

Abstract
Background: Elevated serum uric acid is associated with several cardiovascular disease (CVD) risk factors such as hypertension, inflammation, endothelial dysfunction, insulin resistance, dyslipidemia, and obesity. However, the role of uric acid as an independent risk factor for CVD is not yet clear.
Objective: The aim of the study was to localize quantitative trait loci regulating variation in serum uric acid and also establish the relationship between serum uric acid and other CVD risk factors in Mexican Americans (n = 848; men = 310, women = 538) participating in the San Antonio Family Heart Study.
Methods: Quantitative genetic analysis was conducted using variance components decomposition method, implemented in the software program SOLAR.
Results: Mean ± sd of serum uric acid was 5.35 ± 1.38 mg/dl. Univariate genetic analysis showed serum uric acid and other CVD risk markers to be significantly heritable (P < 0.005). Bivariate analysis showed significant correlation of serum uric acid with body mass index, waist circumference, waist/hip ratio, total body fat, plasma insulin, serum triglycerides, high-density lipoprotein cholesterol, C-reactive protein, and granulocyte macrophage-colony stimulating factor (P < 0.05). A genome-wide scan for detecting quantitative trait loci regulating serum uric acid variation showed a significant logarithm of odds (LOD) score of 4.72 (empirical LOD score = 4.62; P < 0.00001) on chromosome 3p26. One LOD support interval contains 25 genes, of which an interesting candidate gene is chemokine receptor 2.
Summary: There is a significant genetic component in the variation in serum uric acid and evidence of pleiotropy between serum uric acid and other cardiovascular risk factors.
There is a significant genetic component in the variation in serum uric acid and evidence of pleiotropy between serum uric acid and other cardiovascular risk factors.
Uric acid is the final product of purine metabolism in humans, and its circulating concentrations are regulated by the balance in its production and excretion (1). Uric acid is primarily excreted through the kidneys. After filtration, uric acid is reabsorbed, secreted, and then reabsorbed in the proximal tubules. Only 10% of the filtered uric acid is excreted in the urine (2). Increased circulating uric acid levels (hyperuricemia) result from increased consumption of alcohol, purine-rich foods, and fructose-containing sugars and defects in purine metabolism, impaired renal function, hyperinsulinemia leading to increased net tubular absorption, certain types of drugs such as diuretics (3), and genetic predisposition (4).
Hyperuricemia is a risk factor for cardiovascular disease (CVD) (5), which is the leading cause of mortality in Mexican Americans. Two of its major risk factors, obesity and type 2 diabetes, have the highest incidence among this population (6). Increased serum uric acid is associated with other CVD risk factors such as hypertension, inflammation, endothelial dysfunction, and dyslipidemia (7). Potential mechanisms by which hyperuricemia confers CVD risk effects are: 1) impaired endothelial function by interfering with the bioavailability of nitric oxide; 2) renal ischemia and hypertension; 3) inflammation; and 4) production of oxygen radicals during the synthesis of uric acid by xanthine oxidase, especially during ischemia (3).
Aggregation of serum uric acid levels in families (4,8) indicates that variation in serum uric acid levels has a genetic component. Family-based studies have reported significant heritabilties for serum uric acid levels (9,10). Few genome-wide scans for localizing quantitative trait loci (QTLs) influencing the variation in serum uric acid have been undertaken. Moreover, no studies have yet reported the genetic influence on the relationship between serum uric acid and CVD risk factors. Both genetic and environmental factors likely contribute to the variation in serum uric acid. The purpose of this study was 2-fold: to determine 1) whether genetics has significant influence on the variation in serum uric acid levels, and 2) whether serum uric acid is genetically associated with CVD risk factors in this population.
Subjects and Methods
Population characteristics
The San Antonio Family Heart Study (SAFHS) is a family-based genetic study in the Hispanic community in San Antonio, Texas. Begun in 1991, this study focuses on the identification of genes impacting risk for the development of CVD. Probands were recruited without regard to any specific disease status and were at least 90% Mexican-American and between 40–60 yr of age. Approximately 1400 members of more than 40 families have been recruited from low-income neighborhoods in San Antonio (11). Informed consent was obtained from all participants of this study, which was approved by the Institutional Review Board at the University of Texas Health Science Center at San Antonio. Genome-wide scans using 414 polymorphic microsatellite markers (average distance, ∼10 cM) have been successfully conducted for several CVD risk factors (12).
Phenotyping
Blood was collected after an overnight fast, and plasma or serum samples were stored at −80 C until analyzed. Phenotypes that were measured using serum or plasma included uric acid, glucose, insulin, and a lipid panel. Fasting serum uric acid was oxidized in the presence of uricase to form hydrogen peroxide, which was measured photometrically (13). Glucose was analyzed by the glucose oxidase method with an Abbott V/P Analyzer (Abbott Laboratories, Abbott Park, IL). Plasma insulin was measured by a commercially available RIA kit (LINCO Research, Inc., St. Charles, MO).
