

SUMMARY
The transcriptional activator PrfA is required for the expression of virulence factors necessary for Listeria monocytogenes pathogenesis. PrfA is believed to become activated following L. monocytogenes entry into the cytosol of infected host cells resulting in the induction of target genes whose products are required for bacterial intracellular growth and cell-to-cell spread. Several mutations have been identified that appear to lock PrfA into its highly activated cytosolic form (known as prfA* mutations). In this study PrfA and five PrfA* mutant proteins exhibiting differing degrees of activity were purified and analyzed to define the influences of the mutations on distinct aspects of PrfA activity. Based on limited proteolytic digestion conformational changes were detected for the PrfA* mutant proteins in comparison to wild type PrfA. For all but one mutant (PrfA Y63C), the DNA binding affinity as measured by electophoretic mobility shift assay (EMSA) appeared to directly correlate with levels of PrfA mutational activation such that the high activity mutants exhibited the largest increases in DNA binding affinity and moderately activated mutants exhibited more moderate increases. Surprisingly, the ability of PrfA and PrfA* mutants to form dimers in solution appeared to inversely correlate with levels of PrfA-dependent gene expression. Based on comparisons of protein activity and structural similarities with PrfA family members Crp and CooA, the prfA* mutations modify distinct aspects of PrfA activity that include DNA binding and protein-protein interactions.
INTRODUCTION
The transcriptional regulator PrfA (positive regulatory factor A) is responsible for regulating the gene expression of nearly all known virulence factors of Listeria monocytogenes (Chakraborty et al., 1992; Gray et al., 2006; Leimeister-Wachter et al., 1990; Miner et al., 2008; Pizarro-Cerda & Cossart, 2006; Scortti et al., 2007). PrfA is a 27 kD protein that recognizes and binds a 14 base pair DNA palindrome present in the promoters of its target genes (Freitag et al., 1992; Mengaud et al., 1989). PrfA regulates the expression of gene products required for L. monocytogenes invasion of host cells, intracellular growth, and cell-to-cell spread, and it is absolutely essential for bacterial virulence (Freitag, 2006; Scortti et al., 2007).
Based on sequence and structural homology, PrfA has been identified as a member of the Crp/Fnr family of transcriptional activators (Eiting et al., 2005; Korner et al., 2003; Ripio et al., 1997). Proteins within this family generally become activated following the binding of small molecule cofactors. Crp, for example, undergoes an allosteric change after binding cAMP and becomes a site-specific DNA binding protein that recognizes target promoters and interacts with RNA polymerase (RNAP) (Busby & Ebright, 1999; Kim et al., 1992; Kolb et al., 1993; Lawson et al., 2004). Crp appears to exist in an equilibrium between an active form that efficiently binds DNA target sequences and an inactive form that does not. Co-factor cAMP binding by Crp shifts the equilibrium toward the active form, either by stabilizing this form or by destabilizing the inactive form of the protein (Youn et al., 2007). PrfA may exist in an analogous equilibrium state such that binding of a co-factor is required to shift PrfA to a high activity form capable of high affinity DNA binding. Although it is generally believed that a PrfA co-factor exists, this co-factor has not yet been identified.
Mutations in crp have been identified that result in an active form of Crp in the absence of cAMP cofactor (Garges & Adhya, 1985; Harman et al., 1986; Kim et al., 1992; Youn et al., 2006; Youn et al., 2007). Structural and functional studies of these mutants (known as Crp* mutants) have led to the identification of regions of Crp that are important for activity, and it has been observed that Crp* mutants exhibit a conformation that resembles that of wild type Crp bound to cofactor (Harman et al., 1986). Similar to crp*, several prfA mutations have been identified that appear to result in activation of PrfA in the absence of cofactor (known as prfA* mutants) (Miner et al., 2008; Mueller & Freitag, 2005; Ripio et al., 1997; Shetron-Rama et al., 2003; Vega et al., 2004; Wong & Freitag, 2004). Strains with prfA* mutations express high levels of PrfA-dependent gene products under conditions in which gene expression is usually repressed. The prfA* mutations identified thus far are not functionally equivalent, and significant differences in bacterial virulence have been reported for L. monocytogenes strains containing different prfA* alleles (Miner et al., 2008; Mueller & Freitag, 2005; Scortti et al., 2007; Shetron-Rama et al., 2003). This study describes a biochemical comparison of wild-type PrfA with five different PrfA* mutants (including a novel prfA* mutation) to elucidate the effects of specific amino acid substitutions on distinct aspects of PrfA function.
METHODS
Bacterial strains and plasmids
The strains and plasmids used in this study are listed in Table 1. L. monocytogenes strains were grown in brain heart infusion (BHI) medium and E. coli strains were grown in Luria Broth (LB) at 37° C with shaking. Strains containing high activity prfA* alleles such as prfA L140F and prfA G145S have previously been proven difficult to construct using standard allelic exchange techniques [(Port & Freitag, 2007; Wong & Freitag, 2004) and unpublished observations]. However, a modified approach for generating isogenic mutants was developed and used successfully as follows: prfA L140F and prfA G145S were introduced into L. monocytogenes NF-L1124 containing a transcriptional fusion of gus and neo (Karow & Piggot, 1995) downstream of actA in the bacterial chromosome. Selection for the mutant strains was then facilitated based on an increased level of neomycin resistance in the presence of the prfA* allele as conferred by the PrfA-dependent promoter actA. In addition, to prevent expression of the introduced prfA* mutations from the plasmid vector used for allelic exchange, prfA coding sequences missing the ATG start codon were amplified by PCR and inserted into the temperature sensitive plasmid shuttle vector pKSV7 (generating plasmid pNF1147) and the L140F and G145S mutations were then separately introduced via QuikChange Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer’s instructions with primers listed in Table 2 [generating plasmids pNF1162 (prfA L140F) and pNF1161 (prfA G145S)]. To enrich for mutants containing the prfA L140F or prfA G145S chromosomal replacement, 5 μg neomycin ml−1 was used to select for prfA*-induced neomycin resistance on the final day of allelic exchange at 40° C. Following allelic exchange, L. monocytogenes strains containing the desired mutations within prfA were confirmed by sequencing of PCR fragments derived from chromosomal DNA.
Table 1.
Bacterial strains used in this work
| Brief description | Source/Ref | |
|---|---|---|
| Strain | ||
| 10403S | Listeria monocytogenes | (Bishop & Hinrichs, 1987) |
| NF-L476 | 10403S actA-gus-plcB | (Shetron-Rama et al., 2002) |
| NF-L924 | NF-L476 prfA E77K | (Shetron-Rama et al., 2003) |
| NF-L1124 | 10403S actA-gus-neo-plcB | This work |
| NF-L1213 | NF-L1124 prfA Y154C | (Miner et al., 2008) |
| NF-L1214 | NF-L1124 prfA Y63C | This work |
| NF-L1166 | NF-L1124 prfA L140F | This work |
| NF-L1177 | NF-L1124 prfA G145S | This work |
| BL21 Star (DE3) | E. coli protein expression strain | Invitrogen |
| Plasmids | ||
| pET100 | Protein expression vector | Invitrogen |
| pNF1280 | pET100- prfA WT | This work |
| pNF1281 | pET100- prfA G145S | This work |
| pNF1282 | pET100- prfA L140F | This work |
| pNF1283 | pET100- prfA Y63C | This work |
| pNF1284 | pET100- prfA Y154C | This work |
| pNF1285 | pET100- prfA E77K | This work |
Table 2.
