

Abstract
Anti-angiogenesis approaches have the potential to be particularly effective in the treatment of glioblastoma (GBM) tumors. These tumors exhibit extremely high levels of neovascularization, which may contribute to their extremely aggressive behavior not only by providing oxygenation and nutrition, but also by establishing a leaky vasculature that lacks a blood-brain barrier. This leaky vasculature enables migration of tumor cells as well as the build up of fluid that exacerbates tissue damage due to increased intracranial pressure. Considerable progress has been made in the identification of the pro- and anti-angiogenic factors produced by GBM tumors and, in many cases, the effects of these molecules have been demonstrated in animal models of the disease. The safety and efficacy of some of these approaches have now been demonstrated in clinical trials; however, the ability of tumors to overcome these therapies and to re-establish angiogenesis requires further clinical research regarding potential multi-modality therapies as well as basic research into the regulation of angiogenesis by as yet unidentified factors. Optimization of non-invasive procedures for monitoring of angiogenesis would greatly facilitate such research.
Keywords: Glioblastoma, angiogenesis, molecular targets, inhibitors of angiogenesis
I. Introduction
A) Overview of the Angiogenic Process
GBM tumors are the most malignant brain tumors; their aggressive nature results in a median survival of 12 – 15 months (1). Notably, they also are one of the most angiogenic of the known solid malignant tumors (2). Angiogenesis, the induction and creation of a neovasculature, allows tumors to overcome the limitations in nutrient and oxygen delivery that develop when the tumor reaches a size of ≈ 1 to 2 mm in diameter (3;4). The new vessel growth occurs as an expansion of the existing vasculature in response to the secretion of pro-angiogenic growth factors by the tumor cells, stromal cells, and/or inflammatory cells. Multiple pro-angiogenic factors have been identified. Vascular endothelial growth factor-A (VEGF-A) is perhaps the best known of these and plays a key role in angiogenesis. VEGF-A is upregulated in GBM tumors in which it is produced by tumor cells as well as stromal and inflammatory cells (see Table I) (5) (reviewed (3)). Once secreted by the tumor, stromal, or inflammatory cells, VEGF-A can be sequestered or tethered in the extracellular matrix due to interactions with proteoglycans or glycosaminoglycans (6;7). The expression of the receptors for VEGF-A (VEGFR1 and VEGFR2) is upregulated on the endothelial cells (ECs) in GBM tumors as compared to its expression on the ECs of normal brain (7-10). VEGFR3 expression previously was thought to be specific for lymphatic ECs and VEGFR3 is not expressed in the normal brain; however, VEGFR3 has now been shown to be expressed in GBM tumors and has been localized to the ECs despite the absence of a lymphatic system in the brain (11;12). Furthermore, the ligands for VEGFR3 (VEGF-C&D) are expressed by multiple cell types that surround the angiogenic vessels in GBM tumors (see Table I) (11), suggesting that a novel pro-angiogenic paracrine signaling pathway exists in these tumors. Similar observations regarding VEGFR3 expression have been made in other tumors (12).
Table I. Examples of Pro-Angiogenic Molecules Expressed in GBM Tumors: Their Binding Partner(s) and Localization.
| Factor | Cells Producing the Factor in GBM Tumors | Binding Partner(s) in GBM Tumors & its Localization | Efficacy of Targeting Shown in an Animal Model of Malignant Glioma or in a Clinical Trial | References |
|---|---|---|---|---|
| VEGF-A | Tumor, Stromal & Host Inflammatory Cells | VEGFR1 & VEGFR2 on ECs | Yes | (5;7-10;103;104) |
| VEGF-C&D | Perivascular Mix of Cells | VEGFR3 on ECs | No | (11;12) |
| bFGF | Primarily Blood Vessels & Focal Tumor Cells | FGFR1 on ECs & FGFR4 on Tumor Cells | Yes | (13-17;200) |
| SDF-1α | Pleomorphic Angiogenic Vessels | CXCR4 on Circulating EPCs and Tumor Vessels | Yes | (36;39-41) |
| PDGF-BB | ECs | PDGFRβ on pericytes | Yes | (13;32;150;151;156) |
| NG2 | Pericytes | Angiostatin; Localization in the Extracellular Environment | Yes | (83;148) |
| Tenascin-C | Vascular Cells | Integrins αvβ3 & α5β1 on Vascular Cells & Tumor Cells | Yes | (165;166;169;170) |
Another pro-angiogenic growth factor that is upregulated in GBM tumors is basic fibroblast growth factor (bFGF). In GBM tumors, bFGF is expressed by vascular cells and focally by the tumor cells (see Table I) (13). Furthermore, a neutralizing antibody directed toward bFGF inhibited tumor growth in a subcutaneous xenograft model of GBM tumor propagated in the nude mouse (14). The receptors for bFGF include FGFR1, FGFR2 and FGFR4. FGFR1 is upregulated in GBM tumors in which it is expressed by both the tumor cells and the tumor ECs, FGFR4 is expressed only by the tumor cells, and FGFR2 is not detected in GBM tumors although it is expressed in the normal brain (14-17). Like VEGF, bFGF can be tethered in the matrix due to an association with proteoglycans or glycosaminoglycans. The glypican family of proteoglycans, which are expressed on the ECs, serve as co-receptors for bFGF (18).
The binding of VEGF or bFGF activates the existing EC. Binding of VEGF to its receptors on the EC typically results in signaling through the phosphatidylinositol-3-hydroxyl kinase (PI3K)/protein kinase B (Akt) pathway, whereas the bFGF receptors signal predominantly through the protein kinase Cα (PKCα) pathway (19;20). In addition, both VEGF and bFGF stimulation can initiate signaling through the extracellular signal-regulated kinase (Erk) pathway (19-21). The activation of the ECs results in upregulation or increased expression of certain cell adhesion receptors, such as integrins αvβ3 and α5β1, respectively, and the activated ECs exhibit increased cell survival, proliferation and migration responses (22-24). In addition to VEGF and bFGF, other pro-angiogenic growth factors may also act to initiate or amplify angiogenesis in GBM tumors. For example, interleukin-8 (IL-8), which is released from macrophages during a host inflammatory response, activates ECs through the chemokine-1 and 2 receptors (CXCR1, and CXCR2) that are expressed on ECs (see Table I) (25). Other examples, that are not discussed further in this review include hepatocyte growth factor, granulocyte-macrophage-colony-stimulating factor, granulocyte-colony-stimulating factor, and urokinase (13;26;27). In addition, micro-environmental signals can act to amplify the signals provided by the pro-angiogenic factors. Notably, rapid tumor growth that exceeds the oxygen supply causes a decrease in intracellular pH, leading to hypoxia-driven expression of pro-angiogenic growth factors, including VEGF, by both tumor cells and the host inflammatory cells (28;29).
For ECs to form a neovasculature the basement membrane that forms a “cage” around the existing vasculature must be degraded (30). The proteolytic degradation of the basement membrane is mediated by proteases, such as the matrix metalloproteinases (MMPs), that are secreted by tumor and stromal cells (30). This exposes the EC to matrix proteins that promote pro- or anti-angiogenic activity, to fragments derived from the proteolytic breakdown of matrix molecules, and to the matrix-tethered growth factors (such as tethered VEGF and bFGF) (6;31). Integrins expressed on the ECs play a critical role in directing the migration of the ECs into the degraded basement membrane (23). Thus, the degradation of the basement membrane creates pathways that both enable and stimulate the migration of ECs from the existing vasculature, which results in the typical ‘sprouting’ of the neovasculature from the existing vasculature.
Concurrently, vascular-associated cells, including smooth muscle cells and pericytes, are recruited to the neovasculature through platelet–derived growth factor (PDGF) signaling (32). ECs synthesize PDGF and this enables the recruitment of pericytes to the neovessels as they express the PDGF receptor β (PDGFRβ). PDGF also acts as a pro-angiogenic growth factor (see Table I) (13). These vascular-associated cells serve to stabilize the neo-endothelium (32). In normal angiogenesis pericytes accomplish this through the angiopoietin/Tie-2 receptor signaling axis, but this is altered in GBM tumors (see part II E below). Pericytes also can stabilize the neo-endothelium through their deposition of matrix proteins that help to form the new basement membrane (33;34). The new basement membrane can influence EC behavior both directly, for example through the engagement of specific integrin receptors, and indirectly through the modulation of growth factor signaling, for example, integrin αvβ5 can cooperate with VEGFR2 and integrin αvβ3 can cooperate with FGFR in promoting EC survival, proliferation, and migration (35). Integrin αvβ3 expression is upregulated on the ECs in GBM tumors (24). Bone marrow-derived progenitor cells, such as endothelial progenitor cells (EPCs), also assist in vascular remodeling and are mobilized from the bone marrow by the cytokine stromal-derived factor-1α (SDF-1α, also known as CXCL12), (see Table I) (36-38). SDF-1α is expressed by the pleomorphic angiogenic vessels in GBM tumors and binds to the G protein-coupled chemokine 4 receptor (CXCR4) expressed on circulating EPCs and on tumor vessels (36;39;40). Furthermore, targeting of this pathway with a CXCR4 antagonist in an orthotopic xenograft model of GBM tumor propagated in the nude mouse resulted in an inhibition of tumor growth (41).
Unlike the blood vessels in normal tissue, the neovasculature in GBM tumors never fully develops into mature vessels, due in part to an imbalance of pro- and anti-angiogenic growth factors in the tumor. This leads to an atypical and constantly remodeling tumor vasculature.
B) Vessel Morphology and Tumor Hypoxia
Angiogenesis in GBM tumors is characterized by a neovasculature that has a glomeruloid-like histology and exhibits an erratic basement membrane, reduced pericyte coverage, decreased astrocytic endfoot association with blood vessels, and altered expression of EC adhesion molecules (2;24;34;42;43). These abnormalities result in excessive leakiness of the neovasculature. Typically there is an increase in microvessel density and in the number of dilated large tortuous vessels in GBM tumor biopsies.
This excessive leakiness is of particular importance in the expansion and spread of GBM tumors throughout the normal brain and in the treatment of these tumors. The abnormal neovasculature associated with the tumors lacks a normal blood brain barrier (BBB). This is thought to lead to a decrease in the osmotic gradient between the vasculature and interstitium, and to an elevation of the interstitial fluid pressure in the tumor (44). The elevated interstitial fluid pressure, together with the frequent reversions and stalling of the blood flow in the contorted neovasculature, can compromise drug delivery to the GBM tumors (see part IV B below). The abnormal vasculature may also result in enhanced tumor cell access to the vasculature, which likely enables tumor cell migration and invasion throughout the previously uninvolved brain tissue. This may be of considerable importance as it may contribute to the aggressive behavior of the GBM tumor by overcoming the limitation on tumor invasion and local migration in the brain that is imposed by the absence of a lymphatic system. The lack of a lymphatic system has been purported to be one reason for the lack of identified extracranial metastases in the vast majority of GBM tumors. Finally, the leaky tumor vasculature amplifies the effects of the tumor mass on the generation of an increased interstitial pressure, measured as increased intracranial pressure. In the enclosed cranium, this has very negative effects on uninvolved brain tissue. Again, this effect is exacerbated by the lack of a lymphatic system, which limits the ability of the brain to relieve the pressure.
GBM tumors frequently exhibit extensive necrotic cores. This exacerbates hypoxia, which can promote tumor angiogenesis by enhancing the production of pro-angiogenic factors. Hypoxia destabilizes the von Hippel Landau (VHL) molecule; and the VHL molecule inhibits the function of hypoxia-inducible factor-1α (HIF-1α), a transcription factor promoting the transcription of several pro-angiogenic growth factors (28). Hypoxia-driven VHL molecule dissociation from HIF-1α causes HIF-1α to bind the promoter of the hypoxia response element (HRE) in several pro-angiogenic factors, including VEGF and SDF-1α, resulting in their transcription (28;36).
II. Opposing Angiogenic Factors in GBM Tumors
A) Thrombospondin-1 and -2 (TSP-1 and 2)
In normal tissues the balance of pro- and anti-angiogenic factors is in equilibrium. In GBM tumors the balance of pro- and anti-angiogenic growth factors is skewed to favor pro-angiogenic factors, which favors neovascularization. Several anti-angiogenic factors have been described. Thrombospondin-1 (TSP-1) is an extracellular anti-angiogenic factor that is synthesized and secreted by multiple normal cells and tissues, such as platelets, endothelial cells, and smooth muscle cells (see Fig. 1 and Table II) (reviewed in (45;46)). The anti-angiogenic effects of TSP-1 treatment have been studied extensively in dermal microvessel ECs propagated in vitro; TSP-1 inhibits the migration and proliferation of these cells, and also induces apoptosis (47-49). The most potent anti-angiogenic effects of TSP-1 are induced through the binding of TSP-1 to CD36, which is expressed on the surface of microvessel ECs. This interaction is mediated by the type 1 repeat domain (TSR) of TSP-1 and the CLESH-1 domain of CD36 (reviewed in (50)). Other proteins can modify the TSP-1 response by cooperating with or modulating CD36 function; for example, integrin αvβ3 modulates CD36 function in macrophages (51). In addition, TSP-1 also can promote an anti-angiogenic effect that is mediated through other domains of TSP-1, or is caspase-independent (45;52;53). Peptides derived from the TSP-1 TSRs have been administered to mice bearing malignant glioma tumors and shown to inhibit tumor angiogenesis and tumor growth, as discussed in more detail in Part III of this review (54). TSP-1 has been shown to act downstream of the tumor suppressor p53 in cells propagated in vitro (55;56). Loss of p53 function is found in some GBM tumors, yet TSP-1 levels are increased in the tumor vasculature, most likely due to a host stromal anti-tumor response (See Table II and Fig. 1) (57;58).
Fig. 1. Examples of Endogenous Inhibitors of Angiogenesis in the Normal Brain or GBM Tumors: The Cell of Origin or Cellular Localization.
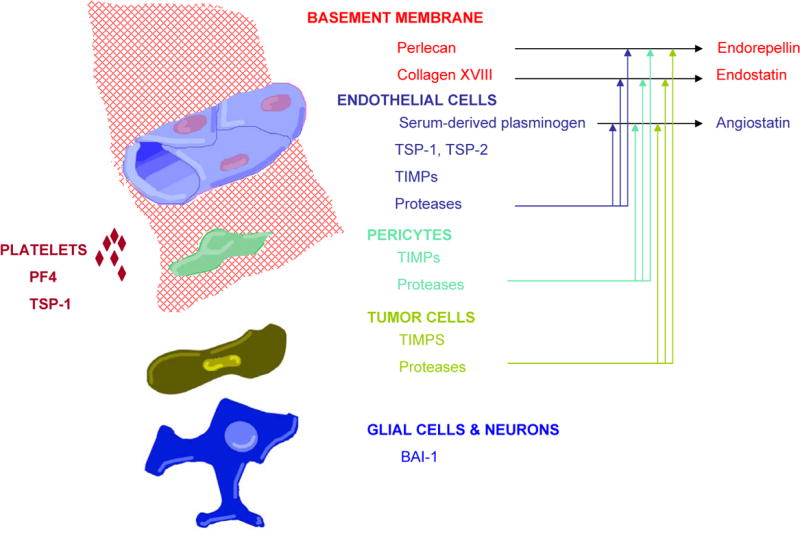
Angiostatin, endorepellin and endostatin are proteolytic cleavage products. Various cells produce proteases but the relative contribution of each of these cellular sources of proteases to the generation of these cleavage products has not yet been defined.
