

Abstract
The E7 oncoprotein of high risk human papillomavirus type 16 (HPV16) binds and inactivates the retinoblastoma (RB) family of proteins. Our previous studies suggested that HPV16 E7 enters the nucleus via a novel Ran-dependent pathway independent of the nuclear import receptors (Angeline et al., 2003). Here, analysis of the localization of specific E7 mutants revealed that the nuclear localization of E7 is independent of its interaction with pRB or of its phosphorylation by CKII. Fluorescence microscopy analysis of enhanced green fluorescent protein (EGFP) and 2xEGFP fusions with E7 and E7 domains in HeLa cells revealed that E7 contains a novel nuclear localization signal (NLS) in the N-terminal domain (aa 1-37). Interestingly, treatment of transfected HeLa cells with two specific nuclear export inhibitors, Leptomycin B and ratjadone, changed the localization of 2xEGFP-E738-98 from cytoplasmic to mostly nuclear. These data suggest the presence of a leucine-rich nuclear export signal (NES) and a second NLS in the C-terminal domain of E7 (aa 38-98). Mutagenesis of critical amino acids in the putative NES sequence (76IRTLEDLLM84) changed the localization of 2xEGFP-E738-98 from cytoplasmic to mostly nuclear suggesting that this is a functional NES. The presence of both NLSs and an NES suggests that HPV16 E7 shuttles between the cytoplasm and nucleus which is consistent with E7 having functions in both of these cell compartments.
Introduction
Infection caused by human papillomaviruses (HPVs) is associated with more than 99% of cervical cancers. Over 200 HPV genotypes have been identified, and 30 are known to infect anogenital mucosal epithelial tissues. Mucosal HPVs have demonstrated different degrees of oncogenic potential, with some classified as “high risk”, such as, types 16, 18, 31 and 45, and others as “low risk”, such as, types 6 and 11. High risk HPVs are frequently detected in invasive cervical carcinomas, whereas the low risk types are more often associated with benign exophytic condylomas (zur Hausen, 2000).
Replication of HPVs is intimately connected to the differentiation program of host epithelial cells. As HPVs depend on the host cell's replication machinery for generation of viral progeny, they have evolved the early proteins E6 and E7 that are able to induce the differentiated cell to maintain active DNA replication machinery which is necessary during viral replication (Jones and Munger, 1996; Rapp and Chen, 1998). In HPV-positive genital cancers or carcinoma cell lines, the integration of the viral genomes HPV16 or 18 into the cellular genome results in the loss of expression of the viral E2 gene and high levels of the E6 and E7 oncoproteins. High risk HPV E6 and E7 proteins can induce cellular immortalization and transformation cooperatively, and are necessary for the induction and maintenance of the transformed state (Rapp and Chen, 1998). Studies in transgenic mice suggest that whereas E7 promotes the formation of benign tumors, E6 acts primarily to accelerate progression of these benign tumors to the malignant stage (Song et al., 2000). Genomic instability is a hallmark of cervical carcinomas and high risk HPV E6 and E7 can rapidly induce numerical and structural chromosome instability (Duensing and Munger, 2004).
High risk HPV16 E7 oncoprotein is a phosphoprotein of 98 amino acids and it is structurally and functionally related to Adenovirus E1A protein. HPV16/18 E7 oncoproteins bind and inactivate several nuclear proteins involved in the control of cell growth including retinoblastoma protein (pRB), and the RB-related pocket proteins, p107 and p130 (Dyson et al., 1992; Dyson et al., 1989; Munger et al., 1989a). E7 oncoproteins also interact with multiple components of the cell cycle machinery, including E2F/cyclin A complex, cyclin E and the cyclin-dependent kinase inhibitors p27 and p21 (Jones and Munger, 1996; Zwerschke and Jansen-Durr, 2000). HPV16 E7 also has target proteins localized in the cytoplasm such as the microtubule-associated N-end rule ubiquitin ligase p600 (Huh et al., 2005), suggesting that E7 has functions mediated in the cytoplasm in addition to its nuclear functions.