Oligonucleotides used in this study
| Primer Name | Sequence | Characteristics |
|---|---|---|
| 5’hly | TCCTATCTTAAAGTGACTTTTATGTT | 5’ hly promoter |
| 3’hly | GCTTCTAAAGATGAAACGCAATATTA | 3’ hly promoter |
| 5’actA | GATGCTTCTAAAAAAGTTGCTGAAGC | 5’ actA promoter |
| 3’actA | TATTCATGAATTATTTTAAGAATATCA | 3’ actA promoter |
| +374 prfA | GCGCTGCAGGAAACTTGTTTTTGTAGGGTTTGG | 3’ truncated prfA ORF |
| 5’ prfA orf | CACCATGAACGCTCAAGCAGAA | 5’ prfA ORF for protein expression |
| 3’ prfA orf | TCCTCATTGAGGAATACTGTT | 3’ prfA ORF for protein expression |
| prfA-trunc- nt3-Fb | GCGCTGCAGGAACGCTCAAGCAGAAG AATTC | 5’ truncated prfA |
| prfA-trunc- nt841-Rb | GCGCTGCAGGGAACAACTATCTGTTGC AGCTC | 3’ truncated prfA ORF |
| prfA-L140F- QkCh-Fb | GATTTTTCGATTAACGGGAAGTTTGGC TCTATTTGCGGTCAAC | 5’ L140F Quikchange |
| prfA-L140F- QkCh-Rb | GTTGACCGCAAATAGAGCCAAACTTCC CGTTAATCGAAAAATC | 3’ L140F Quikchange |
| prfA-G145S- QkCh-Fb | GGAAGCTTGGCTCTATTTGCAGTCAAC TTTTAATCCTGACC | 5’ G145S Quikchange |
| prfA-G145S- QkCh-Rb | GGTCAGGATTAAAAGTTGACTGCAAAT AGAGCCAAGCTTCC | 3’ G145S Quikchange |
Genetic selection for prfA mutations that lead to enhanced actA expression following prfA plasmid mutagenesis in XL1 Red E. coli
Plasmid pNF1019 containing prfA under the control of the prfAP1, prfAP2, and plcA promoters in the integrative plasmid vector pPL2 pNF1019 (Wong & Freitag, 2004) was transformed into chemically competent XL1 Red E. coli hypermutator bacterial cells (Stratagene). Selected transformants were inoculated into LB at 1:1000 dilution and grown with shaking to stationary phase at 37° C. Cultures were repeatedly diluted and grown to stationary phase for a total of 10 cycles. The pNF1019 plasmid was then purified from XL1 Red and introduced via electroporation into conjugation competent SM10 cells. Transfer of pNF1019 from SM10 into L. monocytogenes ΔprfA was carried out as described previously (Wong & Freitag, 2004). Transconjugant prfA mutants exhibiting enhanced actA expression were identified as blue colonies on BHI plates containing 7.5 ug chloramphenicol ml−1, 200 ug streptomycin ml−1, 50 ug 5-bromo-4-chloro-3-indolyl-beta-D-glucuronic acid (X-gluc) ml−1, and 5 ug neomycin ml−1.
Generation and purification of recombinant PrfA* proteins
DNA fragments containing prfA and prfA* ORFs were amplified using PCR of L. monocytogenes genomic DNA isolated from either NF-L1124 (prfA WT), NF-L1177 (prfA G145S), NF-L1166 (prfA L140F), NF-L1214 (prfA Y63C), NF-L1213 (prfA Y154C), or NF-L924 (prfA E77K) using primers listed in Supplementary Table S1. The PCR fragments were then cloned into pET100 using Champion pET Directional TOPO Expression Kit (Invitrogen) per the manufacturer’s instructions. Plasmids containing the prfA and prfA* ORFs were transformed into BL21 Star (DE3) expression cells and PrfA/PrfA* protein production was induced by addition of 1 mM IPTG for 1.5 hours. Protein extracts containing recombinant PrfA/PrfA* proteins were passed over a nickel column, and PrfA was eluted with 200–500 mM imidazole buffer and dialyzed into phosphate buffered saline (137 mM NaCl, 10 mM Phosphate, 2.7 mM KCl, pH 7.4) (PBS) with 10% glycerol (v/v). Purified protein was visualized and assessed for purity following separation on SDS-PAGE gels and Coomassie staining, and also confirmed by western analysis using a α-PrfA polyclonal antibody (Greene & Freitag, 2003).
Limited proteolysis
One microgram of purified wild-type PrfA and each PrfA mutant was incubated with 300 ng trypsin (Sigma) or 250 ng subtilisin (Sigma) in Sigma 10X Multicore buffer for the indicated times at 37° C. Reactions were terminated by the addition of 1 ul phenylmethanesulfonylfluoride and samples were then boiled for 5 minutes and run on 12% acrylamide gels in MES Buffer (50 mM MES, 50 mM Tris base, 0.1% SDS, 1 mM EDTA, pH 7.3) (Invitrogen) for small band separation and visualized by Coomassie stain.
Electrophoretic mobility shift assays (EMSA)
Primers used to amplify DNA fragments (~100 bp) containing the hly and actA promoters from L. monocytogenes genomic DNA using PCR are listed in Table 2. Primers were purchased labeled with cy5.5 label on the 5’ends (Operon Biotechnologies, Inc.) To generate a DNA fragment for use as a non-specific competitor for DNA binding assays, primers were used to amplify the prfA open reading frame (~370 bp), as this region lacks PrfA binding sites (primer sequences listed in Table 2.) Extracts from NF-L890 (ΔprfA) were made as follows: bacteria from 1 L cultures grown to mid-log phase in BHI were collected using centrifugation for 10 minutes at 6,000 x g, resuspended in 20 ml ice cold PBS, and bacterial cells were disrupted by a triple passage through a French Press. Electrophoretic mobility shift assays (EMSA) were performed as previously described (Bockmann et al., 2000). EMSA reaction mixtures consisted of the following: 40 ng labeled DNA probe (hlycy5.5 or actAcy5.5), PrfA protein (as indicated),1 ug BSA ml−1, and 50 mM dIdC in TB buffer (10mM Tris HCl pH 8, 10mM MgCl2, 5mM CaCl2, 1mM EDTA, 0.2mM DTT, 10% glycerol [v/v]) in a final reaction volume of 20 ul. For experiments including cell extracts, 1 ul (3 ug) extract was added to each reaction mixture. Sample reactions containing all components except labeled DNA were incubated for 15 minutes at room temperature. The labeled DNA probe was then added and samples were incubated for 3 minutes at 37°, followed by 27 minutes on ice. Samples were then loaded onto a 5% acrylamide gel (0.5X TBE, Tris-boric acid-EDTA) and run at a constant current of 20 mA for approximately 3 hours in the dark at 4° C. Gels were then visualized as in-gel westerns using the Odyssey Imager (Li-Cor Biosciences) with the cy5.5-labeled fluorescent probes visualized at 700 nm. The His- and Express-tags were found to have minimal impact on PrfA or PrfA* protein function in comparison to purified PrfA protein without the tags (M. Miner, unpublished data), as has been previously reported (Bockmann et al., 2000; Böckmann et al., 1996).