Table II. Examples of Anti-Angiogenic Molecules Expressed in GBM Tumors: Their Binding Partner(s) and Localization.
| Factor | Cells Producing The Factor in GBM Tumors | Binding Partner(s) in GBM Tumors & its Localization | Ability to Inhibit Tumor Growth Shown in an Animal Model of Malignant Glioma | References |
|---|---|---|---|---|
| TSP-1 (TSP-1 TSR Peptidomimetic – ABT-510) | Vascular Cells: Produce TSP-1 | CD36 on ECs | Yes | (53;54;57;58;111-113) |
| BAI-1 | Neurons and glia | Integrin αvβ5 (? on Adjacent Cells) | Yes | (62-66) |
| Endostatin | Proteolytic Fragment of Collagen | Integrin α5β1; VEGFR2, Tropomyosin, & Other Proteins on ECs | Yes | (67;68;73;74) |
| Angiostatin | Proteolytic Fragment of Plasminogen | Integrin αvβ3; ATP synthase; & Other Proteins on ECs | Yes | (80;81;83) |
| PF4 | Platelets & Megakaryocytes | VEGFR on ECs | Yes | (93;94) |
| TIMP-1 | Vascular Cells And Tumor Cells | MMP on Vascular Cells | No | (92) |
| Endorepellin (fragment of perlecan) | Vascular Cells | Integrin α2β1 on ECs | No | (95;96) |
TSP-2 is highly homologous to TSP-1 and also exhibits anti-angiogenic properties that are due in part to the TSRs (45;50;59). TSP-2 is expressed by ECs and other cells in the normal mouse brain (60), and its expression is downregulated in GBM tumors as compared to normal brain (see Fig. 1) (61). Furthermore, mouse malignant glioma tumors propagated orthotopically in the TSP-2-null mouse showed an increased tumor size and an increased microvessel density as compared to tumors propagated in the wild-type mouse (60), supporting a host anti-tumor and anti-angiogenesis role for TSP-2 in the brain.
B) Brain Angiogenesis Inhibitor-1 (BAI-1)
BAI-1 is a brain-specific anti-angiogenic molecule (see Table II and Fig. 1) (62). It is a cell membrane protein that can be cleaved releasing the extracellular domain, the extracellular domain contains five TSRs and is also known as vasculostatin (62;63). In certain experimental conditions BAI-1 may be a p53 target gene (62;64). However, BAI-1 expression in GBM tumors is p53-independent (64). BAI-1 is expressed differentially in normal brain and GBM tumors. In the normal brain there is a high expression of BAI-1 in normal glia and neurons but no expression in blood vessels. In GBM tumors, BAI-1 expression is decreased likely due to the loss of neurons and normal glia (64). Despite the TSR domains, BAI-1 functions independently of CD36 expressed on the ECs and it appears to inhibit cell survival through an interaction of the TSRs with integrin αvβ5 (65). Expression of either full-length BAI-1, or the extracellular domain of BAI, in human glioma cells that were propagated in nude mice resulted in a significant inhibition of tumor growth and tumor angiogenesis as compared to similarly propagated control glioma cells (63;66). This suggests that BAI-1 has an anti-angiogenic effect in vivo, and that the TSRs of BAI-1 are likely necessary for this anti-angiogenic effect.
C) Endostatin and Tumstatin
Anti-angiogenic molecules, including endostatin, can be produced during the proteolytic cleavage of the basement membrane in GBM tumors (see Table II and Fig. 1) (67;68). Endostatin is a C-terminal cleavage product of collagen type XVIII by elastase, cathepsin-L and an MMP (reviewed in (69)) (70). Endostatin appears to be capable of interacting with several different receptors or binding partners on the ECs and to inhibit angiogenesis through multiple mechanisms, including blockade of VEGFR2, binding to integrin α5β1, blockade of focal adhesion kinase (FAK)-mediated EC migration and survival, binding to tropomyosin, and altered regulation of multiple other targets (such as through the nucleolin receptor) (reviewed in (69)) (71;72). Endostatin also can reduce expression of anti-apoptotic Bcl2 family members (reviewed in (69)). In addition, endostatin binds the pro-angiogenic protease MMP-2 and blocks its activation and catalytic activity (reviewed in (69)). Endostatin is expressed in GBM tumors (67;68), and a higher level of endostatin expression was found in GBM tumors than lower grade glioma tumor biopsies on examination by immunoblotting combined with immunohistochemistry (68). This suggests that endostatin expression is part of a host anti-tumor response. Several studies have shown that endostatin inhibits malignant glioma growth in rodent models, for example, when recombinant endostatin is delivered to tumors propagated in an ectopic or an orthotopic manner, or when the endostatin gene is delivered to orthotopic tumors (73;74).
Another anti-angiogenic molecule derived from the proteolytic cleavage of the basement membrane is tumstatin. Tumstatin is generated by the cleavage of collagen type IV by MMPs, and is thought to inhibit neo-vessel formation through an interaction with integrin αvβ3 resulting in blockade of FAK, and the P13K signaling pathway (72;75-77).
D) Angiostatin
One of the best known of the endogenous anti-angiogenic proteins is angiostatin (see Table II and Fig. 1). Angiostatin is made up of the first four kringle (K1-4) domains of plasminogen (78). Plasminogen is primarily synthesized and secreted by the liver and is present in high concentrations in the plasma; it can be proteolytically digested by proteases, such as cathepsin-D and MMPs, to generate angiostatin (reviewed in (79)). The anti-angiogenic activity of angiostatin was first demonstrated in mice bearing subcutaneous Lewis lung carcinoma tumors (78). Multiple studies have now shown that angiostatin inhibits malignant glioma growth in rodent models, for example, when angiostatin is administered to ectopic or orthotopic tumors propagated in immunodeficient mice (80;81). Angiostatin appears to mediate its anti-angiogenic effects through binding to several different receptors or binding partners on the EC, including binding to integrin αvβ3, and ATPsynthase (reviewed in (79)), (82;83). Angiostatin has also been shown to upregulate pro-apoptotic proteins in activated or proliferating endothelial cells (reviewed in (79)).
The fifth kringle domain (K5) of plasminogen also has been reported to have anti-angiogenic activity (84); however, generation of K5 in vivo though the proteolytic cleavage of plasminogen has not been reported. K5 has been shown to promote several anti-angiogenic effects, such as cell cycle arrest, inhibition of migration, and induction of apoptosis, in non-brain EC propagated in vitro (84-87). The anti-angiogenic effects of K5 appear to require binding to cell surface glucose-regulated protein 78 (GRP78), a heat shock protein family member (85). When glioma cells stably expressing K5 are propagated in nude mice the tumors are smaller and demonstrate a reduced angiogenic response (88).
E) Angiopoietins
The angiopoietins can exhibit pro- or anti-angiogenic activities and have diverse roles in the brain and in the neovasculature of GBM tumors. In normal blood vessels angiopoietin-1 is expressed primarily by pericytes and binds the Tie-2 receptor expressed on ECs, leading to an increased association of pericytes with ECs, and to a less leaky, normalized vasculature (32;89). If this occurred in GBM tumors it could be of importance therapeutically as normalization of the vasculature may improve anti-tumor drug delivery and radiation effectiveness. However, in GBM tumors there is increased expression of both angiopoietin-1 and -2, and angiopoietin-2 is expressed by the ECs whereas angiopoietin-1 is expressed by the tumor cells and is not expressed at the blood vessels (90). Angiopoietin-2 is thought to serve as a Tie-2 receptor antagonist (33;90;91). These observations have led investigators to hypothesize that angiopoietin-2 may have a pro-angiogenic function in the presence of VEGF, such as in angiogenic vessels in which VEGF and angiopoietin-2 are both expressed.
Other anti-angiogenic proteins expressed in the brain or GBM tumors, which are not discussed here include, tissue inhibitors of matrix metalloproteinases (TIMP), such as TIMP-1 (92), platelet factor 4 (93;94), endorepellin – a fragment of perlecan (95;96), PEX – a fragment of MMP-2, (97), and pigment epithelium-derived factor (98;99) (see Table II and Fig. 1).
III. Targets for Anti-Angiogenic Therapy
A) ECs
(1) VEGF/VEGFR
ECs offer the most logical target for anti-angiogenic therapy due to their relative genetic stability as compared to the tumor, and their direct contact with blood which could facilitate drug delivery. VEGF-A and the VEGFR2 receptor are the targets of multiple new anti-angiogenic drugs, including antibodies directed toward VEGF-A, the targeting of active VEGFR2 with blocking antibodies or small molecule tyrosine kinase inhibitors, and chimeric VEGFR2-like molecules or Traps (see Fig 2 and Table III) (100) (reviewed in (101;102)). Anti-angiogenic drugs targeting this pathway in a mouse model of GBM tumor and in patients with recurrent GBM tumor decreases the diameter of enlarged tumor vessels and the overall blood vessel size, decreases the thickness of the basement membrane and increases pericyte coverage, resulting in decreased tumor edema (103;104)(reviewed in (101;102)). These data suggest inhibition of the VEGF/VEGFR pathway has a more pronounced effect on the large or dilated vessels in GBM tumors. As inhibition of the VEGF/VEGFR signaling axis in GBM tumors has been reviewed recently (102), we will not discuss this approach further here. Therapeutic inhibition of VEGFR2 also may be achieved, in part, through the administration of endostatin, which interferes with VEGFR2 signaling (reviewed in (69)). The FGFR1 receptor and IL-8 receptors (CXCR1 and CXCR2) offer similar opportunities for therapeutic targeting, although the specificity of such targeting to angiogenic ECs in GBM tumors has yet to be thoroughly explored (14;25;105).
Fig. 2. Examples of Localized Angiogenic Targets in GBM Tumors.
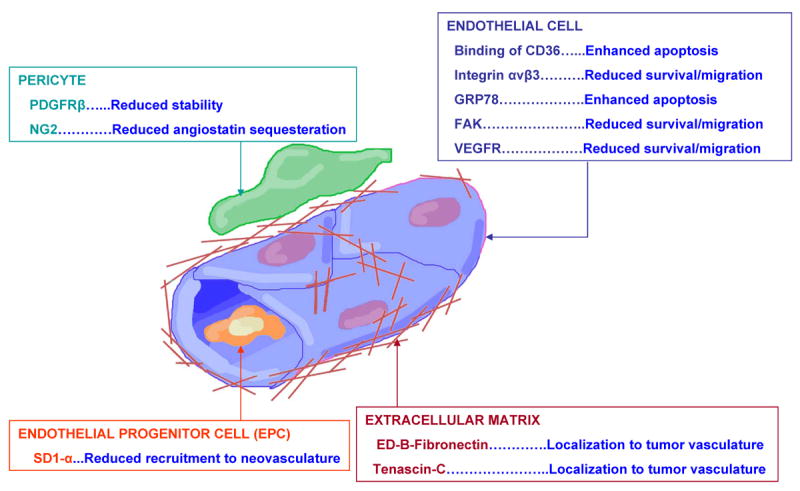
A schematic of the new targets of anti-angiogenic therapy, as discussed in the text, in the neovasculature of GBM tumors.
Table III. Clinical Trials with Anti-Angiogenic Agents Targeting Malignant Gliomas.
| Target | Drug | Drug Type |
|---|---|---|
| CD36 | ABT-510 | Peptide |
| VEGF * | Bevacizumab, Aflibercept | mAb, Trap fusion protein |
| RTK | AEE78, Vatalanib, Sunitinib, Sorafenib, Cediranib, Cetuximab, Erlotinib | Multiple drug types |
| Unclear: thought to be due to decreased growth factor mediated neovascularization | Thalidomide | Synthetic Sedative |
| alphaV Integrins | Cilengitide | Peptide |
| PKC beta | Enzastaurin | Peptide |
| COX-2 | Celecoxib | NSAID |
The information in this table was extracted from the National Institutes of Health (NIH) website http://www.clinicaltrials.gov searching for “glioma, brain cancer, glioblastoma and angiogenesis”. Clinical trials that are open, closed, enrolling, or completed are included in this table. Irrelevant drug listings were excluded. Many drugs have been utilized in multiple concurrent clinical trials. The primary investigator and clinical trial information can be found at this website. Abbreviations: COX-2, cyclooxygenase-2; NSAID, non-steroidal anti-inflammatory drug; mAb, monoclonal antibody; PKC, protein kinase C; RTK, receptor tyrosine kinase *VEGF/VEGFR inhibitors in clinical trials are not discussed in this review, as they were recently reviewed (101;102).
(2) Binding or Activating CD36 (TSR peptides derived from TSP-1)
Binding or activating CD36 is another possible therapeutic target on angiogenic ECs (see Fig. 2 and Table III) (50). TSP-1 binds CD36, resulting in potent anti-angiogenic effects on microvessel ECs. Full-length TSP-1 is an improbable therapeutic agent due to its size, complex domain structure, and multiple biologic activities (45;46); however, peptides derived from the TSRs of TSP-1 have proven to be an effective anti-angiogenic therapy in several mouse models of malignant tumors (54;106;107). Peptidomimetics from the TSRs of TSP-1, such as ABT-510 (NAcSarGlyValDalloIleThrNvaIleArgProNHE), induce apoptosis of human brain microvascular ECs propagated in vitro and inhibit bFGF-stimulated tubulomorphogenesis of these cells propagated in collagen gels (54). CD36 was required for the inhibition of tubulomorphogenesis by ABT-510. Furthermore, ABT-510 inhibits malignant glioma growth in two orthotopic intracerebral mouse models of malignant glioma, and this was shown to be mediated through an induction of apoptosis in the tumor ECs (see Fig. 2) (54). The latter observations were made in a syngeneic mouse model of malignant glioma, in which GL261 mouse malignant glioma cells were propagated in the brains of C57BL/6 mice, and in a xenograft model, in which U-251MG human malignant glioma cells were propagated in the nude mouse brain. The EC death in tumors observed after ABT-510 administration results in a decreased tumor microvessel density, and thus nutrient deprivation of the tumor. It is important to note that although ABT-510 induces apoptosis of primary human brain EC propagated in vitro, its effects in vivo appear to be specific for tumor ECs as we have not detected changes in the normal brain ECs (54). CD36 is expressed on the microvessel ECs in the normal brain (108-110); however, the activation of CD36, the conformation of CD36, the binding partner(s) for CD36, or the conformation of its binding partners may be different in the tumor-associated ECs thereby promoting the tumor-specific effect of ABT-510. Our results regarding the specificity of ABT-510 are consistent with those reported by other investigators concerning the effects of ABT-510 on bladder and Lewis lung carcinoma tumors propagated in mice. All of these studies indicate that ABT-510 inhibits angiogenesis (measured as microvessel density) and tumor growth but does not affect ECs in normal tissues (54;106;107).
The effects of expression of TSR-containing fragments of TSP-1 that do not bind CD36, or the expression of intact TSP-1 on glioma growth in vivo also have been reported. In one study, a peptide from a different region of the TSR that does not bind CD36 was found to inhibit the growth of C6 and 9L gliomas propagated in rats (111). In another study, expression of an N-terminal TSP-1 construct (containing the two cysteine residues involved in disulfide bridging, the pro-collagen-homology domain and the three TSRs) in rat C6 glioma cells resulted in an increase in the growth of the tumors when the cells were propagated in nude mice (112), suggesting that the pro-collagen-homology domain may have unexpected pro-tumor effects. Lastly, in support of our study (54), investigators propagated human LN-229 malignant glioma cells overexpressing intact TSP-1 subcutaneously in nude mice and found a significant reduction in tumor size and vascular density (113).