Anthropometric measurements included height, weight, waist circumference, waist to hip ratio, and body mass index (BMI). Weight was measured to the nearest 0.1 lb, and then to the nearest 0.1 kg, using an ISO-9001 certified Scale-Tronix electronic scale with a capacity of 880 lb (400 kg) (Scale-Tronix, White Plains, NY). Standing height was measured twice, to the nearest 0.1 cm, using a SECA wall-mounted stadiometer (Seca Corp., Hanover, MD). Waist circumference was measured twice, in millimeters, at the level of the umbilicus. The hip circumference was also measured twice, in millimeters, at the widest circumference. These measurements were taken with the participant standing with feet together, arms at their sides, and abdomen relaxed. The average of the two measurements for each variable was then used in the analyses.
BMI was calculated by dividing weight (kilograms) by height (meters) squared. Body fat composition was estimated by bioelectrical impedance technology (Valhalla Scientific, San Diego, CA). Blood pressure was measured three times, with an appropriate arm cuff, using a Random-Zero sphygmomanometer (Gelman-Hawksley Ltd., Sussex, UK). The first measurement of the systolic and diastolic pressures was discarded, and the mean of the second and third readings was used in the analyses.
Genotyping
Genotyping was conducted with DNA isolated from lymphocytes. All members were genotyped for short tandem repeat markers that were spaced at an average interval of 10 cM. All microsatellite markers in the SAFHS were generated using PCR primers from the Research Genetics MapPairs Human screening set versions 6 and 8 (Research Genetics, Inc., Huntsville, AL), and alleles were called using the ABI Prism 377 DNA sequencer and Genescan and Genotyper software (Applied Biosystems, Foster City, CA).
Statistical analyses
Quantitative genetic analyses
Univariate.
A multipoint linkage analysis was employed to detect the presence of a putative QTL or QTLs that might affect serum uric acid. This technique is implemented in the software program SOLAR (14). It is an extension of the variance components approach in which variance due to a specific QTL is added to the basic model. It is based on estimating the effect of a specific QTL on the variation in phenotype and can be modeled as a function of the identity by descent relationship at the marker locus between family members. The phenotypic correlations between family members can be described as the cumulative effect of a specific QTL associated with a marker and residual genetic and environmental effects (15). A model under the null hypothesis in which the additive genetic variance for a specific QTL equals zero was tested against a model under an alternate hypothesis in which the additive variance was estimated. This is known as a likelihood ratio test, and the resultant likelihood ratio test statistic is in this particular case distributed asymptotically as a ½:½ mixture of a χ2 variable with one degree of freedom and a point mass at zero (16). Traditionally, a logarithm of odds (LOD) score, which is computed directly from the likelihood ratio test, is reported in linkage analyses (14).
Bivariate.
Phenotypic, genetic, and environmental correlations were calculated between serum uric acid and other cardiovascular risk factors as summarized by the following model:
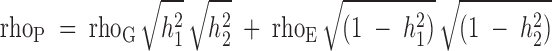 |
where h12 and h22 are heritabilities of the two phenotypes being studied, and rhoG and rhoE are the additive genetic and environmental correlations between the traits, respectively (17).
A model in which all parameters are estimated is compared with a model in which the genetic correlation is constrained to zero. To test for complete pleiotropy between the two traits, a model in which the genetic correlation is constrained to one is compared with a model in which all parameters are estimated. Twice the difference of logarithm likelihood of the two models asymptotically yields a distribution of χ2 with 1 df (16). Evidence of pleiotropy (a common set of genes influencing more than one trait) is indicated by a genetic correlation significantly different from zero.
Prioritization of positional candidate genes
Genesniffer (www.genesniffer.org) was specifically developed to assist in the prioritization of positional candidate genes based on previously detected linkages and one-LOD support chromosomal regions (18). This program provides a quantitative measure of the relevance of the gene to the phenotype of interest. It downloads appropriate web pages (OMIM, PubMed databases, etc.) to locate genes within the given genomic region. This search is based on keywords provided by the investigator related to the biological significance of the particular trait. A cumulative hit score is calculated for each gene. Observed hit scores are compared with the distribution of hit scores generated by random sampling of genes under the null hypothesis of no correlation. Thus, a statistically meaningful measure is given to each gene within the interval.
Results
Quantitative genetic analysis was conducted to localize a QTL regulating the variation in serum uric acid in 848 individuals participating in the SAFHS. Mean ± sd age and serum uric acid levels of these individuals were 47.87 ± 14.8 yr and 5.35 ± 1.38 mg/dl, respectively, with men (n = 310) having higher levels of serum uric acid than women (n = 538) (6.1 ± 1.3 mg/dl vs. 4.94 ± 1.3 mg/dl) (Table 1). Because compromised renal function might cause elevated serum uric acid, we tested traits related to renal function and diabetes that might influence serum uric acid and included significant covariates in our final model. Also, we tested various environmental factors as possible covariates for serum uric acid and found smoking, alcohol intake, and blood pressure medication to be significant. Significant heritability was detected for serum uric acid (h2 = 0.39; P = 2.3 × 10−8) with age, sex, interaction between age and sex, waist to hip ratio, systolic blood pressure, plasma triglyceride, plasma high-density lipoprotein (HDL), serum creatinine, blood pressure medications, alcohol intake, smoking status, and diabetes status as covariates. We also found significant heritabilities for other CVD risk factors (Table 2).
Table 1.