Measurement of β-glucoronidase activity
β-glucuronidase (GUS) activity was measured as previously described (Shetron-Rama et al., 2003) with minor changes. Briefly, L. monocytogenes cultures grown overnight at 37º C in BHI were diluted 1:50 and grown with shaking at 37º C for 8 hours. OD600 was measured for each time point and two 500 μl culture aliquots were collected for all strains except for the prfA L140F (NF-L1166), prfA G145S (NF-L1177), and prfA Y63C (NF-L1214) mutant strains for which two 50 μl aliquots were collected (reflective of the increased GUS activity present in these three highly activated prfA* strains). Bacterial cells were recovered by microcentrifugation and the supernatants were removed,. Bacterial pellets were resuspended in 100 μl (aliquot 1) or 1 ml (aliquot 2) ABT buffer (0.1M potassium phosphate, pH 7.0, 0.1M NaCl, 0.1% Triton). GUS activity was measured as described with the substitution of 4-methylumbelliferyl-β-D-glucuronide in place of 4-methylumbilliferyl-β-D-galactoside (Sigma) (Youngman, 1987). Data were derived from duplicate samples taken from three independent experiments.
Measurement of hemolytic activity
Hemolytic activity was measured as previously described with minor modifications (Camilli et al., 1989). Briefly, bacteria were grown without shaking overnight in BHI at 30° C, the bacterial supernatants were recovered following centrifugation, and two-fold serial dilutions of the supernatants were incubated with PBS-washed sheep red blood cells (0.3% to 10%) for 30 minutes at 37° C. After incubation, RBCs were recovered by centrifugation to measure 50% lysis and supernatants were read in a spectrophotometer plate reader at OD450.
Protein chemical crosslinking
500 ng of purified proteins were incubated with either 10uM sulfo-ethylene glycol bis[succinimidylsuccinate] (S-EGS) or Bis[sulfosuccinimidyl] suberate (BS3) in 0.2M triethylamine (TEA), pH 8.0 for 1 hour at room temperature. Samples were then heated at 85° C for 10 minutes in SDS sample buffer (1% SDS, 10% glycerol, 10 mM Tris-Cl, pH 6.8, 1 mM ethylene diamine tetraacetic acid (EDTA), and 0.05 mg bromphenol blue dye ml−1) containing 5% BME, run on SDS-PAGE and transferred to nitrocellulose. Rabbit polyclonal antibody directed against PrfA was used for western analysis at 1:4000 dilution followed by incubation with goat-anti-rabbit-IRDye 680 at 1:10000 (Li-cor Biosciences). Membranes were visualized on Odyssey Imager (Li-cor Biosciences).
RESULTS
Isolation of a novel mutationally activated L. monocytogenes prfA* mutant (prfA Y63C)
As part of a study designed to identify mutations within prfA that result in increased PrfA-dependent virulence gene expression in L. monocytogenes strains grown in culture, promoterless copies of the genes encoding for neomycin resistance (neo) and β-glucuronidase (gus) were introduced in single copy into the bacterial chromosome downstream of actA in a ΔprfA strain. The actA-gus-neo-plcB transcriptional fusion within the L. monocytogenes chromosome was used for the identification of prfA* mutations based on the enhanced expression of neomycin resistance and blue colony color in the presence of the GUS substrate 5-bromo-4-chloro-3-indolyl-beta-D-glucuronic acid (X-gluc) on indicator plates. Plasmid pPL2-prfA containing a copy of wild type prfA and its promoters was propagated in the E. coli mutator strain XL1-Red as described in Methods and then transformed into conjugation-competent E. coli SM10 cells for conjugal transfer into ΔprfA/actA-gus-neo-plcB L. monocytogenes. Transconjugants with prfA* mutations were selected based on enhanced neomycin resistance and dark blue colony color on plates containing neomycin and X-gluc. Out of approximately 40,000 transconjugants screened, two mutants were identified with approximately 200-fold and 185-fold higher levels of actA expression (based on GUS activity in broth culture) in comparison to the wild-type prfA strain. DNA sequencing of the mutant prfA alleles revealed a leucine to phenylalanine substitution at amino acid position 140 [prfA L140F, a previously described mutation (Wong & Freitag, 2004)], and a novel tyrosine to cysteine amino acid substitution at residue 63 (PrfA Y63C). The position of the Y63C substitution with respect to the three dimensional structure of PrfA and in relation to other identified prfA* mutations is shown in Fig. 1.
Figure 1. Location of prfA* mutations on the PrfA crystal structure.
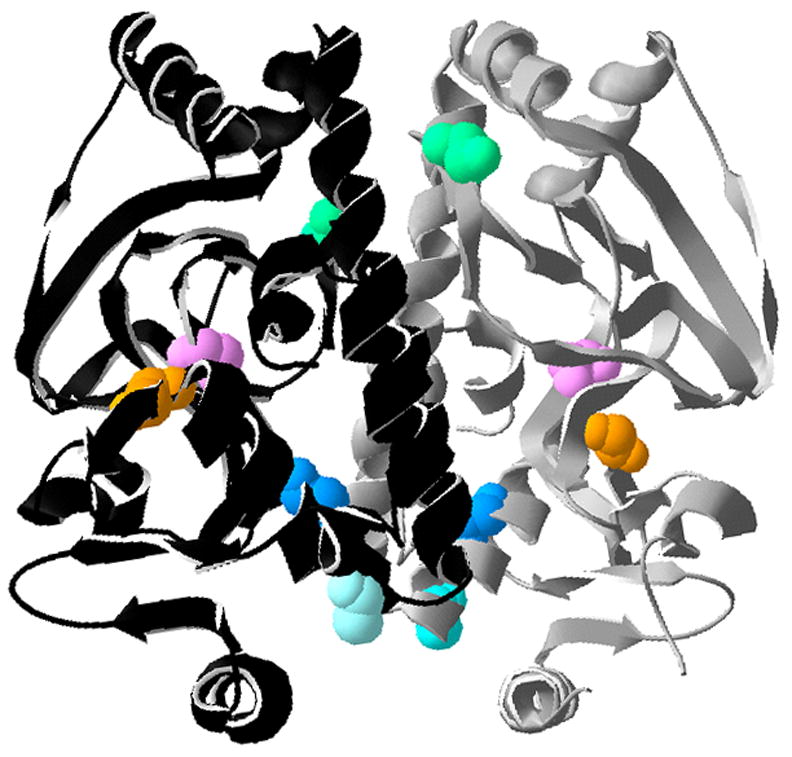
Crystal structure of the PrfA dimer (monomers in black and grey) adapted from Eiting et al (Eiting et al., 2005). Locations of the five PrfA* mutations used in this study are shown on each monomer and color-coded: Y63C (pink), E77K (green), L140F (light blue), G145S (darker blue), and Y154C (orange). Structural motifs discussed in the text are indicated with white letters (αC and αD) or yellow numbers (β5, β6, and β7) and derived from Eiting et al., 2005).