Combining ABT-510 with valproic acid or with metronomic doxorubicin treatment has been shown to increase the effectiveness of ABT-510 therapy, and resulted in an enhanced anti-tumor effect on neuroblastoma cells as well as on other carcinoma cells (Lewis lung, bladder, and prostate) propagated in mouse models (114;115). The mechanism by which valproic acid, a histone deacetylase inhibitor, enhanced the anti-angiogenic and anti-tumor effect of ABT-510 was not examined in the above studies. Other investigators have shown recently that the histone deacetylase inhibitor, trichostatin A, increases TSP-1 expression in cells propagated in vitro (116). This suggests the possibility that the increased effectiveness of ABT-510 administered with valproic acid in the above mouse models of cancer could be due to an increased expression of TSP-1 in the tumor vascular cells induced by valproic acid. These results highlight the need to combine anti-angiogenic therapies.
A phase I clinical trial with ABT-510 monotherapy in 39 patients with advanced solid malignancy that were refractory to standard therapy, excluding primary brain tumors and metastases to the brain, was performed (117). In this study six patients had stable disease lasting for six cycles or more of ABT-510 therapy. Also, the median serum level of bFGF for all patients decreased from 14.1 pg/ml to 3.2 pg/ml after 56 days of treatment, supporting the anti-angiogenic effect of ABT-510. Importantly, as reported for animal studies with ABT-510, no indication of toxicity or damage to normal ECs was observed. A toxicity due to repeated subcutaneous injection of ABT-510 at the same site was reported, and on skin biopsy of one patient a perivascular inflammatory cell infiltrate was seen but no vasculitis or EC death. Recently, the results of a phase II clinical trial with ABT-510 monotherapy were reported. In 21 patients with metastatic melanoma treated with ABT-510, three patients appeared to have a partial response at 18 weeks (118). Importantly, eight of 11 patients who had received six cycles of ABT-510 monotherapy had decreased numbers of circulating EPCs and six patients had decreased plasma levels of VEGF-A, supporting the anti-angiogenic effects of ABT-510 therapy. These data support the contention that anti-angiogenic therapy should be combined with a cytotoxic therapy and/or a secondary anti-angiogenic therapy.
(3) Integrin αvβ3
The expression of matrix-binding adhesion molecules on the EC surface is altered in GBM tumors. The integrin family of cell adhesion receptors has been characterized. Targeting the tumor-associated upregulation of an integrin on ECs, such as the upregulation of integrin αvβ3 on the ECs in GBM tumors (24;119), would likely serve to block the pro-survival signal transmitted to basement membrane bound ECs, as well as to inhibit signaling that promotes EC migration and proliferation (see Fig. 2 and Table III). This could effectively inhibit angiogenesis. The integrin αvβ3 binding molecule, cyclic RGD-based peptide (Cilengitide), has been used for this purpose in a phase I clinical trial and has shown no significant toxicity when administered to 37 patients with surgically-resected and radiated GBM tumors and to 11 patients with anaplastic astrocytoma tumors that were also surgically resected and radiated (120). In this study there were two complete responses, one in a patient with a GBM tumor and one in a patient with an anaplastic astrocytoma tumor, and three patients demonstrated a partial response, suggesting that a subpopulation of patients with malignant glioma tumors respond favorably to the cyclic RGD-peptide. The administration of RGD-peptide also has been shown to inhibit human malignant glioma growth in mice (121;122). Supporting the above results, the targeting of integrin αvβ3 in other types of cancers has proven effective in inhibiting tumor growth in mouse models (123;124). Integrin αvβ3 also may be targeted by therapy with angiostatin, as angiostatin inhibits angiogenesis in part by binding to integrin αvβ3 (82). Lastly, integrin αvβ3 may be targeted by tumstatin, which binds integrin αvβ3 and induces EC death (72;76).
(4) GRP78
Recently, it has been shown that GRP78 is a cell surface binding protein for K5 on non-brain MvEC, and is necessary for the pro-apoptotic effect of K5 in these cells (85). GRP78 is a member of the heat shock protein 70 (HSP70) family and normally functions as an ER-resident chaperone protein (125). Stress, such as hypoxia, can cause an upregulation of GRP78 (reviewed in (125)) GRP78 is upregulated during the unfolded protein response, which is part of the general cellular defense mechanism of stressed cells (reviewed in (125)). It has been shown that GRP78 is upregulated and expressed on the surface of tumor cells in patient biopsies; including biopsies of lung, breast, colon, and prostate cancers (126-128) (reviewed in (125)). The expression of GRP78 and other ER chaperone proteins on the surface of tumor cells, and potentially on tumor EC, could be used as a novel target for therapy (see Fig. 2). Also, as a hypoxic environment is found in GBM tumors, the targeting of GPR78 in these tumors with K5 could be efficacious.
(5) FAK
A potential intracellular target in the EC is FAK (see Fig. 2). FAK is a cytoplasmic non-receptor tyrosine kinase that is necessary for vasculogenesis (129). The embryos of FAK-null mice are not viable due to severe cardiovascular anomalies (129). Additional evidence supporting a role for FAK in angiogenesis is the observation that dominant-negative FAK constructs, such as the FAK-related non-tyrosine kinase construct (FRNK) which corresponds to the C-terminus of the FAK protein, inhibits EC tube formation and branching in tubulomorphogenesis assays (130). FAK promotes two functions essential for neovascularization, cell proliferation and migration (reviewed in (131;132)). It promotes cell cycle progression by activating Erk, and inducing expression of the transcription factor krupple-like factor 8 (KLF8) that binds and promotes cyclin D1 transcription (133-135). Evidence that FAK promotes cell migration is abundant and includes studies using FAK-null mouse embryo fibroblasts that demonstrated reduced cell motility (129), which was rescued by the re-expression of FAK (136). In multiple cell types, including brain ECs and GBM tumor cells, dominant-negative FAK constructs inhibit cell migration whereas wild-type FAK constructs promote cell migration (130;137) (reviewed in (131))(132). Small molecule inhibitors of FAK, such as TAE226 (138) and PF-573,228 (139), are becoming available, and both of these inhibitors significantly block the migration and invasion of cells propagated in vitro. The TAE226 inhibitor also blocks cell proliferation in vitro. A FAK family member Pyk2 has also been shown to promote migration and invasion of GBM tumor cells propagated in vitro (140;141). It is worth noting that different experimental conditions were utilized for the Pyk2 studies as compared to the conditions utilized to test the necessity of FAK for GBM cell migration (137;140;141). The FAK inhibitor PF-573,228 appears to be specific for FAK as it is 250-fold more sensitive to FAK (IC50 = 4 nM), as compared to Pyk2 (IC50 = 1 μM) (139). The effect of the FAK inhibitor TAE226 on Pyk2 has not been reported. These inhibitors offer promise for future animal studies and clinical trials, and suggest that the targeting of FAK in the tumor ECs could be an effective anti-angiogenic therapy (see Fig. 2).
(6) Tie-2 Receptor
Specific inhibition of the pericyte signaling, which supports EC survival in the GBM neovasculature, through targeting of the angiopoietin/Tie-2 signaling axis is another potential therapy (142). However, the neovasculature in GBM tumors has significantly reduced pericyte coverage, thus the effectiveness of this approach in GBM tumors must be determined. Tie-2 expression in GBM tumors is upregulated on the ECs (91), and the increased angiopoietin-2 found in GBM tumors is localized to ECs and associated with angiogenesis; thus, investigators have hypothesized that angiopoietin-2 promotes angiogenesis in the presence of VEGF (91;143;144). The angiopoietin-1 expressed by tumor cells, but not by the ECs, in GBM biopsies may act to promote cell adhesion as angiopoietin-1 and -2 can promote adhesion mediated by integrin α5β1, independently of Tie-2 expression (145). Thus, potential therapies that block Tie-2 signaling in GBM tumors require further investigation.
B) Pericytes
(1) NG2
A possible target on pericytes is the NG2 chondroitin sulfate proteoglycan, which is a transmembrane protein and a marker of pericytes in angiogenic vessels (see Table I and Fig. 2) (146) (reviewed in (32) and (147)). Expression of NG2 is upregulated on pericytes in GBM tumors as compared to pericytes in normal brain (148). NG2 appears to promote a pro-angiogenic effect through its binding and sequestration of angiostatin (83). Human U-251MG malignant glioma cells over-expressing NG2 and propagated in nude rats exhibited increased tumor growth and increased tumor angiogenesis as compared to similarly propagated wild-type tumor cells (83), supporting the potential pro-angiogenic role of NG2 in GBM tumors. This suggests that the vascular supporting cells in GBM tumors may be induced by the tumor to upregulate expression of the pro-angiogenic protein NG2.
(2) PDGFRβ
Another target on pericytes is PDGFRβ, the receptor for PDGF (see Fig. 2). Expression of PDGFRβ on pericytes is responsible for pericyte recruitment to blood vessels (149;150). PDGFRβ is also expressed on some tumor cells in GBM tumors and it can cooperate with integrin αvβ3 in promoting the migration of GBM cells (151;152). PDGFRβ can be targeted through specific tyrosine kinase inhibitors, or through antibodies directed toward PDGF-BB or PDGFRβ. Blocking either the receptor or ligand could theoretically inhibit other key downstream signaling events that promote EC survival as well as inhibit tumor cell migration (150;152). In a study of pancreatic islet cell carcinoma the investigators found an enhanced anti-angiogenic effect when combining VEGF and PDGF antagonists that resulted in anti-EC and anti-pericyte effects (153). In another study investigators also targeted the PDGF and VEGF signaling pathways simultaneously in B16 melanoma tumors propagated in C57BL/6 mice with a small molecule inhibitor of VEGFR (PTK787) and of PDGFR (STI576) (154). In this study a reduction in tumor size, likely due to a reduction in tumor vessel density and tumor vessel size, was found as well as a reduction in pericytes that were loosely attached to the tumor vessels, but no change in the number of pericytes tightly associated with the vessels was observed. Consistent with the above animal studies, in a phase I/II clinical study of patients with recurrent malignant glioma, a PDGFR inhibitor (imatinib) administered alone did not appear to be effective (155); however, when imatinib was combined with hydroxyurea in a phase II clinical study of patients with recurrent malignant glioma the results were encouraging (156). Collectively, these studies underscore the importance of evaluation of anti-angiogenesis therapy as a component of multi-modality strategies.
C) Circulating Progenitor Cells – CXCR4
The circulating progenitor cells that are recruited to angiogenic vessels and promote angiogenesis are of great interest (157-159). Whether the recruited progenitor cells are solely endothelial progenitors or are also progenitors of supportive smooth muscle cells and pericytes is an area of active research. The CXCR4 receptor expressed on circulating EPCs is recognized by SDF-1α; thus, the CXCR4 on these EPCs could be targeted therapeutically with the goal of inhibiting EPC mobilization from the bone marrow (see Fig. 2) (160). As CXCR4 is expressed on smooth muscle cell progenitors, this approach could also be used to target the recruitment of these cells to angiogenic vessels (160). Inhibition of SDF-1α signaling is likely to have multiple effects; for example, circulating EPCs also can express SDF-1α and can therefore recruit additional EPCs from the bone marrow. The importance of this signaling pathway in vasculogenesis is suggested by the severe vascular abnormalities and associated embryonic lethality exhibited by both SDF-1α and CXCR4 knockout mice (161). Interestingly, SDF-1α is expressed by the pleomorphic angiogenic vessels in the GBM tumors (39;40) suggesting that this neovasculature may recruit circulating EPCs, which would support the continued neovascularization described above (see Table I). A reduction in the number of circulating EPCs and CECs was observed when mice bearing grafted Lewis lung carcinoma tumors were administered the TSP-1 TSR mimetic, ABT-510 (157), supporting the concept that circulating EPCs contribute to angiogenesis in tumors. Thus, further investigation of the relative numbers of circulating EPCs, their biologic role in angiogenesis, and the potential efficacy of agents that target these cells in GBM tumors is warranted.
D) EC Matrix/Basement Membrane of the GBM Tumor Vasculature
(1) ED-B Isoform of Fibronectin
Matrix proteins, and fragments of matrix proteins that regulate angiogenesis, are expressed differentially in the neovasculature of GBM tumors as compared to the normal brain (162) (reviewed in (147)). Thus, these matrix proteins and fragments may be extremely useful in the directed delivery of therapies. For example, the oncofetal isoform of fibronectin (the ED-B isoform) is expressed only during embryogenesis and angiogenesis, and is a clear marker of the angiogenic vasculature in GBM tumors (162). An antibody to such a matrix molecule could be used to deliver a cytotoxic conjugate to the GBM tumor vasculature (see Fig. 2) (163;164). This could be a highly effective therapeutic approach as immunorecognition of proteins in the EC basement membrane is likely increased by the exposure of the EC basement membrane that occurs during basement membrane degradation by proteases prior to EC sprouting and by the erratic coverage of the basement membrane by ECs in angiogenic vessels.
(2) Tenascin-C
The matrix protein tenascin-C appears to play an important role in angiogenesis (see Table I and Fig. 2). In GBM tumors, tenascin-C is expressed at sites of neovascularization by vascular cells, including those at the invasive edge of the tumor, but it is not expressed in the normal brain (165). Tenascin-C promotes brain microvascular EC migration and it is synthesized by migrating brain microvascular ECs, suggesting that ECs can promote their own migration through this autocrine loop (166). In addition, tenascin-C can promote FAK phosphorylation and upregulate VEGF expression (166-168), as well as induce proliferative signals that appear to be mediated through its interactions with integrin α5β1 and syndecan-4 (167). These data suggest that the vascular cells in GBM tumors and in other malignant tumors are induced or co-opted by the tumor to synthesize and secrete the pro-angiogenic matrix protein tenascin-C.
In a phase II clinical trial, a 131I-labeled anti-tenascin-C monoclonal antibody (81C6) has been injected into the surgical resection cavity of newly-diagnosed GBM patients, followed by conventional radiotherapy and chemotherapy, and this treatment significantly increased the median survival of the GBM patients (169). In a phase II study, intra-cavitary administration of 131I-labeled anti-tenascin-C antibody (81C6) to patients with recurrent malignant glioma followed by chemotherapy also resulted in a median survival that was greater than that of historical controls treated with surgery and 125I-brachytherapy (170). As tenascin-C has multiple functional domains (171), the physiologic implications of targeting tenascin-C in GBM tumors are likely multifactorial and probably not completely understood. Nevertheless, the current and potential future use of tenascin-C as a homing signal for conjugated cytotoxic agents or anti-angiogenic agents, for radiotherapy, or for radioimaging offers intriguing possibilities (see Fig. 2). The above data suggest that the targeting of matrix proteins associated with the angiogenic vasculature in GBM tumors is feasible and can be therapeutically useful.
IV. Discussion: Clinical Implication/Applications
A) Quantitative Measures of Angiogenesis
The monitoring of anti-angiogenic therapeutic agents and delineation of their mechanism of action requires assays that can be used to accurately quantitate changes in the tumor neovasculature. Angiogenesis can be quantified in biopsies of tumor tissue using direct counts of the vessel surface area including small and large vessels or of the microvessel density after immunostaining the tissue section with an antibody directed toward von Willebrand factor, CD31, or factor VIII (4;54;88;106;107). Angiogenesis can also be assessed in biopsies of GBM tumor tissue by visualizing the decreased pericyte coverage or decreased pericyte density after immunostaining the tissue section with an antibody directed toward NG2, desmin, or smooth muscle actin, all of which are markers of pericytes (32). Methods for the assessment of angiogenesis in the tumor when biopsy tissue is not available are still under development, however, and this complicates the assessment of the clinical efficacy of anti-angiogenic therapy. A radiolabeled anti-fibronectin ED-B isoform antibody (163;164) and a radiolabeled RGD-peptide (124) have been tested as in vivo markers of the neovasculature. A promising approach is the quantitation of indirect markers of tumor angiogenesis that can be assessed non-invasively, such as the quantitation of circulating endothelial cells (CECs) and circulating EPCs (157;172). The criteria for the identification of CEC's and circulating EPCs are still being standardized, however. CEC's are thought to reflect EC damage and increased numbers are seen with tumor angiogenesis. The numbers of circulating EPCs are thought to correlate with ongoing angiogenesis and recruitment from the bone marrow. Currently, multi-channel flow cytometry analyzing a combination of markers (CD45-, CD34+, CD133+, and VEGFR2+) is used to identify circulating EPCs (157;158;172-174). Some investigators include the expression of c-kit (CD117) to identify circulating EPCs (157). Alterations in the numbers of circulating EPCs has been used with some success to measure the vascular response to treatment with the TSP-1 TSR mimetic ABT-510, to treatment with a monoclonal antibody to VEGFR2, and to treatment with a monoclonal antibody to VEGF-A (100;118;157). Quantitation of circulating EPCs and CECs also may offer a measure of the direct effect of anti-angiogenic agents on the mobilization of EPCs from the bone marrow and the sloughing of ECs from angiogenic tumor vessels, respectively.