Characteristics of study participants
| Variables | Women | Men |
|---|---|---|
| Age (yr) | 48.37 ± 14.6 | 47.02 ± 15.1 |
| BMI (kg/m2) | 32.47 ± 7.6 | 30.52 ± 6.2 |
| Waist circumference (cm) | 106.43 ± 17.4 | 103.84 ± 16.3 |
| Waist to hip ratio | 0.94 ± 0.07 | 0.98 ± 0.09 |
| Systolic blood pressure (mm Hg) | 123.36 ± 19.9 | 126.19 ± 17.3 |
| Diastolic blood pressure (mm Hg) | 68.55 ± 9.5 | 71.94 ± 11.2 |
| Serum (mg/dl) | ||
| Creatinine | 0.76 ± 0.5 | 1.01 ± 0.9 |
| Glucose | 102.57 ± 39.2 | 110.36 ± 49.7 |
| Triglycerides | 122.33 ± 67 | 141.11 ± 112 |
| HDL | 50.32 ± 13.1 | 44.61 ± 13.7 |
| Uric acid | 4.94 ± 1.3 | 6.10 ± 1.3 |
| Individuals who (%) | ||
| Smoke | 10 | 10 |
| Take alcohol | 22 | 21 |
| Are diabetic | 13 | 9 |
| Take hypertension medication | 17 | 8 |
| Have hyperuricemia | 13 | 9 |
Data are presented as mean ± sd.
Table 2.
Phenotypic, genetic, and environmental correlation of serum uric acid with circulating levels of CVD risk factors in the SAFHS
| Phenotype | h2 | rhoG | rhoE | rhoP |
|---|---|---|---|---|
| BMI | 0.547 (0.07)a | 0.204 (0.14) | 0.341 (0.09)a | 0.27a |
| Waist circumference (cm) | 0.541 (0.08)a | 0.280 (0.13)b | 0.286 (0.09)a | 0.28a |
| Total body fat (g) | 0.618 (0.08)a | 0.294 (0.13)b | 0.035 (0.10) | 0.17b |
| Waist-hip ratio | 0.348 (0.09)a | 0.289 (0.17) | 0.157 (0.08)c | 0.20b |
| Glucose (mg/dl) | 0.320 (0.07)a | 0.103 (0.18) | −0.10 (0.08) | NS |
| Insulin (μIU/ml) | 0.458 (0.09)a | 0.148 (0.16) | 0.235 (0.08)a | 0.20b |
| Triglycerides (mg/dl) | 0.535 (0.08)a | 0.184 (0.14) | 0.234 (0.09)b | 0.21a |
| HDL cholesterol (mg/dl) | 0.315 (0.07)a | −0.158 (0.17) | −0.13 (0.07)c | −0.18b |
| Total cholesterol (mg/dl) | 0.445 (0.08)a | 0.007 (0.15) | 0.064 (0.09) | NS |
| Systolic blood pressure (mm Hg) | 0.280 (0.08)a | 0.332 (0.21) | −0.05 (0.08) | 0.08c |
| Diastolic blood pressure (mm Hg) | 0.218 (0.07)a | 0.233 (0.21) | 0.01 (0.07) | NS |
| CRP (ng/ml) | 0.222 (0.15)a | −0.09 (0.44) | 0.495 (0.15)a | 0.31a |
| GM-CSF (pg/ml) | 0.327 (0.17)a | −0.18 (0.39) | 0.404 (0.23)c | 0.19b |
Data in parentheses represent se. h2, Heritability; rhoG, genetic correlation; rhoE, environmental correlation; rhoP, phenotypic correlation; NS, not significant.
P < 0.005.
P < 0.05.
P < 0.07.
To understand the relationship between serum uric acid and CVD risk factors, we conducted bivariate analyses. Table 2 depicts genetic as well as phenotypic correlations between serum uric acid and CVD risk factors. Genetic correlation of serum uric acid was significant only with waist circumference and total body fat. Significant and positive phenotypic correlations were obtained for BMI, waist circumference, total body fat, waist-hip ratio, plasma insulin, serum triglycerides, C-reactive protein (CRP), and granulocyte macrophage-colony stimulating factor (GM-CSF). Serum uric acid levels were negatively associated with HDL cholesterol.
Multipoint linkage analysis revealed a QTL influencing serum uric acid levels on chromosome 3p with a significant LOD score of 4.72 (empirical LOD score = 4.62; P < 0.00001; Fig. 1). The peak LOD score for linkage occurred between markers D3S2387 and D3S3050 on chromosome 3p26 with the one-LOD support interval spanning about 7 Mb and containing 25 genes. In our previous study (19), we had evidence of suggestive linkage (LOD = 2.2) on chromosome 3p that overlapped with our present linkage signal (Fig. 2). For this analysis, 4092 relative pairs were used (Table 3). A majority of them were first and second cousins, followed by avuncular, parent-offspring, and sibling relationships. We used the software program Genesniffer to narrow the number of potential positional candidate genes based on their potential relevance to the focal phenotype. Results from this analysis are shown on Table 4 with their respective scores. In our one-LOD support interval (∼7 Mb), there are about 25 known genes. Of these, 12 were given scores by the Genesniffer program, and two [chemokine receptor 2 (CCR2) and CCR5] were selected by the available positional data.
Figure 1.

Summary of the univariate linkage analysis for serum uric acid by each chromosome. Chromosomal location (cM) is represented on the x-axis, and the LOD score shown on the y-axis.