prfA* mutations are not equivalent in their ability to activate PrfA
Direct comparison of L. monocytogenes prfA* mutants has proven challenging for strains containing high activity prfA alleles (prfA L140F and prfA G145S) as it has not been previously possible to construct isogenic chromosomal mutant strains using allelic exchange. Both prfA L140F and prfA G145S strains exhibit subtle fitness defects during bacterial growth in broth culture that have hampered efforts to introduce these mutation into the L. monocytogenes chromosome [(Port & Freitag, 2007; Wong & Freitag, 2004) and unpublished observations]. However, the use of the NF-L1124 strain containing the actA-gus-neo-plcB transcriptional fusion enabled selection of isogenic chromosomal mutants using enhanced neo expression to select for prfA* alleles (see Methods). As a result, it was possible to directly compare in vitro expression of PrfA dependent genes and gene products for all five prfA* strains along with the wild-type parent strain. For the actA gene product, wild-type L. monocytogenes is known to express low-to-undetectable levels of actA during growth in BHI broth culture whereas expression levels increase over 200-fold upon PrfA activation in the host cell cytosol (Bubert et al., 1999; Freitag & Jacobs, 1999; Moors et al., 1999; Shetron-Rama et al., 2002). High level actA expression in broth culture was observed for L. monocytogenes strains containing the prfA G145S, prfA L140F, and prfA Y63C alleles (approximately 200-fold higher than the levels expressed by strains containing wild type prfA) (Fig. 2a); in comparison moderate actA expression was observed for the prfA E77K and prfA Y154C mutant strains (approximate 10-fold increase in expression over wild type).
Figure 2. prfA* strains exhibit increased expression of PrfA-dependent gene products in broth culture.
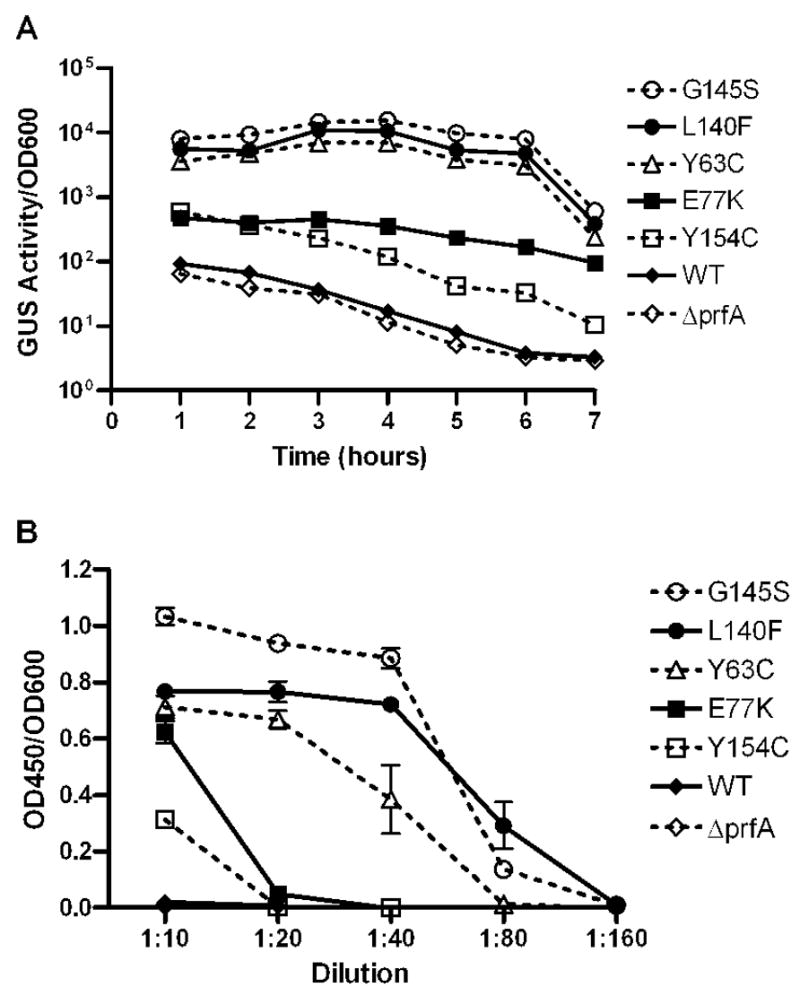
A. Levels of actA expression measured by monitoring GUS activity. Each time point represents the mean +/− the SEM of duplicate samples, and the data is representative of three independent experiments. B. Secreted hemolytic activity as measured by erythrocyte (RBC) lysis. Serial dilutions of bacterial supernatants were incubated with RBCs and cell lysis was determined by measuring absorbance at 450 nm (OD450). Each point represents the mean +/− SD, and the data is derived from three independent experiments.
To compare levels of activation at an additional PrfA-regulated promoter, hly, the expression of listeriolysin O (LLO, encoded by hly) was measured. Bacterial supernatants derived from broth cultures of each mutant strain were incubated with red blood cells and cell lysis was measured (Fig. 2b). Consistent with the observed increases in actA expression, the prfA G145S, prfA L140F, and prfA Y63C strains exhibited the highest levels of LLO activity, followed by prfA E77K and prfA Y154C (Fig. 2b). Taken together, these data indicate a hierarchy of prfA* mutant activity based on the patterns of PrfA-dependent gene expression: high activity mutants (prfA G145S, prfA L140F and prfA Y63C) and moderate activity mutants (prfA E77K and prfA Y154C).
PrfA* mutants are conformationally distinct from the wild type protein
Limited proteolytic digestion of proteins serves as a useful tool for rapid detection of protein conformational changes, and it has been used to distinguish between active and inactive forms of Crp (Harman et al., 1986; Tan et al., 1991). Limited protease digestion of Crp* mutants results in cleavage patterns that resemble those observed for Crp when bound to cAMP (Harman et al., 1986; Tan et al., 1991). To detect any conformational alterations associated with PrfA* mutations, each mutant protein was purified and subjected to limited trypsin digestion (Fig. 3). PrfA G145S protein served as a positive control for the assay as a conformational change in this protein has already been demonstrated by crystal structure analysis (Eiting et al., 2005). As anticipated, PrfA G145S was found to exhibit enhanced susceptibility to protease digestion in comparison to wild-type PrfA (Fig. 3). Similar to PrfA G145S, PrfA Y63C and PrfA E77K exhibited similar patterns of enhanced susceptibility to proteolysis. Interestingly, the highly activated PrfA L140F did not exhibit enhanced susceptibility to proteolysis, but the substitution of phenylalanine for leucine in this mutant occurs adjacent to a trypsin cleavage site, and may thus interfere with protease digestion. These results strongly suggest that the presence of the prfA* mutations alters PrfA conformation.