Serum/plasma levels of certain pro-angiogenic factors, such as bFGF and SDF-1α, could also potentially be useful markers of angiogenesis. Recently Batchelor et al. (103) reported a decrease in the levels of bFGF and SDF-1α associated with anti-angiogenic therapy with AZD2171, a pan-VEGFR tyrosine kinase inhibitor. The expression of these two pro-angiogenic growth factors is upregulated on the vessels or on the pleomorphic angiogenic vessels in GBM tumors (13;39;40).
Perfusion MRI, also known as dynamic contrast-enhanced MRI, is a relatively new imaging technique that can be used for the assessment of tumor angiogenesis (reviewed in (175)). Using this technique, images can be acquired in a relatively short period of time when a patient presents for a standard contrast-enhanced MRI. Recent reports suggest this technique may be useful in monitoring neovascularization, as altered perfusion parameters have been demonstrated in brain tumors after anti-angiogenic therapy with the RGD-peptide Cilengitide (120;176).
B) The Blood Brain Barrier (BBB) and Anti-Angiogenic Therapy
The BBB is a unique functional unit that limits passive, active and facilitated transport of molecules between the blood and the brain (177;178). Key components of the BBB include, the tight junctions between endothelial cells, the pericytes in the basement membrane, and the astrocyte endfoot processes (177;178). There are a reduced number of fenestrations in brain capillaries as compared to capillaries in other organs that are responsible for uptake of molecules, and this limits transport across the BBB (177). In addition, brain endothelial cells express relatively high levels of the P-glycoprotein (Pgp/MDR-1) drug efflux pump that controls the transport of many chemotherapeutic agents, and this limits active transport across the BBB (179).
In GBM tumors the BBB is alternately intact or disrupted depending on whether the vessels are part of the tumor neovasculature or are co-opted vessels (178;180). The status of the BBB may also be dependent on the spatial localization of the vessels in the tumor, in areas lacking a neovasculature (for example some areas of the invasive margin) the BBB is intact; whereas in areas of the tumor where there is a neovasculature the vessels are leaky (disrupted BBB) (181). A disrupted BBB likely promotes access of large molecules to the tumor. Disruption of the BBB should also result in an initial increase in passive transport of small molecules to the tumor; however, passive transport is likely diminished as the disrupted BBB becomes chronic due to an equalization of the osmotic gradient. These observations result in difficulty in targeting the entire tumor with one drug at any given time, as different properties are required for the drug to gain access to the tumor depending on whether there is a disrupted or intact BBB.
Certain anti-angiogenic agents, such as inhibitors of the VEGF/VEGFR signaling pathway, cause a “normalization” of the BBB; normalization is defined as a reduction of vessel leakage and a concomitant reduction of interstitial fluid pressure in the brain parenchyma as assessed by perfusion or contrast-enhanced MRI (103). In a recent study by Batchelor et al. (2007) normalization of the neovasculature occurred post-therapy with a VEGFR inhibitor and correlated with an increased pericyte recruitment to neovessels in the tumor, suggesting pericyte coverage could be an anatomic correlate of vascular normalization in GBM tumors (103). Other anti-angiogenic agents have been shown to reduce vascular permeability associated with disruption of the BBB. For example, endostatin therapy reduced vessel density, vessel diameter, and vessel permeability in malignant glioma tumors, as well as induced a collapse of tumor vessels in a syngeneic intracerebral rat model of malignant glioma (73). This reduction in vessel permeability may have been due to an increase in pericyte coverage of tumor vessels as the authors reported an increase in pericyte number with endostatin therapy; however, pericyte coverage was not shown and the quantitation of pericytes was not described. Also, the anti-angiogenic compound thalidomide administered at teratogenic doses that inhibit angiogenesis (182) may decrease brain vessel permeability induced by stimulation with a β-amyloid peptide (183), although thalidomide used at a similar teratogenic dose was reported to induce increased EC fenestrations and thinned EC processes that correlated with inhibition of bFGF-induced neovascularization in the rabbit corneal micropocket model of angiogenesis (182).
Normalization of the tumor neovasculature with VEGF or VEGFR inhibitors may potentially alter the effectiveness of certain chemotherapeutic agents. Recently, Claes et al. (180) reported a decreased tumor response to temozolomide therapy when temozolomide was administered simultaneously with a VEGFR2 inhibitor in an intracerebral xenograft model of malignant glioma. This was interpreted to be due to a decreased localization of temozolomide to the tumor, although temozolomide levels in the tumor were not measured. Also, the anti-angiogenic compound O-(N-chloroacetyl-carbamoyl)-funagillol (TNP-470) may affect the BBB as a decreased concentration of temozolomide in the tumor was reported when TNP-470 was administered to an orthotopic mouse model of malignant glioma followed by temozolomide therapy (184). However, other reports indicate that simultaneous administration of an anti-angiogenic agent and a chemotherapeutic agent has beneficial results in orthotopic rodent models of malignant glioma. For example, thalidomide and cisplatin combination therapy resulted in a greater inhibition of tumor growth than either agent alone (185), a synthetic fragment of endostatin and carmustine combination therapy increased survival in a synergistic manner (186), and a novel small molecule tyrosine kinase inhibitor (AEE788) that blocks the activity of VEGFR2 and EGFR combined with a mTOR inhibitor (RAD001) yielded greater increases in mouse survival than either agent alone (187). Also, some clinical trial studies of patients with malignant glioma or GBM tumors indicate simultaneous combination therapy with an anti-angiogenic agent and a chemotherapeutic agent yields promising results; for example, the study utilizing a monoclonal antibody to VEGF-A (bevacizumab) and irinotecan (188), the study utilizing thalidomide and temozolomide (189), and the study utilizing thalidomide and carmustine (190). Therefore, the potential of certain anti-angiogenic agents to restore the BBB and reduce the tumor localization of certain chemotherapeutic agents suggests the target of specific anti-angiogenic therapy, the effects of different anti-angiogenic therapies on the BBB, and the time course of these effects must be determined to optimally combine anti-angiogenic therapy with chemotherapy. Agents that normalize the tumor neovasculature could be used following cytotoxic therapy, if the cytotoxic agent of choice cannot cross the BBB post-anti-angiogenic therapy. Also, one could potentially disrupt the BBB transiently for chemotherapeutic delivery purposes, such as with ultrasound, convection enhanced delivery or hypertonic solutions, post-anti-angiogenic therapy (191). One caveat of the latter approach is that there is the potential for neurotoxicity or an inflammatory response with disruption of the BBB, as it allows a broad spectrum of plasma proteins to cross into the brain.
C) Developed Resistance to Anti-Angiogenic Therapy
GBM tumors are able to escape the effectiveness of anti-angiogenic monotherapy, even though the ECs are relatively genetically stable. For example, one study showed that when dermal fibroblasts overexpressing intact TSP-1 were injected subcutaneously along with rat C6 glioma cells into nude mice, there was an initial inhibition of tumor growth and angiogenesis (192). However, prolonged exposure of the tumors to TSP-1 resulted in an increased expression of pro-angiogenic growth factors by the tumor that counter-balanced the inhibitory effect of TSP-1 on neovascularization (192). Also, in an orthotopic model of gliosarcoma propagated in the rat, downregulation of VEGF-A resulted in an increase in the expression of VEGF-D (193), and a study in patients with brain tumors treated with a VEGF inhibitor showed only a temporary normalization of the tumor vasculature (reviewed in (102)). In addition, in a study of 16 patients with recurrent GBM tumors treated with AZD2171 monotherapy (directed toward VEGFR), the 11 patients who experienced disease progression had significantly elevated levels of serum bFGF and SDF-1α (103). These reports highlight the need to move from single anti-angiogenic agent therapy to combined anti-angiogenic therapy. Moreover, anti-angiogenic therapies should be a component of a multi-modality strategy (i.e., cytotoxic and anti-angiogenic), as progressive tumor growth can occur after anti-angiogenic therapy alone despite reduced angiogenesis (194). The latter observation may be due to a co-option of the existing blood vessels by the tumor. In support of this hypothesis treatment with a monoclonal antibody toward VEGFR2 inhibited glioma angiogenesis in an orthotopic mouse model of malignant glioma; however, numerous satellite tumors arose around the primary tumor and these satellite tumors had a central vessel core (195). These data support the need to combine anti-angiogenic therapy with other treatment modalities.
Possible strategies for combining anti-angiogenic therapy with other treatment modalities would be to use agents that target more than one pathway (e.g., targeting the fibronectin ED-B isoform in the EC basement membrane or VEGFR2 on the EC, with an anti-angiogenic or cytotoxic conjugate coupled to an antibody). Alternatively, targeting small and dilated vessels in the tumor with the concurrent use of two anti-angiogenic drugs, one that targets microvessels, such as the TSP-1 TSR mimetic ABT-510, and a VEGF/VEGFR inhibitor that in GBM tumors has a more pronounced effect on enlarged (dilated) vessels could be highly efficacious. Targeting of both ECs and circulating EPCs (e.g., an antibody directed toward CXCR4 together with a TSP-1 TSR mimetic such as ABT-510) warrants consideration. Finally, two recent studies, an animal study targeting VEGF signaling and the clinical trial with AZD2171 in patients with recurrent GBM tumors, suggests that anti-angiogenic agents inhibit angiogenesis temporarily but on cessation of the therapy renewed tumor neovascularization occurs (103;196). This would indicate that cycling of administration of anti-angiogenic agents may be required. In conjunction with conventional chemotherapies and surgery, combinatorial anti-angiogenic therapies could prove efficacious, especially if used simultaneously thereby disallowing tumor adaptation and thus drug resistance.
V. Summary and Outstanding Research Questions
There are opposing actions in GBM tumors, some pro- and others anti-angiogenic, that affect the angiogenic response. Control of the angiogenic response most likely occurs at the cellular level and thus the localization of pro- and anti-angiogenic factors is important (Tables I & II and Figs. 1 & 2). The tumor overcomes the influence of the anti-angiogenic factors produced by the host stromal cells through multiple mechanisms that include: 1) Tumor cell secretion of pro-angiogenic growth factors, such as VEGF; 2) Tumor cell expression of pro-angiogenic growth factor receptors, such as PDGFR and FGFR4, that take advantage of the pro-angiogenic growth factors in the tumor microenvironment to promote their own proliferation and migration; 3) Tumor-induced expression of pro-angiogenic matrix proteins by host stromal cells, such as increased pericyte expression of NG2 and vascular cell expression of tenascin-C; and 4) Tumor-induced downregulation of anti-angiogenic factors by stromal cells, such as the downregulation of TSP-2. Thus, the mechanisms by which GBM tumors “co-opt” the host stromal cells and induce their expression of pro-angiogenic molecules or down-regulate their expression of anti-angiogenic molecules is of great interest. A new model of GBM tumors (197) derived from the PDGF-induced transformation of adult brain progenitor cells has shown that transformed progenitor cells induce proliferative changes in the resident non-transformed progenitor cells. This model could be used to study how GBM tumor cells “co-opt” the host stromal cells to enhance tumor cell growth, survival, and invasion.
Anti-angiogenic therapy appears to be an important new adjuvant therapy for GBM tumors and a number of clinical trials with anti-angiogenic agents are ongoing (see Table III); however, several practical considerations must be addressed. Most investigators agree that anti-angiogenic therapy should be combined with a cytotoxic agent for maximal benefit, but questions still remain regarding which cytotoxic agent to combine with which specific anti-angiogenic agent and whether they should be administered concurrently or in a staggered manner if the anti-angiogenic therapy restores the BBB (104;198). Similarly, questions remain concerning whether two anti-angiogenic agents with different molecular targets should be administered simultaneously and which method of delivery of anti-angiogenic agents is most effective (199). In addition, the most effective schedule for re-administration of anti-angiogenic therapy must be determined. To address these issues effectively it will be necessary to further develop techniques for measuring the response of the neovasculature in tumors during and after anti-angiogenic therapy. Re-biopsy of the tumor is costly and invasive; thus, additional assays for measurement of angiogenesis are necessary. The quantitation of CECs and circulating EPCs from blood samples pre- and post-therapy are promising new assays for assessment of tumor angiogenesis or treatment response, but the markers used for identification of these cells from blood must be optimized and standardized. The recent evidence that perfusion MRI could be used as a measure of tumor angiogenesis is exciting but could be expensive (175;176). Thus, the successful application of anti-angiogenic therapies for the treatment of GBM tumors will require additional translational research, including animal studies and phase I/II clinical trials, over the next several years.
Considerable progress has been made in developing new drugs that target the known pro-angiogenic growth factors, their receptors, and signaling molecules, as well as in the identification of endogenous inhibitors of angiogenesis that could be exploited for therapeutic purposes. Our knowledge of angiogenesis and its regulation is still in its infancy, however, especially in terms of the identification of endogenous inhibitors and their modes of action. Continued basic research is necessary and will likely provide valuable insights into how the tumors overcome the anti-angiogenic effects of endogenous inhibitors of angiogenesis, and how they adapt to anti-angiogenic therapy.
Acknowledgments
Mrs. Rhonda Carr is thanked for secretarial assistance in preparing this manuscript. We apologize for not including all relevant references due to space constraints. This work was supported by grant # CA97110 from the National Institutes of Health, National Cancer Institute to C.L.G., and an institutional T32 grant # NS048039 from the National Institute of Neurological Diseases and Stroke (JCA).