Figure 2.

Plot of chromosome 3 depicting the overlap between the present study and our previous study. Chromosomal location (cM) is represented on the x-axis, and the LOD score shown on the y-axis.
Table 3.
Relative pairs used in this study
| Relationship | No. of pairs |
|---|---|
| Parent-offspring | 302 |
| Siblings | 386 |
| Grandparent-grandchild | 79 |
| Avuncular | 692 |
| Half-siblings | 74 |
| Great grandparent-grandchild | 2 |
| Grand avuncular | 107 |
| Half avuncular | 57 |
| First cousins | 933 |
| Half grand avuncular | 8 |
| First cousins, once removed | 820 |
| Half first cousins | 51 |
| First cousins, twice removed | 22 |
| Half first cousins, once removed | 21 |
| Second cousins | 392 |
| Second cousins, once removed | 140 |
| Half second cousins | 4 |
| Half first cousins and second cousins | 1 |
| Identical sib-pair | 1 |
| Total | 4092 |
Table 4.
Serum uric acid candidate genes in the chromosome 3p one-LOD support interval identified by Genesniffer
| Location (Kb)a | Gene symbol | Size (Kb) | Grand total hit score |
|---|---|---|---|
| 35.202 | CCR2 | 17.805 | NA |
| 54.222 | CCR5 | 27.202 | NA |
| 159.458 | CHL1 | 607.749 | 9680 |
| 767.998 | CNTN6 | 1000.424 | 2621 |
| 1768.951 | CNTN4 | 1311.000 | 939 |
| 3080.551 | IL5RA | 54.108 | 27 |
| 3507.805 | LRRN1 | 580.014 | 115 |
| 4094.770 | SETMAR | 260.975 | 137 |
| 4356.154 | SUMF1 | 140.633 | 56 |
| 4498.408 | ITPR1 | 430.597 | 967 |
| 4930.735 | BHLHB2 | 139.473 | 65 |
| 5071.208 | ARL8B | 129.575 | 968 |
| 5201.799 | EDEM1 | 855.476 | 60 |
| 6057.552 | GRM7 | 941.575 | 2035 |
NA, Not available.
Position based on chromosome coordinate.
Discussion
In this study, we have shown a strong genetic component for the variation of serum uric acid. Serum uric acid was heritable, and a QTL regulating the variation in its levels was localized. The strong signal was in the region of 3p26 and its surrounding one −LOD support interval spans about 7 Mb and contains 25 genes. Heritability of serum uric acid found in our study was similar to those reported by several family-based studies, with estimates ranging from 25 to 73% (9,10). However, few genome-wide scans for localizing QTLs influencing variation in serum uric acid have been undertaken.
Yang et al. (20) reported a genome-wide scan for serum uric acid in a Caucasian population from the Framingham Heart Study with a LOD score of 3.3 on chromosome 15q. In another study where subjects were recruited partly from the Framingham Heart study, serum uric acid was studied as a part of the metabolic syndrome where the variation in serum uric acid was localized to a QTL on chromosome 2q (21). Early studies on the genetics of serum uric acid proposed a major gene for serum uric acid (8), although later studies (segregation analyses) could not support that theory and instead proposed that serum uric acid is affected by several genes (9,10). Although genome-wide linkage studies with serum uric acid have found a number of QTLs on various chromosomes, none of these studies have been replicated. In our previous study (19), we found strong evidence of linkage to a QTL on chromosome 6. However, we also had a suggestive linkage on chromosome 3p, which is about 25 cM distance from our present signal (Fig. 2). The signal that we are reporting in this manuscript represents a significant refinement of our previous marginal signal as a result of the addition of 200 individuals. It is true that there is overlap between the two studies; however, addition of individuals can significantly change family pedigree structures. Increasing family members will affect a network of relationships and their contribution toward the linkage signal. Variation in position for linkages can be substantial (tens of centimorgans) in case of complex disorders, when trying to replicate previous studies (22). Increase in the number of individuals is just one of many factors that can affect genetic information, thereby affecting both location and strength of a linkage signal. In a recently conducted genome-wide association study, variants in solute carrier family 2, member 9 protein (SLC2A9/GLUT9) were strongly associated with serum uric acid (23). This gene, located at 4p15, seems to be involved in fructose as well as uric acid transport and excretion (24).
The region of p26 on chromosome 3 has also been reported to harbor QTLs related to CVD risk factors. In a genome-wide linkage scan in a population comprising African-Americans and Whites, evidence for suggestive linkage was found for resting blood pressure in the same region near the marker D3S2387 (25). The same marker was also associated with BMI (LOD = 3.7) in a cross-sectional family study of African-Americans and Hispanic-Americans (26). In another study, the Hypertension Genetic Epidemiology Network study, genome scan was conducted on factors derived from factor analysis for metabolic syndrome. Significant linkage was found for obesity-insulin factor on chromosome 3p26 (27). In the present study, evidence for suggestive linkage was found on chromosomes 1, 4, and 15 in addition to the strong signal on chromosome 3. In genetic studies exploring cardiovascular risk factors, suggestive linkages were found in the same region of chromosome 1 for metabolic syndrome components (28) and significant linkage for resting heart rate on chromosome 4 (29).