Figure 3. Protease sensitivity of WT PrfA and PrfA* mutants.

A. Wild type (W) and PrfA G145S (G) recombinant proteins were digested for 10, 30, 60, 90, and 120 minutes with trypsin, subjected to SDS-PAGE and visualized by Coomassie stain. Minus symbol denotes protein without trypsin, plus symbol denotes trypsin digestion of denatured protein. Lane M contains molecular weight markers. B. Wild type and PrfA* proteins were treated with trypsin for 10, 30, 60, and 120 minutes. W, wild type; L, L140F; Y1, Y63C, Y2, Y154C; E, E77K. Minus symbol indicates samples without trypsin, plus symbol indicates trypsin treated denatured protein. Numbers on left represent molecular weight in kD. Gel is representative of three similar experiments.
PrfA* mutants appear to exhibit reduced dimer formation in vitro
PrfA has been shown to crystallize as a homodimer by two independent groups (Eiting et al., 2005; Velge et al., 2007), and it was recently shown that wild-type PrfA (27 kD) migrates in SDS-PAGE gels at a molecular weight of approximately 60 kD following incubation with a chemical crosslinking agent (Velge et al., 2007). To assess the ability of the different PrfA* proteins to form dimers, purified proteins were incubated with two distinct chemical crosslinking agents and analyzed on SDS polyacrylamide gels. Two crosslinkers were used: sulfo-ethylene glycol bis[succinimidylsuccinate] (S-EGS) and Bis[sulfosuccinimidyl] suberate (BS3), both of which react with free amine groups but differ in the lengths of the linker arms (16 angstroms for S-EGS and 11 angstroms for BS3). Interestingly, wild-type PrfA was found to form dimers more readily than any of the PrfA* mutants, with the ratio of dimer to monomer for S-EGS and BS3 being 0.27 and 0.44 respectively (Fig. 4). The moderately active PrfA Y154C and PrfA E77K showed reduced dimer formation in comparison to wild type, with dimer to monomer ratios of approximately 0.05 for S-EGS and 0.11 for BS3 (Fig. 4). Unexpectedly, the most highly active PrfA* mutants PrfA G145S, PrfA L140F and PrfA Y63C exhibited the lowest ratio of dimer to monomer following chemical crosslinking (Fig. 4).
Figure 4. Dimer formation of PrfA and PrfA* proteins.

A. Purified wild type and PrfA* protein (500 ng) was incubated with 10uM EGS (E) or BS3 (B) or without crosslinking agent (-) for one hour at room temperature and subjected to SDS-PAGE and Western analysis with polyclonal antibody directed against PrfA (Greene & Freitag, 2003). Arrows indicate molecular weight in kD. B. Ratio of dimer to monomer as determined from three independent experiments. Ratios were calculated by measuring the intensity of the peptide bands and dividing the monomer values by the dimer values, and include standard deviations.
High activity PrfA* mutant proteins exhibit enhanced binding to the hly and actA promoters
It has been previously demonstrated by EMSA that the PrfA G145S mutant protein binds to the hly promoter with higher affinity than wild-type PrfA (Mauder et al., 2006; Vega et al., 2004). Subsequent to these studies, Eiting et al (Eiting et al., 2005) demonstrated by surface plasmon resonance that PrfA G145S binds to the hly promoter with an 18-fold higher affinity than wild-type. Consistent with these observations, both of the highly activated PrfA* mutants PrfA G145S and PrfA L140F were observed to readily bind DNA fragments containing the hly promoter (Fig. 5). Wild-type PrfA was found to bind only weakly to the hly promoter with detectable binding observed in the presence of 2 ug PrfA protein and no binding evident in the presence of 500 ng protein (Fig. 5). In contrast, the highly active PrfA* mutants PrfA G145S and PrfA L140F bound target DNA with very high affinity such that all available DNA was bound at low protein concentrations (500 ng) (Fig. 5). PrfA Y63C, PrfA Y154C, and PrfA E77K also demonstrated higher affinity DNA binding in comparison to wild-type protein, but to a lesser degree than either PrfA G145S or PrfA L140F (Fig. 5). Binding in all cases was specific as the addition of cold specific competitor DNA but not non-specific competitor DNA eliminated the PrfA-dependent mobility shift (M. Miner, unpublished data).
Figure 5. In vitro DNA binding activity of WT PrfA and PrfA* proteins.

A. Purified protein was incubated with labeled hly or actA DNA fragments. All reactions contained 500 ng protein except those denoted with * which contained 10 ng. B. Increasing amounts of purified protein were incubated with hly DNA fragments. Amounts of protein are denoted above the lane. Gels are representative of three experiments. fp, free probe; CIII, PrfA-bound DNA [CIII designation derived from (Bockmann et al., 2000)].
Purified PrfA protein has been previously shown to bind the hly promoter with higher affinity than the actA promoter (Böckmann et al., 1996). Consistent with the promoter preference previously observed for the wild-type protein, the mutant PrfA proteins were observed to bind the actA promoter with a lower apparent affinity than the hly promoter, however the relative binding affinity hierarchy for the proteins was similar to that observed for hly DNA (Fig. 5). Binding was detectable with significantly lower amounts of both PrfA G145S and PrfA L140F protein in comparison to wild-type PrfA.
Examination of PrfA*-DNA-RNA polymerase complex formation in bacterial cell extracts
Activation of target gene expression requires both binding of PrfA to target promoter sites and recruitment of RNA polymerase (RNAP). To examine the ability of PrfA* mutants to form complexes with target promoter DNA fragments and RNAP, purified PrfA and PrfA* mutant proteins were incubated with DNA in the presence of cell extracts derived from a L. monocytogenes prfA deletion strain. As previously mentioned for purified protein incubated with DNA, wild-type PrfA exhibited weak binding of the hly promoter in comparison to PrfA* mutants (Eiting et al., 2005; Mauder et al., 2006; Vega et al., 2004) (Fig. 6a, CIII complexes). However, in the presence of bacterial cell extracts wild-type PrfA formed DNA-RNAP complexes with an affinity apparently equal to that of the PrfA* proteins (Fig. 6a, CI complexes). These results suggest that PrfA binding to the hly promoter is enhanced by the presence of RNAP and/or other components within bacterial cell extracts. The PrfA* mutants appeared to form PrfA-DNA-RNAP complexes that were roughly equivalent in amount to those formed using wild type PrfA (Fig. 6a, CI complexes), and the absence of visible CII bands (RNAP-DNA complexes) suggests that RNAP is limiting under these assay conditions. Similar results were observed with the actA promoter (Fig. 6b).
Figure 6. PrfA and PrfA* formation of DNA-RNAP complexes.

Purified wild type or PrfA* proteins (500 ng) were incubated with hly (A.) or actA (B.) DNA fragments in the presence of cell extracts derived from a ΔprfA strain (NF-L890). Gels are representative of three experiments. fp, free probe; CI, PrfA + DNA; CII, RNAP + DNA,; CIII, PrfA-RNAP-DNA [CI, CII, and CIII designations are derived from (Bockmann et al., 2000)].