Abbreviations
- ABT-510
NAcSarGlyValDalloIleThrNvaIleArgProNHE
- Akt
protein kinase β
- BAI-1
brain angiogenesis inhibitor-1
- bFGF
basic fibroblast growth factor
- BBB
blood brain barrier
- CXCR
chemokine receptor
- CECs
circulating endothelial cells
- EC
endothelial cell
- EPC
endothelial progenitor cell
- ER
endoplasmic reticulum
- Erk
extracellular signal-regulated kinase
- FAK
focal adhesion kinase
- FGFR
fibroblast growth factor receptor
- GBM
glioblastoma tumor
- GRP78
glucose-regulated protein 78
- HIF1α
hypoxia-inducible factor 1α
- HRE
hypoxia response element
- HSP70
heat shock protein 70
- IL-8
interleukin-8
- KLF8
krupple-like factor 8
- MMP
matrix metalloproteinases
- MRI
magnetic resonance imaging
- NIH
National Institutes of Health
- PDGF
platelet-derived growth factor
- PDGFR
PDGF receptor
- Pgp
P-glycoprotein or MDR-1
- PI3K
phosphatidylinositol-3-hydroxyl kinase
- PKC
protein kinase C
- SDF-1α
stromal derived factor 1α
- TSP-1
thrombospondin-1
- TIMP
tissue inhibitor of matrix metalloproteinase
- TSR
type 1 repeat domain
- VEGF
vascular endothelial growth factor
- VEGFR
VEGF receptor
- VHL
von Hippel-Landau molecule
- vWf
von Willebrand factor
Reference List
- 1.Nabors LB, Fiveash J. Treatment of adults with recurrent malignant glioma. Expert Rev Neurother. 2005 Jul;5(4):509–14. doi: 10.1586/14737175.5.4.509. [DOI] [PubMed] [Google Scholar]
- 2.Kleihues P, Louis DN, Scheithouer BW, Rorck LLB, Reifenberg G, Burger PC, et al. The WHO classification of tumors in the nervous system. J Neuropathol Exp Neurol. 2002;60(3):215–225. doi: 10.1093/jnen/61.3.215. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Folkman J. Patterns and emering mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 4.Folkman J. Angiogenesis. Annu Rev Med. 2006;57:1–18. doi: 10.1146/annurev.med.57.121304.131306. [DOI] [PubMed] [Google Scholar]
- 5.Goldman CK, Kim J, Wong WL, Brock TA, Gillespie GY. Epidermal growth factor stimulates vascular endothelial growth factor production by human malignant glioma cells. Mol Cell Biol. 1993;4:121–33. doi: 10.1091/mbc.4.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whitelock JM, Murdoch AD, Iozzo RV, Underwood PA. The degradation of human endothelial cell-derived perlecan and release of bound basic fibroblast growth factor by stromelysin, collagenase, plasmin, and heparanases. J Biol Chem. 1996 Apr 26;271(17):10079–86. doi: 10.1074/jbc.271.17.10079. [DOI] [PubMed] [Google Scholar]
- 7.Klagsbrun M, D'Amore PA. Vascular endothelial growth factor and its receptors. Cytokine Growth Factor Rev. 1996 Oct;7(3):259–70. doi: 10.1016/s1359-6101(96)00027-5. [DOI] [PubMed] [Google Scholar]
- 8.Kukk E, Lymboussaki A, Taira S, Kaipainen A, Jeltsch M, Joukov V, et al. VEGF-C receptor binding and pattern of expression with VEGFR-3 suggests a role in lymphatic vascular development. Development. 1996 Dec;122(12):3829–37. doi: 10.1242/dev.122.12.3829. [DOI] [PubMed] [Google Scholar]
- 9.Stratmann A, Machein MR, Plate KH. Anti-angiogenic gene therapy of malignant glioma. Acta Neurochir Suppl. 1997;68:105–10. doi: 10.1007/978-3-7091-6513-3_20. [DOI] [PubMed] [Google Scholar]
- 10.Witmer AN, Dai J, Weich HA, Vrensen GF, Schlingemann RO. Expression of vascular endothelial growth factor receptors 1, 2, and 3 in quiescent endothelia. J Histochem Cytochem. 2002 Jun;50(6):767–77. doi: 10.1177/002215540205000603. [DOI] [PubMed] [Google Scholar]
- 11.Grau SJ, Trillsch F, Herms J, Thon N, Nelson PJ, Tonn JC, et al. Expression of VEGFR3 in glioma endothelium correlates with tumor grade. J Neurooncol. 2007 Apr;82(2):141–50. doi: 10.1007/s11060-006-9272-4. [DOI] [PubMed] [Google Scholar]
- 12.Witmer AN, van Blijswijk BC, Dai J, Hofman P, Partanen TA, Vrensen GF, et al. VEGFR-3 in adult angiogenesis. J Pathol. 2001 Nov;195(4):490–7. doi: 10.1002/path.969. [DOI] [PubMed] [Google Scholar]
- 13.Karcher S, Steiner HH, Ahmadi R, Zoubaa S, Vasvari G, Bauer H, et al. Different angiogenic phenotypes in primary and secondary glioblastomas. Int J Cancer. 2006 May 1;118(9):2182–9. doi: 10.1002/ijc.21648. [DOI] [PubMed] [Google Scholar]
- 14.Morrison RS, Yamaguchi F, Bruner JM, Tang M, McKeehan W, Berger MS. Fibroblast growth factor receptor gene expression and immunoreactivity are elevated in human glioblastoma multiforme. Cancer Res. 1994 May 15;54(10):2794–9. [PubMed] [Google Scholar]
- 15.Morrison RS, Yamaguchi F, Saya H, Bruner JM, Yahanda AM, Donehower LA, et al. Basic fibroblast growth factor and fibroblast growth factor receptor I are implicated in the growth of human astrocytomas. J Neurooncol. 1994;18(3):207–16. doi: 10.1007/BF01328955. [DOI] [PubMed] [Google Scholar]
- 16.Ueba T, Takahashi JA, Fukumoto M, Ohta M, Ito N, Oda Y, et al. Expression of fibroblast growth factor receptor-1 in human glioma and meningioma tissues. Neurosurgery. 1994 Feb;34(2):221–5. doi: 10.1227/00006123-199402000-00003. [DOI] [PubMed] [Google Scholar]
- 17.Yamada SM, Yamada S, Hayashi Y, Takahashi H, Teramoto A, Matsumoto K. Fibroblast growth factor receptor (FGFR) 4 correlated with the malignancy of human astrocytomas. Neurol Res. 2002 Apr;24(3):244–8. doi: 10.1179/016164102101199864. [DOI] [PubMed] [Google Scholar]
- 18.Qiao D, Meyer K, Mundhenke C, Drew SA, Friedl A. Heparan sulfate proteoglycans as regulators of fibroblast growth factor-2 signaling in brain endothelial cells. Specific role for glypican-1 in glioma angiogenesis. J Biol Chem. 2003 May 2;278(18):16045–53. doi: 10.1074/jbc.M211259200. [DOI] [PubMed] [Google Scholar]
- 19.Pintucci G, Moscatelli D, Saponara F, Biernacki PR, Baumann FG, Bizekis C, et al. Lack of ERK activation and cell migration in FGF-2-deficient endothelial cells. FASEB J. 2002 Apr;16(6):598–600. doi: 10.1096/fj.01-0815fje. [DOI] [PubMed] [Google Scholar]
- 20.Simons M. Integrative signaling in angiogenesis. Mol Cell Biochem. 2004 Sep;264(12):99–102. doi: 10.1023/b:mcbi.0000044379.25823.03. [DOI] [PubMed] [Google Scholar]
- 21.Gliki G, Wheeler-Jones C, Zachary I. Vascular endothelial growth factor induces protein kinase C (PKC)-dependent Akt/PKB activation and phosphatidylinositol 3′-kinase-mediates PKC delta phosphorylation: role of PKC in angiogenesis. Cell Biol Int. 2002;26(9):751–9. doi: 10.1016/s1065-6995(02)90926-1. [DOI] [PubMed] [Google Scholar]
- 22.Kim S, Bell K, Mousa SA, Varner JA. Regulation of Angiogenesis in Vivo by Ligation of Integrin alpha5beta1 with the Central Cell-binding Domain of Fibronectin. Am J Pathol. 2000;156(4):1345–1362. doi: 10.1016/s0002-9440(10)65005-5. April 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brooks PC, Clark RAF, Cheresh DA. Requirement of vascular integrin αvβ3 for angiogenesis. Science. 1994;264:569–71. doi: 10.1126/science.7512751. [DOI] [PubMed] [Google Scholar]
- 24.Gladson CL. Expression of integrin alpha v beta 3 in small blood vessels of glioblastoma tumors. J Neuropathol Exp Neurol. 1996 Nov;55(11):1143–9. doi: 10.1097/00005072-199611000-00005. [DOI] [PubMed] [Google Scholar]
- 25.Brat DJ, Bellail AC, Van Meir EG. The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro Oncol. 2005 Apr;7(2):122–33. doi: 10.1215/S1152851704001061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gondi CS, Lakka SS, Dinh DH, Olivero WC, Gujrati M, Rao JS. Intraperitoneal injection of a hairpin RNA-expressing plasmid targeting urokinase-type plasminogen activator (uPA) receptor and uPA retards angiogenesis and inhibits intracranial tumor growth in nude mice. Clin Cancer Res. 2007 Jul 15;13(14):4051–60. doi: 10.1158/1078-0432.CCR-06-3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lakka SS, Gondi CS, Rao JS. Proteases and glioma angiogenesis. Brain Pathol. 2005 Oct;15(4):327–41. doi: 10.1111/j.1750-3639.2005.tb00118.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ, Van Meir EG. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol. 2005 Apr;7(2):134–53. doi: 10.1215/S1152851704001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu L, Simon MC. Regulation of transcription and translation by hypoxia. Cancer Biol Ther. 2004 Jun;3(6):492–7. doi: 10.4161/cbt.3.6.1010. [DOI] [PubMed] [Google Scholar]
- 30.Overall CM, Kleifeld O. Tumour microenvironment - opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat Rev Cancer. 2006 Mar;6(3):227–39. doi: 10.1038/nrc1821. [DOI] [PubMed] [Google Scholar]
- 31.Belotti D, Paganoni P, Manenti L, Garofalo A, Marchini S, Taraboletti G, et al. Matrix metalloproteinases (MMP9 and MMP2) induce the release of vascular endothelial growth factor (VEGF) by ovarian carcinoma cells: implications for ascites formation. Cancer Res. 2003 Sep 1;63(17):5224–9. [PubMed] [Google Scholar]
- 32.Bergers G, Song S. The role of pericytes in blood-vessel formation and maintenance. Neuro-oncol. 2005;7(4):452–464. doi: 10.1215/S1152851705000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Metheny-Barlow LJ, Li LY. The enigmatic role of angiopoietin-1 in tumor angiogenesis. Cell Res. 2003 Oct;13(5):309–17. doi: 10.1038/sj.cr.7290176. [DOI] [PubMed] [Google Scholar]
- 34.Baluk P, Morikawa S, Haskell A, Mancuso M, McDonald DM. Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am J Pathol. 2003 Nov;163(5):1801–15. doi: 10.1016/S0002-9440(10)63540-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hood JD, Frausto R, Kiosses WB, Schwartz MA, Cheresh DA. Differential alphav integrin-mediated Ras-ERK signaling during two pathways of angiogenesis. J Cell Biol. 2003 Sep 1;162(5):933–43. doi: 10.1083/jcb.200304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004 Aug;10(8):858–64. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- 37.Annabi B, Naud E, Lee YT, Eliopoulos N, Galipeau J. Vascular progenitors derived from murine bone marrow stromal cells are regulated by fibroblast growth factor and are avidly recruited by vascularizing tumors. J Cell Biochem. 2004 Apr 15;91(6):1146–58. doi: 10.1002/jcb.10763. [DOI] [PubMed] [Google Scholar]
- 38.Aghi M, Cohen KS, Klein RJ, Scadden DT, Chiocca EA. Tumor stromal-derived factor-1 recruits vascular progenitors to mitotic neovasculature, where microenvironment influences their differentiated phenotypes. Cancer Res. 2006 Sep 15;66(18):9054–64. doi: 10.1158/0008-5472.CAN-05-3759. [DOI] [PubMed] [Google Scholar]
- 39.Salvucci O, Yao L, Villalba S, Sajewicz A, Pittaluga S, Tosato G. Regulation of endothelial cell branching morphogenesis by endogenous chemokine stromal-derived factor-1. Blood. 2002 Apr 15;99(8):2703–11. doi: 10.1182/blood.v99.8.2703. [DOI] [PubMed] [Google Scholar]
- 40.Rempel SA, Dudas S, Ge S, Gutierrez JA. Identification and localization of the cytokine SDF1 and its receptor, CXC chemokine receptor 4, to regions of necrosis and angiogenesis in human glioblastoma. Clin Cancer Res. 2000 Jan;6(1):102–11. [PubMed] [Google Scholar]
- 41.Rubin JB, Kung AL, Klein RS, Chan JA, Sun Y, Schmidt K, et al. A small-molecule antagonist of CXCR4 inhibits intracranial growth of primary brain tumors. Proc Natl Acad Sci U S A. 2003 Nov 11;100(23):13513–8. doi: 10.1073/pnas.2235846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vitolo D, Paradiso P, Uccini S, Ruco LP, Baroni CD. Expression of adhesion molecules and extracellular matrix proteins in glioblastomas: relation to angiogenesis and spread. Histopathology. 1996 Jun;28(6):521–8. doi: 10.1046/j.1365-2559.1996.d01-471.x. [DOI] [PubMed] [Google Scholar]
- 43.Lossinsky AS, Mossakowski MJ, Pluta R, Wisniewski HM. Intercellular adhesion molecule-1 (ICAM-1) upregulation in human brain tumors as an expression of increased blood-brain barrier permeability. Brain Pathol. 1995 Oct;5(4):339–44. doi: 10.1111/j.1750-3639.1995.tb00614.x. [DOI] [PubMed] [Google Scholar]
- 44.Boucher Y, Leunig M, Jain RK. Tumor angiogenesis and interstitial hypertension. Cancer Res. 1996 Sep 15;56(18):4264–6. [PubMed] [Google Scholar]
- 45.Adams JC, Lawler J. The thrombospondins. Int J Biochem Cell Biol. 2004;36:961–968. doi: 10.1016/j.biocel.2004.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lawler J. Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J Cell Mol Med. 2002 Jan;6(1):1–12. doi: 10.1111/j.1582-4934.2002.tb00307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jimenez B, Volpert OV, Crawford SE, Febbraio M, Silverstein RL, Bouck N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat Med. 2000;6:41–48. doi: 10.1038/71517. [DOI] [PubMed] [Google Scholar]
- 48.Volpert OV, Zaichuk T, Zhou W, Reiher F, Ferguson TA, Stuart PM, et al. Inducer-stimulated Fas targets activated endothelium for destruction by anti-angiogenic thrombospondin-1 and pigment epithelium-derived factor. Nat Med. 2002 Apr;8(4):349–57. doi: 10.1038/nm0402-349. [DOI] [PubMed] [Google Scholar]
- 49.Nor JE, Mitra RS, Sutorik MM, Mooney DJ, Castle VP, Polverini PH. Thrombospondin-1 induces endothelial cell apoptosis and inhibits angiogenesis by activating the caspase death pathway. J Vasc Res. 2000;37:209–218. doi: 10.1159/000025733. [DOI] [PubMed] [Google Scholar]
- 50.Simantov R, Silverstein RL. CD36: critical anti-angiogenic receptor. Front Biosci. 2003;8:s874–882. doi: 10.2741/1168. [DOI] [PubMed] [Google Scholar]