In the one-LOD support interval surrounding our QTL on chromosome 3p26, 14 positional candidate genes have been identified. There are two important clusters of genes in this region. One cluster encodes chemokine receptors (CCR2, CCR3, CCR5, CCR8, CCR9, and CCRL2) and the other, contactins (CNTN4 and CNTN6). Genes CCR2 and CCR5 were not identified by the Genesniffer, possibly because of the confusion about the physical location of these genes. According to NCBI Mapviewer and OMIM web sites, the physical location of these two genes is 3p21-22. However, base pair positions of these genes show that these should be near the telomere and within our one-LOD-support interval. Because these genes are relevant to cardiovascular risk, they are included in the potential candidate genes. CCR2 and CCR5 are receptors for monocyte chemoattractant protein 1 (MCP1), which plays a major role in the recruitment of monocytes from the blood in the early stages of atherosclerosis (30) and “regulated upon activation, normal T cell expressed, and secreted” (RANTES) or CCL5, protein, respectively. Both MCP1 and RANTES have been implicated in the development of atherosclerotic lesions, leukocyte adhesion (31), and host defense (32). Given the role of serum uric acid in inflammation as well as studies showing that uric acid stimulates MCP1 production in smooth muscle cells (33), this location of chromosome 3p where the gene for MCP1 receptor resides combines to support our argument. Other relevant candidate genes in this region are contactins, part of the Ig superfamily (34), and inositol 1, 4, 5 triphosphate receptor 1 (ITPR1), which is known to play a role in cardiac contraction and is known to be affected by diabetes because its mRNA expression was significantly reduced in diabetic compared with nondiabetic subjects (35).
Epidemiological studies have shown that increased serum uric acid concentration is associated with several CVD risk factors such as hypertension, inflammation, endothelial dysfunction, dyslipidemia, and obesity. In this study, we found significant positive correlations between serum uric acid and several phenotypes associated with obesity, adiposity, inflammation, and hypertension. In the National Health and Nutrition Examination Survey follow-up study, 1971–1992, increased serum uric acid levels were found to be positively and significantly associated with CVD mortality (7). It is not yet clear whether serum uric acid is an independent risk factor for CVD or whether it is merely associated with other CVD risk factors. However, a recently published study by Strasak et al. (36) shows that serum uric acid is an independent predictor for all major forms of CVD death. This was a 21-yr prospective follow-up study. Longitudinal studies provided a platform for such studies. In the SAFHS, we have data spanning 15 yr. We are planning to measure uric acid in blood samples collected during earlier phases and investigate whether serum uric acid can indeed predict CVD events and/or mortality. Also, serum uric acid was found to be an independent predictor of hypertension incidence and longitudinal blood pressure progression in a study spanning 4 yr in individuals who had no hypertension, gout, heart, or renal problems at baseline (37). In our present study, we found a positive correlation of serum uric acid with systolic blood pressure, but not with diastolic blood pressure. In another study over 9 yr of follow-up, serum uric acid was positively associated with incident hypertension (38). In addition, childhood serum uric acid concentration is known to predict adult blood pressure (39). Hyperuricemia is known to predict CVD events, particularly in hypertensive individuals (3). It is present in 25% of untreated hypertensive subjects and in more than 75% of those with malignant hypertension (40). Whether hyperuricemia is causally related to hypertension remains to be determined. Uric acid, however, causes endothelial dysfunction, and oxygen radicals generated by xanthine oxidase contribute to the pathogenesis of hypertension (41).
Obesity is characterized by excess body fat, and this excess body fat induces a variety of CVD risk factors such as insulin resistance, hypertension, dyslipidemia, and endothelial dysfunction (42). In this study, we found significant genetic correlation of serum uric acid with waist circumference (visceral adiposity) and total body fat. A significant genetic correlation indicates a shared genetic effect between serum uric acid and body fat (total and visceral). It means that common sets of genes are regulating both serum uric acid and body fat. Studies have shown a positive correlation between visceral fat accumulation and serum uric acid (43). Waist circumference is considered a surrogate measure for visceral fat accumulation. Increased free fatty acid production due to an increase in visceral fat might indirectly affect uric acid production through common pathways involving nicotinamide adenine dinucleotide phosphate. It is also believed that development of insulin resistance due to increased visceral fat might contribute to decreased uric acid excretion and increased serum uric acid concentrations (44). This is significant because body fat (particularly visceral) is a major CVD risk factor, and obese individuals are more likely to have hyperuricemia than their lean counterparts. Because higher uric acid levels have been associated with most of the components of the metabolic syndrome (45,46), it has been suggested that hyperuricemia should be added to the cluster of abnormalities associated with the metabolic syndrome (47). Moreover, the prevalence of the metabolic syndrome is higher in individuals with hyperuricemia (45). We also found a positive association of serum uric acid with several components of the metabolic syndrome in this study and a negative association with HDL cholesterol, implying that an increase in serum uric acid is associated with an increase in all metabolic syndrome components with the exception HDL cholesterol. We found a positive correlation between proinflammatory cytokines CRP and GM-CSF and serum uric acid. Coutinho et al. (46) found a similar association between uric acid and CRP. Uric acid has also been reported to regulate inflammatory pathways in vascular smooth muscle cells (32) and in endothelial cells (41). One limitation of this study is that we did not have individual medication data, such as losartan, allopurinol, etc.; instead we had medication classes, and we could include only those in our analysis. However, not accounting for such covariates could lead to a significant attenuation of the linkage signal; it would not lead to a fake genetic signal. Similarly, we did not have data on urinary pH, which is an important feature in the formation of uric acid stones (48).