DISCUSSION
Despite its critical role in promoting the pathogenesis of L. monocytogenes, the mechanism by which PrfA becomes activated in the host cell cytosol remains undefined. However, the isolation and characterization of mutationally activated prfA alleles has helped to define the consequences of PrfA activation on L. monocytogenes physiology and pathogenesis (Mauder et al., 2006; Miner et al., 2008; Mueller & Freitag, 2005; Ripio et al., 1997; Scortti et al., 2007; Shetron-Rama et al., 2003; Vega et al., 2004; Wong & Freitag, 2004). This study represents the first biochemical comparison of multiple PrfA* mutant proteins with differing levels of activation. Our results suggest that prfA* mutations have the capacity to activate PrfA via a variety of structural and functional modifications.
Overall, the expression levels of PrfA-dependent gene products in vitro appeared in this study to correlate most strongly with the binding affinity of PrfA for target DNA (Fig. 5). prfA* mutations that conferred the highest levels of PrfA-dependent gene expression in vitro exhibited the highest affinity of DNA binding as detected by EMSA, with moderately-active prfA mutant alleles correspondingly exhibiting more moderate increases in DNA binding. Interestingly, although wild type PrfA formed very low amounts of protein-DNA complexes with either the hly or actA promoter fragments in comparison to PrfA* proteins (Fig. 5), PrfA-RNAP-DNA complexes were readily formed for both promoter fragments with RNAP present in cell extracts (Fig. 6). Previous studies by Mauder et al (Mauder et al., 2006) have suggested that the binding efficiency of PrfA to its binding site alone (CIII formation) does not necessarily indicate its potential to initiate transcription at a PrfA dependent promoter. Their conclusions were based on in vitro transcription assays using purified PrfA proteins (including PrfA G145S) and partially purified RNAP with linear DNA templates. However, substantially less PrfA G145S is required to form either PrfA-DNA or PrfA-RNAP-DNA complexes than wild type PrfA [(Mauder et al., 2006) and Fig. 5], thus it seems reasonable to speculate that under conditions in which PrfA concentrations are limiting, activated PrfA or PrfA* mutants with increased DNA binding affinity would be better able to stimulate the formation of active transcription complexes with RNAP.
Mutations that enhance PrfA-dependent gene expression in vitro have been isolated in multiple regions of the protein (Fig. 1). PrfA G145S and PrfA L140F map within the αD α-helix of PrfA, with G145S positioned near the center of the helix and L140F located at one end [(Eiting et al., 2005) and Fig. 1]. The mutations are positioned near what corresponds to the hinge region of Crp, a region believed to mediate communication between the C and N terminal domains of the protein (Garges & Adhya, 1985; Harman et al., 1986; Kim et al., 1992; Youn et al., 2006). Selected mutations in the Crp hinge region lead to the constitutive activation of Crp in the absence of cAMP via a change in secondary structure that enhances the solvent exposure of the DNA-binding helix. Eiting et al (Eiting et al., 2005) reported a similar structural change occurring in PrfA G145S mutants. Based on the functional similarities of the L140F mutant with G145S, most notably a large increase in DNA binding affinity, the PrfA L140F mutation may mediate a comparable structural change. While the conformational changes imparted by the L140F mutation as detected by limited proteolysis indicated that the PrfA L140F protease susceptibility was most similar to that of the wild type protein (Fig. 3), this result may be misleading as the L140F mutation is located near a trypsin cleavage site (K139) which could likely influence the efficiency of trypsin cleavage at this position.
Other prfA* mutations with the potential for distinct structural influences include the PrfA E77K, Y63C, and the Y154C mutations. The E77K mutation lies between β6 and β7 in a region near the central C helices [(Eiting et al., 2005) and Fig. 1]. This mutation enhanced PrfA DNA binding to a lesser extent than that of the G145S and L140F mutations, which could suggest E77K has either a more modest effect on the repositioning of the central C helices or that E77K enhances PrfA-dependent gene expression through a different mechanism. The E77K mutation is located near a region of PrfA that corresponds with an area of Crp and CooA known to interact with RNA polymerase (AR2) (Leduc et al., 2001; Niu et al., 1996). AR2 is comprised of a patch of positively charged residues that contact an acidic patch on α-NTD of RNAP. As the PrfA E77K substitution adds a positively charged lysine residue within a potential similarly located AR2 region, it is possible that the additional positive charge enhances PrfA interactions with RNAP.
Y154C and Y63C map within regions of PrfA (αD and β5 respectively) that are associated with a structural tunnel that may serve as a binding pocket for PrfA co-factor (Eiting et al., 2005). Y154C is located at the very end of the αD helix whereas Y63C is located within the β5 domain (Fig. 1). Despite the similar chemical nature of the substitutions, these mutations have dramatically different affects on PrfA function. The Y154C mutation slightly enhanced PrfA-dependent gene expression in broth culture and exhibited a modest but reproducible increase in apparent DNA binding affinity (Fig. 5). Interestingly, this mutation impedes PrfA-dependent gene expression in cytosolic L. monocytogenes, suggesting that the Y154C mutation may interfere with the shift of PrfA to a fully activated state (Miner et al., 2008). In contrast, Y63C dramatically increased PrfA-dependent gene expression in broth culture but did not dramatically increase DNA binding affinity (Fig. 2 and Fig. 5). Several possibilities exist that could account for the effects of these mutations on PrfA function. The mutations could either: (1) inhibit (Y154C) or enhance (Y63) PrfA co-factor binding; (2) stabilize the low (Y154C) or high (Y63C) activity form of PrfA; or (3) result in the formation of disulfide bridges that serve to lock PrfA in either a low activity (Y154C) or high activity (Y63) state. Whereas we cannot differentiate between these possibilities at this time, we favor the Y63C mutation enhancing co-factor binding for the simple reason that no significant increase in DNA binding was observed for this mutant in vitro, suggesting that its high activity is not due to increased accessibility of the PrfA DNA binding helix-turn-domain.
The apparent inverse correlation that was found to exist between the ability of the PrfA* mutants to form dimers and their ability to activate gene expression was unexpected. Crp has been long known to form dimers as an active transcription factor, and Fnr is believed to form dimers when active and be monomeric when inactive (Lazazzera et al., 1993). While the chemical crosslinking experiments presented here suggest that PrfA dimerization inversely correlates with DNA binding and activation of target gene expression, the crosslinking agents used were specifically reactive for amine groups and it is possible that these moieties are less available as a result of conformational changes resulting from the prfA* mutations.