- 51.Bottcher A, Gaipl US, Furnrohr BG, Herrmann M, Girkontaite I, Kalden JR, et al. Involvement of phosphatidylserine, alphavbeta3, CD14, CD36, and complement C1q in the phagocytosis of primary necrotic lymphocytes by macrophages. Arthritis Rheum. 2006 Mar;54(3):927–38. doi: 10.1002/art.21660. [DOI] [PubMed] [Google Scholar]
- 52.Elzie CA, Murphy-Ullrich JE. The N-terminus of thrombospondin: the domain stands apart. Int J Biochem Cell Biol. 2004 Jun;36(6):1090–101. doi: 10.1016/j.biocel.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 53.Armstrong LC, Bjorkblom B, Hankenson KD, Siadak AW, Stiles CE, Bornstein P. Thrombospondin 2 inhibits microvascular endothelial cell proliferation by a caspase-independent mechanism. Mol Biol Cell. 2002 Jun;13(6):1893–905. doi: 10.1091/mbc.E01-09-0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anderson JC, Grammer JR, Wang W, Nabors LB, Henkin J, Stewart JE, Jr, et al. ABT-510, a Modified Type 1 Repeat Peptide of Thrombospondin, Inhibits Malignant Glioma Growth In Vivo by Inhibiting Angiogenesis. Cancer Biol Ther. 2007 Mar 26;6(3) doi: 10.4161/cbt.6.3.3630. [DOI] [PubMed] [Google Scholar]
- 55.Tenan M, Fulci G, Albertoni M, Diserens AC, Hamou MF, El Atifi-Borel M, et al. Thrombospondin-1 is downregulated by anoxia and suppresses tumorigenicity of human glioblastoma cells. J Exp Med. 2000 May 15;191(10):1789–98. doi: 10.1084/jem.191.10.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harada H, Nakagawa K, Saito M, Kohno S, Nagato S, Furukawa K, et al. Introduction of wild-type p53 enhances thrombospondin-1 expression in human glioma cells. Cancer Lett. 2003 Feb 28;191(1):109–19. doi: 10.1016/s0304-3835(02)00592-x. [DOI] [PubMed] [Google Scholar]
- 57.Pijuan-Thompson V, Grammer JR, Stewart J, Silverstein RL, Pearce SF, Tuszynski GP, et al. Retinoic acid alters the mechanism of attachment of malignant astrocytoma and neuroblastoma cells to thrombospondin-1. Exp Cell Res. 1999 May 25;249(1):86–101. doi: 10.1006/excr.1999.4458. [DOI] [PubMed] [Google Scholar]
- 58.Kawataki T, Naganuma H, Sasaki A, Yoshikawa H, Tasaka K, Nukui H. Correlation of thrombospondin-1 and transforming growth factor-beta expression with malignancy of glioma. Neuropathology. 2000 Sep;20(3):161–9. doi: 10.1046/j.1440-1789.2000.00327.x. [DOI] [PubMed] [Google Scholar]
- 59.Streit M, Riccardi L, Velasco P, Brown LF, Hawighorst T, Bornstein P, et al. Thrombospondin-2: a potent endogenous inhibitor of tumor growth and angiogenesis. Proc Natl Acad Sci U S A. 1999 Dec 21;96(26):14888–93. doi: 10.1073/pnas.96.26.14888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fears CY, Grammer JR, Stewart JE, Jr, Annis DS, Mosher DF, Bornstein P, et al. Low-density lipoprotein receptor-related protein contributes to the antiangiogenic activity of thrombospondin-2 in a murine glioma model. Cancer Res. 2005 Oct 15;65(20):9338–46. doi: 10.1158/0008-5472.CAN-05-1560. [DOI] [PubMed] [Google Scholar]
- 61.Kazuno M, Tokunaga T, Oshika Y, Tanaka Y, Tsugane R, Kijima H, et al. Thrombospondin-2 (TSP2) expression is inversely correlated with vascularity in glioma. Eur J Cancer. 1999 Mar;35(3):502–6. doi: 10.1016/s0959-8049(98)00374-8. [DOI] [PubMed] [Google Scholar]
- 62.Nishimori H, Shiratsuchi T, Urano T, Kimura Y, Kiyono K, Tatsumi K, et al. A novel brain-specific p53-target gene, BAI1, containing thrombospondin type 1 repeats inhibits experimental angiogenesis. Oncogene. 1997 Oct;15(18):2145–50. doi: 10.1038/sj.onc.1201542. [DOI] [PubMed] [Google Scholar]
- 63.Kaur B, Brat DJ, Devi NS, Van Meir EG. Vasculostatin, a proteolytic fragment of brain angiogenesis inhibitor 1, is an antiangiogenic and antitumorigenic factor. Oncogene. 2005 May 19;24(22):3632–42. doi: 10.1038/sj.onc.1208317. [DOI] [PubMed] [Google Scholar]
- 64.Kaur B, Brat DJ, Calkins CC, Van Meir EG. Brain angiogenesis inhibitor 1 is differentially expressed in normal brain and glioblastoma independently of p53 expression. Am J Pathol. 2003 Jan;162(1):19–27. doi: 10.1016/S0002-9440(10)63794-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Koh JT, Kook H, Kee HJ, Seo YW, Jeong BC, Lee JH, et al. Extracellular fragment of brain-specific angiogenesis inhibitor 1 suppresses endothelial cell proliferation by blocking alphavbeta5 integrin. Exp Cell Res. 2004 Mar 10;294(1):172–84. doi: 10.1016/j.yexcr.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 66.Kang X, Xiao X, Harata M, Bai Y, Nakazaki Y, Soda Y, et al. Antiangiogenic activity of BAI1 in vivo: implications for gene therapy of human glioblastomas. Cancer Gene Ther. 2006 Apr;13(4):385–92. doi: 10.1038/sj.cgt.7700898. [DOI] [PubMed] [Google Scholar]
- 67.Strik HM, Schluesener HJ, Seid K, Meyermann R, Deininger MH. Localization of endostatin in rat and human gliomas. Cancer. 2001 Mar 1;91(5):1013–9. [PubMed] [Google Scholar]
- 68.Morimoto T, Aoyagi M, Tamaki M, Yoshino Y, Hori H, Duan L, et al. Increased levels of tissue endostatin in human malignant gliomas. Clin Cancer Res. 2002 Sep;8(9):2933–8. [PubMed] [Google Scholar]
- 69.Folkman J. Antiangiogenesis in cancer therapy--endostatin and its mechanisms of action. Exp Cell Res. 2006 Mar 10;312(5):594–607. doi: 10.1016/j.yexcr.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 70.Heljasvaara R, Nyberg P, Luostarinen J, Parikka M, Heikkila P, Rehn M, et al. Generation of biologically active endostatin fragments from human collagen XVIII by distinct matrix metalloproteases. Exp Cell Res. 2005 Jul 15;307(2):292–304. doi: 10.1016/j.yexcr.2005.03.021. [DOI] [PubMed] [Google Scholar]
- 71.Shi H, Huang Y, Zhou H, Song X, Yuan S, Fu Y, et al. Nucleolin is a receptor that mediates antiangiogenic and antitumor activity of endostatin. Blood. 2007 Oct 15;110(8):2899–906. doi: 10.1182/blood-2007-01-064428. [DOI] [PubMed] [Google Scholar]
- 72.Sudhakar A, Sugimoto H, Yang C, Lively J, Zeisberg M, Kalluri R. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by alpha v beta 3 and alpha 5 beta 1 integrins. Proc Natl Acad Sci U S A. 2003 Apr 15;100(8):4766–71. doi: 10.1073/pnas.0730882100. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 73.Barnett FH, Scharer-Schuksz M, Wood M, Yu X, Wagner TE, Friedlander M. Intra-arterial delivery of endostatin gene to brain tumors prolongs survival and alters tumor vessel ultrastructure. Gene Ther. 2004 Aug;11(16):1283–9. doi: 10.1038/sj.gt.3302287. [DOI] [PubMed] [Google Scholar]
- 74.Sorensen DR, Read TA, Porwol T, Olsen BR, Timpl R, Sasaki T, et al. Endostatin reduces vascularization, blood flow, and growth in a rat gliosarcoma. Neuro Oncol. 2002 Jan;4(1):1–8. doi: 10.1215/15228517-4-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maeshima Y, Colorado PC, Torre A, Holthaus KA, Grunkemeyer JA, Ericksen MB, et al. Distinct antitumor properties of a type IV collagen domain derived from basement membrane. J Biol Chem. 2000 Jul 14;275(28):21340–8. doi: 10.1074/jbc.M001956200. [DOI] [PubMed] [Google Scholar]
- 76.Maeshima Y, Colorado PC, Kalluri R. Two RGD-independent alpha vbeta 3 integrin binding sites on tumstatin regulate distinct anti-tumor properties. J Biol Chem. 2000 Aug 4;275(31):23745–50. doi: 10.1074/jbc.C000186200. [DOI] [PubMed] [Google Scholar]
- 77.Mundel TM, Kalluri R. Type IV collagen-derived angiogenesis inhibitors. Microvasc Res. 2007 May 25; doi: 10.1016/j.mvr.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.O'Reilly MS, Holmgren L, Shing Y, Chen C, Rosenthal RA, Moses M, et al. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell. 1994 Oct 21;79(2):315–28. doi: 10.1016/0092-8674(94)90200-3. [DOI] [PubMed] [Google Scholar]
- 79.Wahl ML, Kenan DJ, Gonzalez-Gronow M, Pizzo SV. Angiostatin's molecular mechanism: aspects of specificity and regulation elucidated. J Cell Biochem. 2005 Oct 1;96(2):242–61. doi: 10.1002/jcb.20480. [DOI] [PubMed] [Google Scholar]
- 80.Kirsch M, Strasser J, Allende R, Bello L, Zhang J, Black PM. Angiostatin suppresses malignant glioma growth in vivo. Cancer Res. 1998 Oct 15;58(20):4654–9. [PubMed] [Google Scholar]
- 81.Joe YA, Hong YK, Chung DS, Yang YJ, Kang JK, Lee YS, et al. Inhibition of human malignant glioma growth in vivo by human recombinant plasminogen kringles 1-3. Int J Cancer. 1999 Aug 27;82(5):694–9. doi: 10.1002/(sici)1097-0215(19990827)82:5<694::aid-ijc12>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 82.Tarui T, Miles LA, Takada Y. Specific interaction of angiostatin with integrin alpha(v)beta(3) in endothelial cells. J Biol Chem. 2001 Oct 26;276(43):39562–8. doi: 10.1074/jbc.M101815200. [DOI] [PubMed] [Google Scholar]
- 83.Chekenya M, Hjelstuen M, Enger PO, Thorsen F, Jacob AL, Probst B, et al. NG2 proteoglycan promotes angiogenesis-dependent tumor growth in CNS by sequestering angiostatin. FASEB J. 2002 Apr;16(6):586–8. doi: 10.1096/fj.01-0632fje. [DOI] [PubMed] [Google Scholar]
- 84.Cao Y, Chen A, An SSA, Ji RW, Davidson D, Cao Y, et al. Kringle 5 of plasminogen is a novel inhibitor of endothelial cell growth. J Biol Chem. 1997;272:22924–8. doi: 10.1074/jbc.272.36.22924. [DOI] [PubMed] [Google Scholar]
- 85.Davidson DJ, Haskell C, Majest S, Kherzai A, Egan DA, Walter KA, et al. Kringle 5 of human plasminogen induces apoptosis of endothelial and tumor cells through surface-expressed glucose-regulated protein 78. Cancer Res. 2005 Jun 1;65(11):4663–72. doi: 10.1158/0008-5472.CAN-04-3426. [DOI] [PubMed] [Google Scholar]
- 86.Lu H, Dhanabal M, Volk R, Waterman MJ, Ramchandran R, Knebelmann B, et al. Kringle 5 causes cell cycle arrest and apoptosis of endothelial cells. Biochem Biophys Res Commun. 1999 May 19;258(3):668–73. doi: 10.1006/bbrc.1999.0612. [DOI] [PubMed] [Google Scholar]
- 87.Zhang D, Kaufman PL, Gao G, Saunders RA, Ma JX. Intravitreal injection of plasminogen kringle 5, an endogenous angiogenic inhibitor, arrests retinal neovascularization in rats. Diabetologia. 2001 Jun;44(6):757–65. doi: 10.1007/s001250051685. [DOI] [PubMed] [Google Scholar]
- 88.Perri SR, Nalbantoglu J, Annabi B, Koty Z, Lejeune L, Francois M, et al. Plasminogen kringle 5-engineered glioma cells block migration of tumor-associated macrophages and suppress tumor vascularization and progression. Cancer Res. 2005 Sep 15;65(18):8359–65. doi: 10.1158/0008-5472.CAN-05-0508. [DOI] [PubMed] [Google Scholar]
- 89.Eklund L, Olsen BR. Tie receptors and their angiopoietin ligands are context-dependent regulators of vascular remodeling. Exp Cell Res. 2006 Mar 10;312(5):630–41. doi: 10.1016/j.yexcr.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 90.Zagzag D, Amirnovin R, Greco MA, Yee H, Holash J, Wiegand SJ, et al. Vascular apoptosis and involution in gliomas precede neovascularization: a novel concept for glioma growth and angiogenesis. Lab Invest. 2000;80:837–49. doi: 10.1038/labinvest.3780088. [DOI] [PubMed] [Google Scholar]
- 91.Stratmann A, Risau W, Plate KH. Cell type-specific expression of angiopoietin-1 and angiopoietin-2 suggests a role in glioblastoma angiogenesis. Am J Pathol. 1998 Nov;153(5):1459–66. doi: 10.1016/S0002-9440(10)65733-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Groft LL, Muzik H, Rewcastle NB, Johnston RN, Knauper V, Lafleur MA, et al. Differential expression and localization of TIMP-1 and TIMP-4 in human gliomas. Br J Cancer. 2001 Jul 6;85(1):55–63. doi: 10.1054/bjoc.2001.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tanaka T, Manome Y, Wen P, Kufe DW, Fine HA. Viral vector-mediated transduction of a modified platelet factor 4 cDNA inhibits angiogenesis and tumor growth. Nat Med. 1997 Apr;3(4):437–42. doi: 10.1038/nm0497-437. [DOI] [PubMed] [Google Scholar]
- 94.Bikfalvi A. Recent developments in the inhibition of angiogenesis: examples from studies on platelet factor-4 and the VEGF/VEGFR system. Biochem Pharmacol. 2004 Sep 15;68(6):1017–21. doi: 10.1016/j.bcp.2004.05.030. [DOI] [PubMed] [Google Scholar]
- 95.Mongiat M, Sweeney SM, San Antonio JD, Fu J, Iozzo RV. Endorepellin, a novel inhibitor of angiogenesis derived from the C terminus of perlecan. J Biol Chem. 2003 Feb 7;278(6):4238–49. doi: 10.1074/jbc.M210445200. [DOI] [PubMed] [Google Scholar]
- 96.Bix G, Fu J, Gonzalez EM, Macro L, Barker A, Campbell S, et al. Endorepellin causes endothelial cell disassembly of actin cytoskeleton and focal adhesions through alpha2beta1 integrin. J Cell Biol. 2004 Jul 5;166(1):97–109. doi: 10.1083/jcb.200401150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Brooks PC, Silletti S, von Schalscha TL, Friedlander M, Cheresh DA. Disruption of angiogenesis by PEX, a noncatalytic metalloproteinase fragment with integrin binding activity. Cell. 1998 Feb 6;92(3):391–400. doi: 10.1016/s0092-8674(00)80931-9. [DOI] [PubMed] [Google Scholar]
- 98.Guan M, Yam HF, Su B, Chan KP, Pang CP, Liu WW, et al. Loss of pigment epithelium derived factor expression in glioma progression. J Clin Pathol. 2003 Apr;56(4):277–82. doi: 10.1136/jcp.56.4.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang T, Guan M, Xu C, Chen Y, Lu Y. Pigment epithelium-derived factor inhibits glioma cell growth in vitro and in vivo. Life Sci. 2007 Sep 29;81(16):1256–63. doi: 10.1016/j.lfs.2007.08.025. [DOI] [PubMed] [Google Scholar]
- 100.Willett CG, Boucher Y, di TE, Duda DG, Munn LL, tong RT, et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med. 2004 Feb;10(2):145–7. doi: 10.1038/nm988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jain RK. Anti-angiogenic therapy for cancer: current and emerging concepts. Oncology. 2005;19:7–16. [PubMed] [Google Scholar]