To summarize, we found a strong genetic component in the variation in serum uric acid levels in Mexican Americans. We also found significant association of serum uric acid with other CVD risk factors, particularly adiposity phenotypes, and identified a QTL that influences serum uric acid levels. The next logical step is to explore the association between variants in positional candidate genes and serum uric acid and other CVD risk factors.
Acknowledgments
We thank all participants of the San Antonio Family Heart Study for their cooperation and generous participation. We also acknowledge the Fredric C. Bartter General Clinical Research Center, University of Texas Health Science Center at San Antonio, San Antonio, Texas (supported by M01-RR01346), for providing clinical support for this project.
Footnotes
This study was supported by Grant PO1 HL4522 from the National Heart, Lung and Blood Institute, Grant MH59490 from the National Institute of Mental Health, the George O'Brien Kidney Research Center Grant P50-DK-61597 from National Institutes of Health (NIH)/National Institute of Diabetes, Digestive and Kidney Diseases, and the Veterans Administration Merit Review (to H.E.A. and N.H.A.). S.D.N. is supported by an American Heart Association Postdoctoral Fellowship (0525206Y). F.T. is supported by the American Heart Association Scientist Development Grant Award (0530059N). This investigation was conducted in facilities constructed with support for the Research Facilities Improvement Program Grants C06 RR017515 and RR013556 from the National Center for Research Resources/NIH.
Disclosure Statement: The authors have nothing to disclose.
First Published Online November 11, 2008
Abbreviations: BMI, Body mass index; CCR, chemokine receptor; CRP, C-reactive protein; CVD, cardiovascular disease; GM-CSF, granulocyte macrophage-colony stimulating factor; HDL, high-density lipoprotein; LOD, logarithm of odds; MCP1, monocyte chemoattractant protein 1; QTL, quantitative trait locus.
References
- Dawson J, Walters M 2006 Uric acid and xanthine oxidase: future therapeutic targets in the prevention of CVD? Br J Clin Pharm 62:633–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alderman MH 2002 Uric acid and cardiovascular risk. Curr Opin Pharmacol 2:126–130 [DOI] [PubMed] [Google Scholar]
- Schachter M 2005 Uric acid and hypertension. Curr Pharm Des 11:4139–4143 [DOI] [PubMed] [Google Scholar]
- Cameron JS, Simmonds HA 2005 Hereditary hyperuricemia and renal disease. Semin Nephrol 25:9–18 [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Kang D-H, Feig D, Sanchez-Lozada LG, Srinivas TR, Ejaz AA, Segal M, Johnson RJ 2006 Unearthing uric acid: an ancient factor with recently found significance in renal and CVD. Kid Int 69:1722–1725 [DOI] [PubMed] [Google Scholar]
- Torres M, Azen S, Varma R, LALES Group 2006 Prevalence of obesity and associated co-morbid conditions in a population-based sample of primarily urban Mexican-Americans. Ethn Dis 16:362–369 [PubMed] [Google Scholar]
- Fang J, Alderman MH 2000 Serum uric acid and cardiovascular mortality. The NHANES1 Epidemiologic follow-up study, 1971–1992. JAMA 283:2404–2410 [DOI] [PubMed] [Google Scholar]
- Stecher RM, Hersh AH, Solomon WM 1949 The heredity of gout and its relationship to familial hyperpuricemia. Ann Intern Med 31:595–614 [DOI] [PubMed] [Google Scholar]
- Rao DC, Laskarzewski PM, Morrison JA, Kelly K, Glueck CJ 1982 The Clinical Lipid Research Clinic Family Study: familial determinants of plasma uric acid. Hum Genet 60:257–261 [DOI] [PubMed] [Google Scholar]
- Rice T, Vogler GP, Perry TS, Laskarzewski PM, Province MA, Rao DC 1990 Heterogeneity in the familial aggregation of fasting uric acid levels in five North American populations: the Lipid Research Clinics Family Study. Am J Med Genet 36:219–225 [DOI] [PubMed] [Google Scholar]
- MacCluer JW, Stern MP, Almasy L, Atwood LA, Blangero J, Comuzzie AG, Dyke B, Haffner SM, Henkel RD, Hixson JE, Kammerer CM, Mahaney MC, Mitchell BD, Rainwater DL, Samollow PB, Sharp RM, VandeBerg JL, Williams JT 1999 Genetics of atherosclerosis risk factors in Mexican Americans. Nutr Rev 57:S59–S65 [DOI] [PubMed] [Google Scholar]