Multiple prfA* mutations have been isolated in L. monocytogenes using a variety of approaches (Miner et al., 2008) (Shetron-Rama et al., 2003; Vega et al., 2004) and the reconstruction of these mutations in isogenic backgrounds has been highly desirable for unambiguous comparison of the effects of the prfA* mutations on L. monocytogenes physiology and pathogenesis. While the moderately active prfA* alleles have been easily introduced into isogenic strains using allelic exchange (Miner et al., 2008; Shetron-Rama et al., 2003; Vega et al., 2004), this approach has not proven feasible for the higher activity prfA* mutants prfA G145S and prfA L140F [(Port & Freitag, 2007; Wong & Freitag, 2004) and unpublished observations]. The prfA G145S and prfA L140F mutations appear to confer a subtle fitness defect upon L. monocytogenes that is not evident in pure cultures of bacteria but which can be detected in mixed cultures when the mutant strains are grown in the presence of wild type bacteria (J. Bruno and N. Freitag, unpublished data). A fitness defect has also been reported for high activity crp* mutants (Youn et al., 2006). To our knowledge, until now the prfA G145S had never been reintroduced into its correct chromosomal location by allelic exchange in any L. monocytogenes strain, including EGD and 10403S. This current work therefore represents a novel method enabling the reconstruction of prfA* isogenic strains with highly active prfA* mutations without the use of plasmids and with prfA* in the proper chromosomal location.
In summary, prfA* mutations appear to activate PrfA through a variety of structural and functional modifications. In general, the prfA* mutations that most dramatically enhanced the binding of PrfA to its DNA recognition sequences resulted in the highest levels of PrfA-dependent gene expression in bacterial cultures. Surprisingly, an apparent inverse correlation appears to exist between the level of PrfA activation conferred by a prfA* mutation and the ability of the purified mutant protein to form a dimer. Future studies focused on three dimensional structural analyses of the mutant proteins will help to further clarify the influences of individual prfA* mutations on PrfA activation.
Acknowledgments
We thank Dr. Patrick Piggot for the pBEST501 plasmid, Dr. Hao Shen and Dr. Jeff Miller for the L. monocytogenes ΔprfA strain, and members of the Freitag lab for helpful discussions. This work was supported by Public Health Service grants AI41816 (N.E.F) from NIAID, by a NIAID Bacterial Pathogenesis training grant fellowship AI55396 (M.D.M), a National Science Foundation Graduate Research Fellowship (NSF-GRF) (G.C.P.), and by the M. J. Murdock Trust. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the funding sources.
References
- Bishop DK, Hinrichs DJ. Adoptive transfer of immunity to Listeria monocytogenes: The influence of in vitro stimulation on lymphocyte subset requirements. J Immunol. 1987;139:2005–2009. [PubMed] [Google Scholar]
- Bockmann R, Dickneite C, Goebel W, Bohne J. PrfA mediates specific binding of RNA polymerase of Listeria monocytogenes to PrfA-dependent virulence gene promoters resulting in a transcriptionally active complex. Mol Microbiol. 2000;36:487–497. doi: 10.1046/j.1365-2958.2000.01868.x. [DOI] [PubMed] [Google Scholar]
- Böckmann R, Dickneite C, Middendorf B, Goebel W, Sokolovic Z. Specific binding of the Listeria monocytogenes transcriptional regulator PrfA to target sequences requires additional factor(s) and is influenced by iron. Mol Microbiolo. 1996;22:643–653. doi: 10.1046/j.1365-2958.1996.d01-1722.x. [DOI] [PubMed] [Google Scholar]
- Bubert A, Sokolovic Z, Chun SK, Papatheodorou L, Simm A, Goebel W. Differential expression of Listeria monocytogenes virulence genes in mammalian host cells. Mol Gen Genet. 1999;261:323–336. doi: 10.1007/pl00008633. [DOI] [PubMed] [Google Scholar]
- Busby S, Ebright RH. Transcription activation by catabolite activator protein (CAP) J Mol Biol. 1999;293:199–213. doi: 10.1006/jmbi.1999.3161. [DOI] [PubMed] [Google Scholar]
- Camilli A, Paynton CR, Portnoy DA. Intracellular methicillin selection of Listeria monocytogenes mutants unable to replicate in a macrophage cell line. Proc Natl Acad Sci USA. 1989;86:5522–5526. doi: 10.1073/pnas.86.14.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty T, Leimeister-Wachter M, Domann E, Hartl M, Goebel W, Nichterlein T, Notermans S. Coordinate regulation of virulence genes in Listeria monocytogenes requires the product of the prfA gene. J Bacteriol. 1992;174:568–574. doi: 10.1128/jb.174.2.568-574.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiting M, Hageluken G, Schubert WD, Heinz DW. The mutation G145S in PrfA, a key virulence regulator of Listeria monocytogenes, increases DNA-binding affinity by stabilizing the HTH motif. Mol Microbiol. 2005;56:433–446. doi: 10.1111/j.1365-2958.2005.04561.x. [DOI] [PubMed] [Google Scholar]
- Freitag NE, Youngman P, Portnoy DA. Transcriptional activation of the Listeria monocytogenes hemolysin gene in Bacillus subtilis. J Bacteriol. 1992;174:1293–1298. doi: 10.1128/jb.174.4.1293-1298.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitag NE, Jacobs KE. Examination of Listeria monocytogenes intracellular gene expression by using the green fluorescent protein of Aequorea victoria. Infect Immun. 1999;67:1844–1852. doi: 10.1128/iai.67.4.1844-1852.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitag NE. From hot dogs to host cells: how the bacterial pathogen Listeria monocytogenes regulates virulence gene expression. Future Microbiol. 2006;1:89–101. doi: 10.2217/17460913.1.1.89. [DOI] [PubMed] [Google Scholar]
- Garges S, Adhya S. Sites of allosteric shift in the structure of cyclic AMP receptor protein. Cell. 1985;41:745–751. doi: 10.1016/s0092-8674(85)80055-6. [DOI] [PubMed] [Google Scholar]
- Gray MJ, Freitag NE, Boor KJ. How the bacterial pathogen Listeria monocytogenes mediates the switch from environmental Dr. Jekyll to pathogenic Mr. Hyde. Infect Immun. 2006;74:2505–2512. doi: 10.1128/IAI.74.5.2505-2512.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene SL, Freitag NE. Negative regulation of PrfA, the key activator of Listeria monocytogenes virulence gene expression, is dispensable for bacterial pathogenesis. Microbiology. 2003;149:111–120. doi: 10.1099/mic.0.25692-0. [DOI] [PubMed] [Google Scholar]
- Harman JG, McKenney K, Peterkofsky A. Structure-function analysis of three cAMP-independent forms of the cAMP receptor protein. J Biol Chem. 1986;261:16332–16339. [PubMed] [Google Scholar]
- Karow M, Piggot PJ. Construction of gusA transcriptional fusion vectors for Bacillus subtilis and their utilization for studies of spore formation. Gene. 1995;163:69–74. doi: 10.1016/0378-1119(95)00402-r. [DOI] [PubMed] [Google Scholar]