- 102.Jain RK, di TE, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nat Rev Neurosci. 2007 Aug;8(8):610–22. doi: 10.1038/nrn2175. [DOI] [PubMed] [Google Scholar]
- 103.Batchelor TT, Sorensen AG, di TE, Zhang WT, Duda DG, Cohen KS, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007 Jan;11(1):83–95. doi: 10.1016/j.ccr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Winkler F, Kozin SV, tong RT, Chae SS, Booth MF, Garkavtsev I, et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell. 2004 Dec;6(6):553–63. doi: 10.1016/j.ccr.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 105.Stefanik DF, Rizkalla LR, Soi A, Goldblatt SA, Rizkalla WM. Acidic and basic fibroblast growth factors are present in glioblastoma multiforme. Cancer Res. 1991 Oct 15;51(20):5760–5. [PubMed] [Google Scholar]
- 106.Reiher FK, Volpert OV, Jimenez B, Crawford SE, Dinney CP, Henkin J, et al. Inhibition of tumor growth by systemic treatment with thrombospondin-1 peptide mimectics. Int J Cancer. 2002;98(5):682–689. doi: 10.1002/ijc.10247. [DOI] [PubMed] [Google Scholar]
- 107.Haviv F, Braadley MF, Kalvin DM, Schneider AJ, Davidson DJ, Majest SM, et al. Thrombospondin-1 mimetic peptide inhibitors of angiogenesis and tumor growth: design, synthesis and optimization of pharmacokinetics and biuological activities. J Med Chem. 2005;48(8):2838–2846. doi: 10.1021/jm0401560. [DOI] [PubMed] [Google Scholar]
- 108.Barnwell JW, Asch AS, Nachman R, Yamaya M, Aikawa M, Ingravaile P. A human 88-kD membrane glycoprotein (CD36) function, in vitro as a receptor for a cytoadherence ligand and Plasmodium falciparon-infected eerythrocytes. J Clin Invest. 1989;84:765–772. doi: 10.1172/JCI114234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Husemann J, Loike JD, Anankov R, Febbraio M, Silverstein SC. Scavenger receptors in neurobiology and neuropathology: their role on microglia and other cells of the nervous system. GLIA. 2002;40:195–205. doi: 10.1002/glia.10148. [DOI] [PubMed] [Google Scholar]
- 110.Febbraio M, Hajjar DP, Silverstein RL. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest. 2001 Sep;108(6):785–91. doi: 10.1172/JCI14006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bogdanov A, Jr, Marecos E, Cheng HC, Chandrasekaran L, Krutzsch HC, Roberts DD, et al. Treatment of experimental brain tumors with trombospondin-1 derived peptides: an in vivo imaging study. Neoplasia. 1999 Nov;1(5):438–45. doi: 10.1038/sj.neo.7900044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.de Fraipont F, Keramidas M, El AM, Chambaz EM, Berger F, Feige JJ. Expression of the thrombospondin 1 fragment 167-569 in C6 glioma cells stimulates tumorigenicity despite reduced neovascularization. Oncogene. 2004 Apr 29;23(20):3642–9. doi: 10.1038/sj.onc.1207438. [DOI] [PubMed] [Google Scholar]
- 113.Kragh M, Quistorff B, Tenan M, Van Meir EG, Kristjansen PE. Overexpression of thrombospondin-1 reduces growth and vascular index but not perfusion in glioblastoma. Cancer Res. 2002 Feb 15;62(4):1191–5. [PubMed] [Google Scholar]
- 114.Yang Q, Tian Y, Liu S, Zeine R, Chlenski A, Salwen HR, et al. Thrombospondin-1 peptide ABT-510 combined with valproic acid is an effective antiangiogenesis strategy in neuroblastoma. Cancer Res. 2007 Feb 15;67(4):1716–24. doi: 10.1158/0008-5472.CAN-06-2595. [DOI] [PubMed] [Google Scholar]
- 115.Quesada A, Nelius T, Yao R, Zaichuk TA, Alfranca A, Filleur A, et al. In vivo up regulation of CD95 and CD95L causes synergistic inhibition of angiogenesis by TSP-1 peptide and metronomic doxorubicin treatment. Cell Death Differentiation. 2005;12:549–558. doi: 10.1038/sj.cdd.4401615. [DOI] [PubMed] [Google Scholar]
- 116.Kang JH, Kim SA, Chang SY, Hong S, Hong KJ. Inhibition of trichostatin A-induced antiangiogenesis by small-interfering RNA for thrombospondin-1. Exp Mol Med. 2007 Jun 30;39(3):402–11. doi: 10.1038/emm.2007.45. [DOI] [PubMed] [Google Scholar]
- 117.Hoekstra R, de Vos FY, Eskens FA, Gietema JA, van der Gaast A, Groen HJ, et al. Phase I saftey, pharmacokinetic and pharmacodynamic study of the thrombospondin-1-mimetic angiogenesis inhibitor ABT-510 in patients with advanced cancer. J Clin Oncol. 2005;23(22):5188–5197. doi: 10.1200/JCO.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 118.Markovic SN, Suman VJ, Rao RA, Ingle JN, Kaur JS, Erickson LA, et al. A phase II study of ABT-510 (thrombospondin-1 analog) for the treatment of metastatic melanoma. Am J Clin Oncol. 2007 Jun;30(3):303–9. doi: 10.1097/01.coc.0000256104.80089.35. [DOI] [PubMed] [Google Scholar]
- 119.Gingras MC, Roussel E, Bruner JM, Branch CD, Moser RP. Comparison of cell adhesion molecule expression between glioblastoma multiforme and autologous normal brain tissue. J Neuroimmunol. 1995 Mar;57(12):143–53. doi: 10.1016/0165-5728(94)00178-q. [DOI] [PubMed] [Google Scholar]
- 120.Nabors LB, Mikkelsen T, Rosenfeld SS, Hochberg F, Akella NS, Fisher JD, et al. Phase I and correlative biology study of cilengitide in patients with recurrent malignant glioma. J Clin Oncol. 2007 May 1;25(13):1651–7. doi: 10.1200/JCO.2006.06.6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chatterjee S, Matsumura A, Schradermeier J, Gillespie GY. Human malignant glioma therapy using anti-alpha(v)beta3 integrin agents. J Neurooncol. 2000;46(2):135–44. doi: 10.1023/a:1006444300504. [DOI] [PubMed] [Google Scholar]
- 122.Yamada S, Bu XY, Khankaldyyan V, Gonzales-Gomez I, McComb JG, Laug WE. Effect of the angiogenesis inhibitor Cilengitide (EMD 121974) on glioblastoma growth in nude mice. Neurosurgery. 2006 Dec;59(6):1304–12. doi: 10.1227/01.NEU.0000245622.70344.BE. [DOI] [PubMed] [Google Scholar]
- 123.Reinmuth N, Liu W, Ahmad SA, Fan F, Stoeltzing O, Parikh AA, et al. Alphavbeta3 integrin antagonist S247 decreases colon cancer metastasis and angiogenesis and improves survival in mice. Cancer Res. 2003 May 1;63(9):2079–87. [PubMed] [Google Scholar]
- 124.Janssen ML, Oyen WJ, Dijkgraaf I, Massuger LF, Frielink C, Edwards DS, et al. Tumor targeting with radiolabeled alpha(v)beta(3) integrin binding peptides in a nude mouse model. Cancer Res. 2002 Nov 1;62(21):6146–51. [PubMed] [Google Scholar]
- 125.Li J, Lee AS. Stress induction of GRP78/BiP and its role in cancer. Curr Mol Med. 2006 Feb;6(1):45–54. doi: 10.2174/156652406775574523. [DOI] [PubMed] [Google Scholar]
- 126.Shin BK, Wang H, Yim AM, Le Naour F, Brichory F, Jang JH, et al. Global profiling of the cell surface proteome of cancer cells uncovers an abumdance of proteins with chaperone function. J Biol Chem. 2003;278(9):7607–7616. doi: 10.1074/jbc.M210455200. [DOI] [PubMed] [Google Scholar]
- 127.Arap MA, Lahdenranta J, Mintz p, Hajitou A, Sarkis AS, Arap W, et al. Cell surface expression of the stress response chaperone GRP78 enables tumor targeting by circulating ligands. Cancer Cell. 2004;6:275–284. doi: 10.1016/j.ccr.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 128.Bini L, Magi B, Marzocchi B, Arcuri F, Tripodi S, Cintorino M, et al. Protein expression profiles in human breast ductal carcinoma and histologically normal tissue. Electrophoresis. 1997 Dec;18(15):2832–41. doi: 10.1002/elps.1150181519. [DOI] [PubMed] [Google Scholar]
- 129.Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, et al. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995 Oct 12;377(6549):539–44. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- 130.Haskell H, Natarajan M, Hecker TP, Ding Q, Stewart J, Jr, Grammer JR, et al. Focal adhesion kinase is expressed in the angiogenic blood vessels of malignant astrocytic tumors in vivo and promotes capillary tube formation of brain microvascular endothelial cells. Clin Cancer Res. 2003 Jun;9(6):2157–65. [PubMed] [Google Scholar]
- 131.Cox BD, Natarajan M, Stettner MR, Gladson CL. New concepts regarding focal adhesion kinase promotion of cell migration and proliferation. J Cell Biochem. 2006 Jul 5; doi: 10.1002/jcb.20956. [DOI] [PubMed] [Google Scholar]
- 132.Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003 Apr 15;116(Pt 8):1409–16. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- 133.Zhao J, Zheng C, Guan J. Pyk2 and FAK differentially regulate progression of the cell cycle. J Cell Sci. 2000 Sep;113(Pt 17):3063–72. doi: 10.1242/jcs.113.17.3063. [DOI] [PubMed] [Google Scholar]
- 134.Zhao J, Bian ZC, Yee K, Chen BP, Chien S, Guan JL. Identification of transcription factor KLF8 as a downstream target of focal adhesion kinase in its regulation of cyclin D1 and cell cycle progression. Mol Cell. 2003 Jun;11(6):1503–15. doi: 10.1016/s1097-2765(03)00179-5. [DOI] [PubMed] [Google Scholar]
- 135.Ding Q, Grammer JR, Nelson MA, Guan JL, Stewart JE, Jr, Gladson CL. p27Kip1 and cyclin D1 are necessary for focal adhesion kinase regulation of cell cycle progression in glioblastoma cells propagated in vitro and in vivo in the scid mouse brain. J Biol Chem. 2005 Feb 25;280(8):6802–15. doi: 10.1074/jbc.M409180200. [DOI] [PubMed] [Google Scholar]
- 136.Owen JD, Ruest PJ, Fry DW, Hanks SK. Induced focal adhesion kinase (FAK) expression in FAK null cells enhances cell spreading and migration requiring bothe auto- and activation lo9op phosphorylation sites and inhibits adhesion-dependent tyrosine phosphorylation of Pyk2. Mol Cell Biol. 1999;19:4806–18. doi: 10.1128/mcb.19.7.4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Natarajan M, Stewart JE, Golemis EA, Pugacheva EN, Alexandropoulos K, Cox BD, et al. HEF1 is a necessary and specific downstream effector of FAK that promotes the migration of glioblastoma cells. Oncogene. 2006 Mar 16;25(12):1721–32. doi: 10.1038/sj.onc.1209199. [DOI] [PubMed] [Google Scholar]
- 138.Shi Q, Hjelmeland AB, Keir ST, Song L, Wickman S, Jackson D, et al. A novel low-molecular weight inhibitor of focal adhesion kinase, TAE226, inhibits glioma growth. Mol Carcinog. 2007 Jun;46(6):488–96. doi: 10.1002/mc.20297. [DOI] [PubMed] [Google Scholar]
- 139.Slack-Davis JK, Martin KH, Tilghman RW, Iwanicki M, Ung EJ, Autry C, et al. Cellular characterization of a novel focal adhesion kinase inhibitor. J Biol Chem. 2007 May 18;282(20):14845–52. doi: 10.1074/jbc.M606695200. [DOI] [PubMed] [Google Scholar]
- 140.Lipinski CA, Tran NL, Bay C, Kloss J, McDonough WS, Beaudry C, et al. Differential role of proline-rich tyrosine kinase 2 and focal adhesion kinase in determining glioblastoma migration and proliferation. Mol Cancer Res. 2003;1:323–32. [PubMed] [Google Scholar]
- 141.Lipinski CA, Tran NL, Menashi E, Rohl C, Kloss J, Bay RC, et al. The tyrosine kinase pyk2 promotes migration and invasion of glioma cells. Neoplasia. 2005 May;7(5):435–45. doi: 10.1593/neo.04712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bach F, Uddin FJ, Burke D. Angiopoietins in malignancy. Eur J Surg Oncol. 2007 Feb;33(1):7–15. doi: 10.1016/j.ejso.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 143.Lobov IB, Brooks PC, Lang RA. Angiopoietin-2 displays VEGF-dependent modulation of capillary structure and endothelial cell survival in vivo. Proc Natl Acad Sci U S A. 2002 Aug 20;99(17):11205–10. doi: 10.1073/pnas.172161899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, ZagZag D, et al. Vessel Cooption, Regression, and Growth in Tumors Mediated by Angiopoietins and VEGF. Science. 1999;284:1994–1998. doi: 10.1126/science.284.5422.1994. 18 June 1999. [DOI] [PubMed] [Google Scholar]
- 145.Carlson TR, Feng Y, Maisonpierre PC, Mrksich M, Morla AO. Direct cell adhesion to the angiopoietins mediated by integrins. J Biol Chem. 2001 Jul 13;276(28):26516–25. doi: 10.1074/jbc.M100282200. [DOI] [PubMed] [Google Scholar]
- 146.Chekenya M, Enger PO, Thorsen F, Tysnes BB, Al-Sarraj S, Read TA, et al. The glial precursor proteoglycan, NG2, is expressed on tumour neovasculature by vascular pericytes in human malignant brain tumours. Neuropathol Appl Neurobiol. 2002 Oct;28(5):367–80. doi: 10.1046/j.1365-2990.2002.00412.x. [DOI] [PubMed] [Google Scholar]
- 147.Wang D, Anderson JC, Gladson CL. The role of the extracellular matrix in angiogenesis in malignant glioma tumors. Brain Pathol. 2005 Oct;15(4):318–26. doi: 10.1111/j.1750-3639.2005.tb00117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Schrappe M, Klier FG, Spiro RC, Waltz TA, Reisfeld RA, Gladson CL. Correlation of chondroitin sulfate proteoglycan expression on proliferation brain capillary endothelial cells wiht the malignant phenotype of astroglial cells. Cancer Res. 1991;51:4986–93. [PubMed] [Google Scholar]
- 149.Hellstrom M, Kalen M, Lindahl P, Abramsson A, Betsholtz C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999 Jun;126(14):3047–55. doi: 10.1242/dev.126.14.3047. [DOI] [PubMed] [Google Scholar]
- 150.Guo P, Hu B, Gu W, Xu L, Wang D, Huang HJ, et al. Platelet-derived growth factor-B enhances glioma angiogenesis by stimulating vascular endothelial growth factor expression in tumor endothelia and by promoting pericyte recruitment. Am J Pathol. 2003 Apr;162(4):1083–93. doi: 10.1016/S0002-9440(10)63905-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hermanson M, Funa K, Koopmann J, Maintz D, Waha A, Westermark B, et al. Association of loss of heterozygosity on chromosome 17p with high platelet-derived growth factor alpha receptor expression in human malignant gliomas. Cancer Res. 1996 Jan 1;56(1):164–71. [PubMed] [Google Scholar]
