu HHK, Takagi S, Quinn JB, Chow LC. Fast-setting calcium phosphate scaffolds with tailored macropore formation rates for bone regeneration. J Biomed Mater Res. 2004;68A:725–34. doi: 10.1002/jbm.a.20093. [DOI] [PubMed] [Google Scholar]
- 26.Merck Index, Merck, Whitehouse, NJ. http://en.wikipedia.org/wiki/Foodadditives; http://vm.cfsan.fda.gov/%7Edms/eafus.html.
- 27.Yokoyama A, Yamamoto S, Kawasaki T, Kohgo T, Nakasu M. Development of calcium phosphate cement using chitosan and citric acid for bone substitute materials. Biomaterials. 2002;23:1091–101. doi: 10.1016/s0142-9612(01)00221-6. [DOI] [PubMed] [Google Scholar]
- 28.Guide to dental materials and devices. 7. American Dental Association; 19741975. ADA Specification No. 9 for Dental Silicate Cement; pp. 194–202. [Google Scholar]
- 29.Burguera EF, Xu HHK, Takagi S, Chow LC. High early-strength calcium phosphate bone cement: effects of dicalcium phosphate dehydrate and absorbable fibers. J Biomed Mater Res. 2005;75A:966–75. doi: 10.1002/jbm.a.30497. [DOI] [PubMed] [Google Scholar]
- 30.Attawia MA, Uhrich KE, Botchwey E, Langer R, Laurencin CT. In vitro bone biocompatibility of poly(anhydride-co-imides) containing pyromellitylimidoalanine. J Orthopaed Res. 1996;14:445–54. doi: 10.1002/jor.1100140315. [DOI] [PubMed] [Google Scholar]
- 31.International Standards Organization. ISO 10993-5. Biological evaluation of medical devices-Part 5: tests for in vitro cytotoxicity. Geneva, Switzerland: ISO; 1999. [Google Scholar]
- 32.Simon CG, Jr, Khatri CA, Wight SA, Wang FW. Preliminary report on the biocompatibility of a moldable, resorbable, composite bone graft consisting of calcium phosphate cement and poly(lactide-co-glycolide) microspheres. J Orthop Res. 2002;20:473–82. doi: 10.1016/S0736-0266(01)00140-1. [DOI] [PubMed] [Google Scholar]
- 33.Xu HHK, Smith DT, Simon CG., Jr Strong and bioactive composites containing nano-silica-fused whiskers for bone repair. Biomaterials. 2004;25:4615–26. doi: 10.1016/j.biomaterials.2003.12.058. [DOI] [PubMed] [Google Scholar]
- 34.Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, Darnell J. Molecular cell biology. 4. chapter 18–19 New York: Freeman and Company; 2000. [Google Scholar]
- 35.White SN, Luo W, Paine ML, Fong H, Sarikaya M, Snead ML. Biological organization of hydroxyapatite crystallites into a fibrous continuum toughens and controls anisotropy in human enamel. J Dent Res. 2001;80:321–6. doi: 10.1177/00220345010800010501. [DOI] [PubMed] [Google Scholar]
- 36.Marshall GW., Jr Dentin: microstructure and characterization. Quintessence Intl. 1993;24:606–17. [PubMed] [Google Scholar]
- 37.El-Ghannam AR, Ducheyne P, Risbud M, Adams CS, Shapiro IM, Castner D, et al. Model surfaces engineered with nanoscale roughness and RGD tripeptides promote osteoblast activity. J Biomed Mater Res. 2004;68A:615–27. doi: 10.1002/jbm.a.20051. [DOI] [PubMed] [Google Scholar]
- 38.Suchanek W, Yoshimura M. Processing and properties of hydroxyapatite-based biomaterials for use as hard tissue replacement implants. J Mater Res. 1998;13:94–117. [Google Scholar]
- 39.Damien CJ, Parsons JR. Bone graft and bone graft substitutes: a review of current technology and applications. J Appl Biomater. 1991;2:187–208. doi: 10.1002/jab.770020307. [DOI] [PubMed] [Google Scholar]
- 40.Silver IA, Murrills RJ, Etherington DJ. Microelectrode studies on the acid microenvironment beneath adherent macrophages and osteoclasts. Exp Cell Res. 1988;175:266–76. doi: 10.1016/0014-4827(88)90191-7. [DOI] [PubMed] [Google Scholar]