- Kim J, Adhya S, Garges S. Allosteric changes in the cAMP receptor protein of Escherichia coli: hinge reorientation. Proc Natl Acad Sci USA. 1992;89:9700–9704. doi: 10.1073/pnas.89.20.9700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb A, Busby S, Buc H, Garges S, Adhya S. Transcriptional regulation by cAMP and its receptor protein. Annu Rev Biochem. 1993;62:749–795. doi: 10.1146/annurev.bi.62.070193.003533. [DOI] [PubMed] [Google Scholar]
- Korner H, Sofia HJ, Zumft WG. Phylogeny of the bacterial superfamily of Crp-Fnr transcription regulators: exploiting the metabolic spectrum by controlling alternative gene programs. FEMS Microbiol Rev. 2003;27:559–592. doi: 10.1016/S0168-6445(03)00066-4. [DOI] [PubMed] [Google Scholar]
- Lawson CL, Swigon D, Murakami KS, Darst SA, Berman HM, Ebright RH. Catabolite activator protein: DNA binding and transcription activation. Curr Opin Struct Biol. 2004;14:10–20. doi: 10.1016/j.sbi.2004.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazazzera BA, Bates DM, Kiley PJ. The activity of the Escherichia coli transcription factor FNR is regulated by a change in oligomeric state. Genes Dev. 1993;7:1993–2005. doi: 10.1101/gad.7.10.1993. [DOI] [PubMed] [Google Scholar]
- Leduc J, Thorsteinsson MV, Gaal T, Roberts GP. Mapping CooA.RNA polymerase interactions. Identification of activating regions 2 and 3 in CooA, the co-sensing transcriptional activator. J Biol Chem. 2001;276:39968–39973. doi: 10.1074/jbc.M105758200. [DOI] [PubMed] [Google Scholar]
- Leimeister-Wachter M, Haffner C, Domann E, Goebel W, Chakraborty T. Identification of a gene that positively regulates expression of listeriolysin, the major virulence factor of Listeria monocytogenes. Proc Natl Acad Sci USA. 1990;87:8336–8340. doi: 10.1073/pnas.87.21.8336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauder N, Ecke R, Mertins S, Loeffler DI, Seidel G, Sprehe M, Hillen W, Goebel W, Muller-Altrock S. Species-specific differences in the activity of PrfA, the key regulator of listerial virulence genes. J Bacteriol. 2006;188:7941–7956. doi: 10.1128/JB.00473-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengaud J, Vicente MF, Cossart P. Transcriptional mapping and nucleotide sequence of the Listeria monocytogenes hlyA region reveal structural features that may be involved in regulation. Infect Immun. 1989;57:3695–3701. doi: 10.1128/iai.57.12.3695-3701.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner M, Port G, Bouwer H, Chang J, Freitag N. A novel prfA mutation that promotes Listeria monocytogenes cytosol entry but reduces bacterial spread and cytotoxicity. Microbial Pathog. 2008 doi: 10.1016/j.micpath.2008.06.006. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moors MA, Levitt B, Youngman P, Portnoy DA. Expression of listeriolysin O and ActA by intracellular and extracellular Listeria monocytogenes. Infect Immun. 1999;67:131–139. doi: 10.1128/iai.67.1.131-139.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller KJ, Freitag NE. Pleiotropic enhancement of bacterial pathogenesis resulting from the constitutive activation of the Listeria monocytogenes regulatory factor PrfA. Infect Immun. 2005;73:1917–1926. doi: 10.1128/IAI.73.4.1917-1926.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu W, Kim Y, Tau G, Heyduk T, Ebright RH. Transcription activation at class II CAP-dependent promoters: two interactions between CAP and RNA polymerase. Cell. 1996;87:1123–1134. doi: 10.1016/s0092-8674(00)81806-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizarro-Cerda J, Cossart P. Subversion of cellular functions by Listeria monocytogenes. J Pathol. 2006;208:215–223. doi: 10.1002/path.1888. [DOI] [PubMed] [Google Scholar]
- Port GC, Freitag NE. Identification of novel Listeria monocytogenes secreted virulence factors following mutational activation of the central virulence regulator, PrfA. Infect Immun. 2007;75:5886–5897. doi: 10.1128/IAI.00845-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripio MT, Dominguez-Bernal G, Lara M, Suarez M, Vazquez-Boland JA. A Gly145Ser substitution in the transcriptional activator PrfA causes constitutive overexpression of virulence factors in Listeria monocytogenes. J Bacteriol. 1997;179:1533–1540. doi: 10.1128/jb.179.5.1533-1540.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scortti M, Monzo HJ, Lacharme-Lora L, Lewis DA, Vazquez-Boland JA. The PrfA virulence regulon. Microbes Infect. 2007;9:1196–1207. doi: 10.1016/j.micinf.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Shetron-Rama LM, Marquis H, Bouwer HGA, Freitag NE. Intracellular induction of Listeria monocytogenes actA expression. Infect Immun. 2002;70:1087 –1096. doi: 10.1128/IAI.70.3.1087-1096.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetron-Rama LM, Mueller K, Bravo JM, Bouwer HG, Way SS, Freitag NE. Isolation of Listeria monocytogenes mutants with high-level in vitro expression of host cytosol-induced gene products. Mol Microbiol. 2003;48:1537–1551. doi: 10.1046/j.1365-2958.2003.03534.x. [DOI] [PubMed] [Google Scholar]
- Tan GS, Kelly P, Kim J, Wartell RM. Comparison of cAMP receptor protein (CRP) and a cAMP-independent form of CRP by Raman spectroscopy and DNA binding. Biochemistry. 1991;30:5076–5080. doi: 10.1021/bi00234a034. [DOI] [PubMed] [Google Scholar]
- Vega Y, Rauch M, Banfield MJ, Ermolaeva S, Scortti M, Goebel W, Vazquez-Boland JA. New Listeria monocytogenes prfA* mutants, transcriptional properties of PrfA* proteins and structure-function of the virulence regulator PrfA. Mol Microbiol. 2004;52:1553–1565. doi: 10.1111/j.1365-2958.2004.04052.x. [DOI] [PubMed] [Google Scholar]
- Velge P, Herler M, Johansson J, et al. A naturally occurring mutation K220T in the pleiotropic activator PrfA of Listeria monocytogenes results in a loss of virulence due to decreasing DNA-binding affinity. Microbiology. 2007;153:995–1005. doi: 10.1099/mic.0.2006/002238-0. [DOI] [PubMed] [Google Scholar]
- Wong KK, Freitag NE. A novel mutation within the central Listeria monocytogenes regulator PrfA that results in constitutive expression of virulence gene products. J Bacteriol. 2004;186:6265–6276. doi: 10.1128/JB.186.18.6265-6276.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn H, Kerby RL, Conrad M, Roberts GP. Study of highly constitutively active mutants suggests how cAMP activates cAMP receptor protein. J Biol Chem. 2006;281:1119–1127. doi: 10.1074/jbc.M509421200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn H, Kerby RL, Koh J, Roberts GP. A C-helix residue, Arg-123, has important roles in both the active and inactive forms of the cAMP receptor protein. J Biol Chem. 2007;282:3632–3639. doi: 10.1074/jbc.M606602200. [DOI] [PubMed] [Google Scholar]
- Youngman P. Plasmid Vectors for Recovering and Exploiting Tn917 Transpositions in Bacillus and Other Gram-Positive Bacteria. In: Hardy K, editor. Plasmids: a Practical Approach. Oxford: IRL Press; 1987. pp. 79–103. [Google Scholar]