- 152.Ding Q, Stewart JE, Jr, Olman MA, Klobe MR, Gladson CL. The pattern of enhancement of Src kinase activity on platelet-derived growth factor stimulation of glioblastoma cells is affected by the integrin engaged. J Biol Chem. 2003;278:39882–39891. doi: 10.1074/jbc.M304685200. [DOI] [PubMed] [Google Scholar]
- 153.Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003 May;111(9):1287–95. doi: 10.1172/JCI17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hasumi Y, Klosowska-Wardega A, Furuhashi M, Ostman A, Heldin CH, Hellberg C. Identification of a subset of pericytes that respond to combination therapy targeting PDGF and VEGF signaling. Int J Cancer. 2007 Dec 15;121(12):2606–14. doi: 10.1002/ijc.22999. [DOI] [PubMed] [Google Scholar]
- 155.Wen PY, Yung WK, Lamborn KR, Dahia PL, Wang Y, Peng B, et al. Phase I/II study of imatinib mesylate for recurrent malignant gliomas: North American Brain Tumor Consortium Study 99-08. Clin Cancer Res. 2006 Aug 15;12(16):4899–907. doi: 10.1158/1078-0432.CCR-06-0773. [DOI] [PubMed] [Google Scholar]
- 156.Reardon DA, Egorin MJ, Quinn JA, Rich JN, Gururangan S, Vredenburgh JJ, et al. Phase II study of imatinib mesylate plus hydroxyurea in adults with recurrent glioblastoma multiforme. J Clin Oncol. 2005 Dec 20;23(36):9359–68. doi: 10.1200/JCO.2005.03.2185. [DOI] [PubMed] [Google Scholar]
- 157.Shaked Y, Bertolini F, Man S, Rogers MS, Cervi D, Foutz T, et al. Genetic heterogeneity of the vasculogenic phenotype parallels angiogenesis; Implications for cellular surrogate marker analysis of antiangiogenesis. Cancer Cell. 2005 Jan;7(1):101–11. doi: 10.1016/j.ccr.2004.11.023. [DOI] [PubMed] [Google Scholar]
- 158.Dome B, Timar J, Dobos J, Meszaros L, Raso E, Paku S, et al. Identification and clinical significance of circulating endothelial progenitor cells in human non-small cell lung cancer. Cancer Res. 2006 Jul 15;66(14):7341–7. doi: 10.1158/0008-5472.CAN-05-4654. [DOI] [PubMed] [Google Scholar]
- 159.Jain RK, Duda DG. Role of bone marrow-derived cells in tumor angiogenesis and treatment. Cancer Cell. 2003 Jun;3(6):515–6. doi: 10.1016/s1535-6108(03)00138-7. [DOI] [PubMed] [Google Scholar]
- 160.Zernecke A, Schober A, Bot I, von HP, Liehn EA, Mopps B, et al. SDF-1alpha/CXCR4 axis is instrumental in neointimal hyperplasia and recruitment of smooth muscle progenitor cells. Circ Res. 2005 Apr 15;96(7):784–91. doi: 10.1161/01.RES.0000162100.52009.38. [DOI] [PubMed] [Google Scholar]
- 161.Tachibana K, Hirota S, Iizasa H, Yoshida H, Kawabata K, Kataoka Y, et al. The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature. 1998 Jun 11;393(6685):591–4. doi: 10.1038/31261. [DOI] [PubMed] [Google Scholar]
- 162.Castellani P, Viale G, Dorcaratto A, Nicolo G, Kaczmarek J, Querze G, et al. The fibronectin isoform containing the ED-B oncofetal domain: a marker of angiogenesis. Int J Cancer. 1994 Dec 1;59(5):612–8. doi: 10.1002/ijc.2910590507. [DOI] [PubMed] [Google Scholar]
- 163.Mariani G, Lasku A, Balza E, Gaggero B, Motta C, Di LL, et al. Tumor targeting potential of the monoclonal antibody BC-1 against oncofetal fibronectin in nude mice bearing human tumor implants. Cancer. 1997 Dec 15;80(12 Suppl):2378–84. doi: 10.1002/(sici)1097-0142(19971215)80:12+<2378::aid-cncr7>3.3.co;2-0. [DOI] [PubMed] [Google Scholar]
- 164.Tarli L, Balza E, Viti F, Borsi L, Castellani P, Berndorff D, et al. A high-affinity human antibody that targets tumoral blood vessels. Blood. 1999 Jul 1;94(1):192–8. [PubMed] [Google Scholar]
- 165.Zagzag D, Friedlander DR, Dosik J, Chikramane S, Chan W, Greco MA, et al. Tenascin-C expression by angiogenic vessels in human astrocytomas and by human brain endothelial cells in vitro. Cancer Res. 1996;56(1):182–9. [PubMed] [Google Scholar]
- 166.ZagZag D, Shiff B, Jallo GI, Greco MA, Blanco C, Cohen H, et al. Tenascin-C promotes microvascular cell migration and phosphorylation of focal adhesion kinase. Cancer Res. 2002 May 1;62(9):2660–8. [PubMed] [Google Scholar]
- 167.Huang W, Chiquet-Ehrismann R, Moyano JV, Garcia-Pardo A, Orend G. Interference of tenascin-C with syndecan-4 binding to fibronectin blocks cell adhesion and stimulates tumor cell proliferation. Cancer Res. 2001 Dec 1;61(23):8586–94. [PubMed] [Google Scholar]
- 168.Orend G, Huang W, Olayioye MA, Hynes NE, Chiquet-Ehrismann R. Tenascin-C blocks cell-cycle progression of anchorage-dependent fibroblasts on fibronectin through inhibition of syndecan-4. Oncogene. 2003 Jun 19;22(25):3917–26. doi: 10.1038/sj.onc.1206618. [DOI] [PubMed] [Google Scholar]
- 169.Akabani G, Reardon DA, Coleman RE, Wong TZ, Metzler SD, Bowsher JE, et al. Dosimetry and radiographic analysis of 131I-labeled anti-tenascin 81C6 murine monoclonal antibody in newly diagnosed patients with malignant gliomas: a phase II study. J Nucl Med. 2005 Jun;46(6):1042–51. [PubMed] [Google Scholar]
- 170.Reardon DA, Akabani G, Coleman RE, Friedman AH, Friedman HS, Herndon JE, et al. Phase II trial of murine (131)I-labeled antitenascin monoclonal antibody 81C6 administered into surgically created resection cavities of patients with newly diagnosed malignant gliomas. J Clin Oncol. 2002 Mar 1;20(5):1389–97. doi: 10.1200/JCO.2002.20.5.1389. [DOI] [PubMed] [Google Scholar]
- 171.Jones PL, Jones FS. Tenascin-C in development and disease: gene regulation and cell function. Matrix Biol. 2000 Dec;19(7):581–96. doi: 10.1016/s0945-053x(00)00106-2. [DOI] [PubMed] [Google Scholar]
- 172.Duda DG, Cohen KS, Scadden DT, Jain RK. A protocol for phenotypic detection and enumeration of circulating endothelial cells and circulating progenitor cells in human blood. Nat Protoc. 2007;2(4):805–10. doi: 10.1038/nprot.2007.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Mancuso P, Burlini A, Pruneri G, Goldhirsch A, Martinelli G, Bertolini F. Resting and activated endothelial cells are increased in the peripheral blood of cancer patients. Blood. 2001 Jun 1;97(11):3658–61. doi: 10.1182/blood.v97.11.3658. [DOI] [PubMed] [Google Scholar]
- 174.Mancuso P, Colleoni M, Calleri A, Orlando L, Maisonneuve P, Pruneri G, et al. Circulating endothelial-cell kinetics and viability predict survival in breast cancer patients receiving metronomic chemotherapy. Blood. 2006 Jul 15;108(2):452–9. doi: 10.1182/blood-2005-11-4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Barrett T, Brechbiel M, Bernardo M, Choyke PL. MRI of tumor angiogenesis. J Magn Reson Imaging. 2007 Aug;26(2):235–49. doi: 10.1002/jmri.20991. [DOI] [PubMed] [Google Scholar]
- 176.Akella NS, Twieg DB, Mikkelsen T, Hochberg FH, Grossman S, Cloud GA, et al. Assessment of brain tumor angiogenesis inhibitors using perfusion magnetic resonance imaging: quality and analysis results of a phase I trial. J Magn Reson Imaging. 2004 Dec;20(6):913–22. doi: 10.1002/jmri.20202. [DOI] [PubMed] [Google Scholar]
- 177.Hawkins BT, Egleton RD. Pathophysiology of the blood-brain barrier: animal models and methods. Curr Top Dev Biol. 2008;80:277–309. doi: 10.1016/S0070-2153(07)80007-X. [DOI] [PubMed] [Google Scholar]
- 178.Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004 Jun;16(1):1–13. doi: 10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 179.Pardridge WM, Golden PL, Kang YS, Bickel U. Brain microvascular and astrocyte localization of P-glycoprotein. J Neurochem. 1997 Mar;68(3):1278–85. doi: 10.1046/j.1471-4159.1997.68031278.x. [DOI] [PubMed] [Google Scholar]
- 180.Claes A, Wesseling P, Jeuken J, Maass C, Heerschap A, Leenders WP. Antiangiogenic compounds interfere with chemotherapy of brain tumors due to vessel normalization. Mol Cancer Ther. 2008 Jan;7(1):71–8. doi: 10.1158/1535-7163.MCT-07-0552. [DOI] [PubMed] [Google Scholar]
- 181.Muldoon LL, Soussain C, Jahnke K, Johanson C, Siegal T, Smith QR, et al. Chemotherapy delivery issues in central nervous system malignancy: a reality check. J Clin Oncol. 2007 Jun 1;25(16):2295–305. doi: 10.1200/JCO.2006.09.9861. [DOI] [PubMed] [Google Scholar]
- 182.D'Amato RJ, Loughnan MS, Flynn E, Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci U S A. 1994 Apr 26;91(9):4082–5. doi: 10.1073/pnas.91.9.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ryu JK, McLarnon JG. Thalidomide inhibition of perturbed vasculature and glial-derived tumor necrosis factor-alpha in an animal model of inflamed Alzheimer's disease brain. Neurobiol Dis. 2008 Feb;29(2):254–66. doi: 10.1016/j.nbd.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 184.Ma J, Pulfer S, Li S, Chu J, Reed K, Gallo JM. Pharmacodynamic-mediated reduction of temozolomide tumor concentrations by the angiogenesis inhibitor TNP-470. Cancer Res. 2001 Jul 15;61(14):5491–8. [PubMed] [Google Scholar]
- 185.Murphy S, Davey RA, Gu XQ, Haywood MC, McCann LA, Mather LE, et al. Enhancement of cisplatin efficacy by thalidomide in a 9L rat gliosarcoma model. J Neurooncol. 2007 Nov;85(2):181–9. doi: 10.1007/s11060-007-9406-3. [DOI] [PubMed] [Google Scholar]
- 186.Pradilla G, Legnani FG, Petrangolini G, Francescato P, Chillemi F, Tyler BM, et al. Local delivery of a synthetic endostatin fragment for the treatment of experimental gliomas. Neurosurgery. 2005 Nov;57(5):1032–40. doi: 10.1227/01.NEU.0000180059.33665.c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Goudar RK, Shi Q, Hjelmeland MD, Keir ST, McLendon RE, Wikstrand CJ, et al. Combination therapy of inhibitors of epidermal growth factor receptor/vascular endothelial growth factor receptor 2 (AEE788) and the mammalian target of rapamycin (RAD001) offers improved glioblastoma tumor growth inhibition. Mol Cancer Ther. 2005 Jan;4(1):101–12. [PubMed] [Google Scholar]
- 188.Vredenburgh JJ, Desjardins A, Herndon JE, Dowell JM, Reardon DA, Quinn JA, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007 Feb 15;13(4):1253–9. doi: 10.1158/1078-0432.CCR-06-2309. [DOI] [PubMed] [Google Scholar]
- 189.Baumann F, Bjeljac M, Kollias SS, Baumert BG, Brandner S, Rousson V, et al. Combined thalidomide and temozolomide treatment in patients with glioblastoma multiforme. J Neurooncol. 2004 Mar;67(12):191–200. doi: 10.1023/b:neon.0000021803.01170.03. [DOI] [PubMed] [Google Scholar]
- 190.Fine HA, Wen PY, Maher EA, Viscosi E, Batchelor T, Lakhani N, et al. Phase II trial of thalidomide and carmustine for patients with recurrent high-grade gliomas. J Clin Oncol. 2003 Jun 15;21(12):2299–304. doi: 10.1200/JCO.2003.08.045. [DOI] [PubMed] [Google Scholar]
- 191.Kemper EM, Boogerd W, Thuis I, Beijnen JH, van TO. Modulation of the blood-brain barrier in oncology: therapeutic opportunities for the treatment of brain tumours? Cancer Treat Rev. 2004 Aug;30(5):415–23. doi: 10.1016/j.ctrv.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 192.Filleur S, Volpert OV, Degeorges A, Voland C, Reiher F, Clezardin P, et al. In vivo mechanisms by which tumors producing thrombospondin 1 bypass its inhibitory effects. Genes Dev. 2001 Jun 1;15(11):1373–82. doi: 10.1101/gad.193501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Moffat BA, Chen M, Kariaapper MS, Hamstra DA, Hall DE, Stojanovska J, et al. Inhibition of vascular endothelial growth factor (VEGF)-A causes a paradoxical increase in tumor blood flow and up-regulation of VEGF-D. Clin Cancer Res. 2006 Mar 1;12(5):1525–32. doi: 10.1158/1078-0432.CCR-05-1408. [DOI] [PubMed] [Google Scholar]
- 194.Leenders WP, Kusters B, Verrijp K, Maass C, Wesseling P, Heerschap A, et al. Antiangiogenic therapy of cerebral melanoma metastases results in sustained tumor progression via vessel co-option. Clin Cancer Res. 2004 Sep 15;10(18 Pt 1):6222–30. doi: 10.1158/1078-0432.CCR-04-0823. [DOI] [PubMed] [Google Scholar]
- 195.Kunkel P, Ulbricht U, Bohlen P, Brockmann MA, Fillbrandt R, Stavrou D, et al. Inhibition of glioma angiogenesis and growth in vivo by systemic treatment with a monoclonal antibody against vascular endothelial growth factor receptor-2. Cancer Res. 2001 Sep 15;61(18):6624–8. [PubMed] [Google Scholar]
- 196.Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005 Oct;8(4):299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 197.Assanah M, Lochhead R, Ogden A, Bruce J, Goldman J, Canoll P. Glial progenitors in adult white matter are driven to form malignant gliomas by platelet-derived growth factor-expressing retroviruses. J Neurosci. 2006 Jun 21;26(25):6781–90. doi: 10.1523/JNEUROSCI.0514-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Bello L, Carrabba G, Giussani C, Lucini V, Cerutti F, Scaglione F, et al. Low-dose chemotherapy combined with an antiangiogenic drug reduces human glioma growth in vivo. Cancer Res. 2001 Oct 15;61(20):7501–6. [PubMed] [Google Scholar]
- 199.Schmidt NO, Ziu M, Carrabba G, Giussani C, Bello L, Sun Y, et al. Antiangiogenic therapy by local intracerebral microinfusion improves treatment efficiency and survival in an orthotopic human glioblastoma model. Clin Cancer Res. 2004 Feb 15;10(4):1255–62. doi: 10.1158/1078-0432.ccr-03-0052. [DOI] [PubMed] [Google Scholar]
- 200.Takahashi JA, Fukumoto M, Kozai Y, Ito N, Oda Y, Kikuchi H, et al. Inhibition of cell growth and tumorigenesis of human glioblastoma cells by a neutralizing antibody against human basic fibroblast growth factor. FEBS Lett. 1991 Aug 19;288(12):65–71. doi: 10.1016/0014-5793(91)81004-r. [DOI] [PubMed] [Google Scholar]