- Comuzzie AG, Mitchell BD, Cole S, Martin LJ, Hsueh W-C, Rainwater DL, Almasy L, Stern MP, J. Hixson, MacCluer JW, Blangero J 2003 The genetics of obesity in Mexican Americans: the evidence from genome scanning efforts in the San Antonio Family Heart Study. Hum Biol 75:635–646 [DOI] [PubMed] [Google Scholar]
- Domagk GF, Schlicke HH 1968 A colorimetric method using uricase and peroxidase for the determination of uric acid. Anal Biochem 22:219–224 [DOI] [PubMed] [Google Scholar]
- Almasy L, Blangero J 1998 Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet 62:1198–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blangero J, Almasy L 1997 Multipoint oligogenic linkage analysis of quantitative traits. Genet Epidemiol 14:959–964 [DOI] [PubMed] [Google Scholar]
- Self S, Liang K 1987 Asymptotic properties of maximum likelihood-ratio tests under nonstandard conditions. J Am Stat Assoc 82:605–610 [Google Scholar]
- Hopper JL, Mathews JD 1982 Extensions to multivariate normal models for pedigree analysis. Ann Hum Genet 46:373–383 [DOI] [PubMed] [Google Scholar]
- Rutherford S, Cai G, Lopez-Alvarenga JC, Kent JW, Voruganti VS, Proffitt JM, Curran JE, Johnson MP, Dyer TD, Jowett JB, Bastarrachea RA, Atwood LD, Goring HH, Maccluer JW, Moses EK, Blangero J, Comuzzie AG, Cole SA 2007 A chromosome 11q quantitative-trait locus influences change of blood-pressure measurements over time in Mexican Americans of the San Antonio Family Heart Study. Am J Hum Genet 81:744–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath SD, Voruganti VS, Arar NH, Thameem F, Lopez-Alvarenga JC, Bauer R, MacCluer JW, Blangero J, Comuzzie AG, Abboud HE 2007 Genome scan for determinants of serum uric acid variability. J Am Soc Nephrol 18:3156–3163 [DOI] [PubMed] [Google Scholar]
- Yang Q, Guo CY, Cupples LA, Levy D, Wilson PWF, Fox CS 2005 Genome-wide search for genes affecting serum uric acid levels: the Framingham Heart Study. Metabolism 54:1435–1441 [DOI] [PubMed] [Google Scholar]
- Tang W, Miller MB, Rich SS, North KE, Panlow JS, Borecki I, Myers RH, Hopkins PN, Leppert M, Arnett DK 2003 Linkage analysis of a composite factor for the multiple metabolic syndrome. The National Heart, Lung and Blood Institute Family Heart Study. Diabetes 52:2840–2847 [DOI] [PubMed] [Google Scholar]
- Roberts SB, MacLean CJ, Neale MC, Eaves LJ, Kendler KS 1999 Replication of linkage studies of complex traits: an examination of variation in location estimates. Am J Hum Genet 65:876–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitart V, Rudan I, Hayward C, Gray NK, Floyd J, Palmer CAN, Knott SA, Kolcic I, Polasek O, Graessler J, Wilson JF, Marinaki A, Riches PL, Shu X, Janicijevic B, Smolj-Narancic N, Gorgoni B, Morgan J, Campbell S, Biloglav Z, Barac-Lauc L, Pericic M, Martinovic Klaric I, Zgaga L, Skaric-Juric T, Wild SH, Richardson WA, Hohenstein P, Kimber CH, Tenesa A, Donnelly LA, Fairbanks LD, Aringer M, McKeigue PM, Ralston SH, Morris AD, Rudan P, Hastie ND, Harry Campbell, Wright AF 2008 SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat Genet 40:437–442 [DOI] [PubMed] [Google Scholar]
- Le MT, Shafiu M, Mu W, Johnson R 2008 SLC2A9—a fructose transporter identified as a novel uric acid transporter. Nephrol Dial Transplant 23:2746–2749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice T, Rankinen T, Chagnon YC, Province MA, Perusse L, Leon AS, Skinner JS, Wilmore JH, Bouchard C, Rao DC 2002 Genomewide linkage scan of resting blood pressure. Hypertension 39:1037 [DOI] [PubMed] [Google Scholar]
- Norris JM, Langefeld CD, Scherzinger AL, Rich SS, Bookman E, Beck SR, Saad MF, Haffner SM, Bergman RN, Bowden DW, Wagenknecht LE 2005 Quantitative trait loci for abdominal fat and BMI in Hispanic-Americans and African-Americans: the IRAS Family Study. Int J Obes 29:67–77 [DOI] [PubMed] [Google Scholar]
- Kraja AT, Hunt SC, Pankow JS, Myers RH, Heiss G, Lewis CE, Rao DC, Province MA 2005 Quantitative trait loci for metabolic syndrome in the Hypertension Genetic Epidemiology Network Study. Obes Res 13:1885–1890 [DOI] [PubMed] [Google Scholar]
- Bosse Y, Despres JP, Chagnon YC, Rice T, Rao DC, Bouchard C, Perusse L, Vohl MC 2007 Quantitative trait locus on 15q for a metabolic syndrome variable derived from factor analysis. Obesity 15:544–550 [DOI] [PubMed] [Google Scholar]
- Martin LJ, Comuzzie AG, Sonnenberg GE, Myklebust J, James R, Marks J, Blangero J, Kissebah A 2004 Major quantitative trait locus for resting heart rate maps to a region on chromosome 4. Hypertension 43:1146–1151 [DOI] [PubMed] [Google Scholar]
- Charo IF, Taubman MB 2004 Chemokines in the pathogenesis of vascular disease. Circ Res 95:858–866 [DOI] [PubMed] [Google Scholar]
- Kuziel WA, Morgan SJ, Dawson TC, Griffen S, Smithies O, Ley K, Maeda N 1997 Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc Natl Acad Sci USA 94:12053–12058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara T, Warr G, Loy J, Bravo R 1997 Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J Exp Med 186:1757–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanellis J, Watanabe S, Li JH, Kang DH, Li P, Nakagawa T, Wamsley A, Sheikh-Hamad D, Lan HY, Feng L, Johnson RJ 2003 Uric acid stimulates monocyte chemoattractant protein-1 production in vascular smooth muscle cells via mitogen-activated protein kinase and cycoloxygenase-2. Hypertension 41:1287–1293 [DOI] [PubMed] [Google Scholar]
- Zeng L, Zhang C, Xu J, Ye X, Wu Q, Dai J, Ji C, Gu S, Xie Y, Mao Y 2002 A novel splice variant of the cell adhesion molecule contactin 4 (CNTN4) is mainly expressed in human brain. J Hum Genet 47:497–499 [DOI] [PubMed] [Google Scholar]
- Guner S, Arioglu E, Tay A, Tasdelen A, Aslamaci S, Bidasee KR, Dincer UD 2004 Diabetes decreases mRNA levels of calcium-releasing channels in human atrial appendage. Mol Cell Biochem 263:143–150 [DOI] [PubMed] [Google Scholar]
- Strasak AM, Kelleher CC, Brant LJ, Rapp K, Ruttmann E, Concin H, Diem G, Pfieffer KP, Ulmer H, VHM, PP Study Group 2008 Serum uric acid as an independent predictor for all major forms of cardiovascular death in 28,613 elderly women: a prospective 21-year follow-up study. Int J Cardiol 125:232–239 [DOI] [PubMed] [Google Scholar]
- Sundstorm J, Sullivan L, D'Agostino RB, Levy D, Kannel WB, Vasan RS 2005 Relations of serum uric acid to longitudinal blood pressure tracking and hypertension incidence. Hypertension 45:28–33 [DOI] [PubMed] [Google Scholar]
- Mellen PB, Bleyer AJ, Erlinger TP, Evans GW, Nieto FJ, Wagenknecht LE, Wofford MR, Herrington DM 2006 Serum uric acid predicts incident hypertension in a biethnic cohort. The Atherosclerosis Risk in Communities Study. Hypertension 48:1037–1042 [DOI] [PubMed] [Google Scholar]
- Alper Jr AB, Chen W, Yau L, Srinivasan SR, Berenson GS, Hamm LL 2005 Childhood uric acid predicts adult blood pressure: The Bogalusa Heart Study. Hypertension 45:34–38 [DOI] [PubMed] [Google Scholar]
- Johnson RJ, Kang DH, Feig D, Kivlighn S, Kanellis J, Watanabe S, Tuttle KR, Rodriguez-Iturbe B, Herrera-Acosta J, Mazzali M 2003 Is there a pathogenetic role for uric acid in hypertension and cardiovascular and renal disease? Hypertension 41:1183–1190 [DOI] [PubMed] [Google Scholar]
- Khosla UM, Zharikov S, Finch JL, Nakagawa T, Roncal C, Mu W, Krotova K, Block ER, Prabhakar S, Johnson RJ 2005 Hyperuricemia induces endothelial dysfunction. Kidney Int 67:1739–1742 [DOI] [PubMed] [Google Scholar]
- Hauner H 2005 Secretory factors from human adipose tissue and their functional role. Proc Nutr Soc 64:163–169 [DOI] [PubMed] [Google Scholar]
- Takahashi S, Yamamoto T, Tsutsumi Z, Moriwaki Y, Yamakita J, Higashino K 1997 Close correlation between visceral fat accumulation and uric acid metabolism in healthy men. Metabolism 46:1162–1165 [DOI] [PubMed] [Google Scholar]
- Hikita M, Ohno I, Mori Y, Ichida K, Yokose T, Hosoya T 2007 Relationship between hyperuricemia and body fat distribution. Intern Med 46:1353–1358 [DOI] [PubMed] [Google Scholar]
- Choi HK, Ford ES 2007 Prevalence of the metabolic syndrome in individuals with hyperuricemia. Am J Med 120:442–447 [DOI] [PubMed] [Google Scholar]
- Coutinho TA, Turner ST, Peyser PA, Bielak LF, Sheedy II PF, Kullo IJ 2007 Associations of serum uric acid with markers of inflammation, metabolic syndrome and subclinical atherosclerosis. Am J Hypertens 20:83–89 [DOI] [PubMed] [Google Scholar]
- Zavaroni I, Mazza S, Fantuzzi M, Dall'Aglio E, Bonora E, Delsignore R, Passeri M, Reaven GM 1993 Changes in insulin and lipid metabolism in males with asymptomatic hyperuricemia. J Intern Med 234:25–30 [DOI] [PubMed] [Google Scholar]
- Maalouf NM, Cameron MA, Moe OW, Adams-Huet B, Sakhee K 2007 Low urine pH: a novel feature of the metabolic syndrome. Clin J Am Soc Nephrol 2:883–888 [DOI] [PubMed] [Google Scholar]