HPV16 E7 consists of three domains: CR1 (aa 1-15), CR2 (aa 16-37) and CR3 (aa 38-98), the C-terminal domain involved in binding Zn and dimerization. The CR2 domain contains both the pRB binding site and the CKII phosphorylation site (Munger et al., 2001). High risk HPV16 E7 is predominantly a nuclear protein in invasive cervical carcinoma in situ, in the CaSki cervical carcinoma cell line and also when expressed transiently in different cell lines like HaCaT and U2OS (Guccione et al., 2002, Fiedler et al., 2004). In cervical biopsies the high levels of nuclear HPV16 E7 correlate with reduced pRB levels during progression to carcinomas, suggesting that E7-dependent pRB degradation occurs in vivo (Fiedler et al., 2004). It was suggested, using transfection assays, that a region of HPV16 E7 (aa 16 – 41) can localize a reporter protein into the nucleus (Fujikawa et al., 1994). We have previously analyzed the nuclear import of HPV16 E7 in import assays in digitonin-permeabilized cells and discovered that it enters the nucleus via a novel Ran-dependent pathway that is independent of karyopherin β import receptors (importins) (Angeline, Merle, and Moroianu, 2003). This is in agreement with HPV16 E7 not having a canonical basic NLS or any other characterized NLS known to interact with karyopherins/importins.
In order to map the NLS of HPV16 E7 and characterize the requirements for nuclear localization we used transfection assays with EGFP and 2xEGFP fusion plasmids containing E7 wild type, or E7 mutants or different E7 domains followed by fluorescence microscopy analysis of the intracellular localization. These in vivo experiments were complemented by in vitro nuclear import assays with GST-fusion proteins containing either wild type E7 or different E7 domains.
Our data revealed that neither binding of pRB nor phosphorylation of E7 by CKII is required for nuclear localization of HPV16 E7. Interestingly, we discovered that HPV16 E7 contains an nNLS in the N-terminal domain (aa 1-37 containing both CR1 and CR2 domains) and both a second cNLS and a leucine-rich nuclear export signal (NES) in the CR3 domain (aa 38-98). The presence of both NLSs and an NES is not surprising as HPV16 E7 is known to interact with proteins localized both in the nucleus and in the cytoplasm.
Results
Binding of E7 to pRB is not required for nuclear localization of HPV16 E7
To analyze the intracellular localization of HPV16 E7, we used transient transfection experiments in HeLa cells with enhanced green fluorescence protein (EGFP) fused to the wild type E7. We made an EGFP-E7 plasmid with the EGFP fused at the N terminus of E7 and upon transfection of HeLa cells the translated EGFP-E7 fusion protein was well expressed and localized mostly in the nucleus (Fig. 1A, panel A), in contrast with the EGFP itself which, as expected, was localized throughout the cell (Fig. 1A, panel C).
Fig. 1.

A. EGFP-16E7 localizes mostly to the nucleus in HeLa cells in vivo. HeLa cells were transfected with either EGFP-E7 (panels A and B), or EGFP (panels C and D) plasmids and examined by fluorescence microscopy at 24 hours post transfection. Panels A and C represent the fluorescence of the EGFP and panels B and D the DAPI staining of the nuclei. B. The HPV16 E7ΔDLYC variant deficient in binding pRB is mostly nuclear in vivo. HeLa cells were transfected with EGFP-E7ΔDLYC plasmid and examined by fluorescence microscopy at 24 hours post transfection. Panel A represents the fluorescence of the EGFP and panel B the DAPI staining of the nuclei.
We also made an E7-EGFP construct where the EGFP was placed at the C terminus of E7. Upon transfection of HeLa cells with this plasmid the resultant E7-EGFP fusion protein was localized in the cytoplasm, suggesting that when the EGFP is located at the C terminus of E7 it blocks the NLS and nuclear localization of E7 (data not shown).
We have previously shown that the E7ΔDLYC variant defective in binding pRB is imported into the nucleus in digitonin-permeabilized cells as the wild type E7 (Angeline, Merle, and Moroianu, 2003). Here, we investigated the localization of the E7ΔDLYC variant in vivo and again found that it has a similar nuclear localization much as the E7 wild type (Fig. 1B, panel A) confirming our previous in vitro data. These results suggest that binding of HPV16 E7 to pRB is not required for the nuclear import and localization of E7 oncoprotein.
CKII phosphorylation of E7 is not required for nuclear localization of HPV16 E7
HPV16 E7 can be phosphorylated at two serines in positions 31/32 by casein kinase II (CKII) (Barbosa et al., 1990). The stretch of five acidic residues on the carboxy-terminal side of the serine in HPV16 E7 (33EEEDE) is required for CKII recognition, and the mutant E7ΔEDE cannot be phosphorylated by CKII (Munger et al., 1989b). We therefore analyzed in vivo the localization of an EGFP-E7ΔEDE mutant in comparison with EGFP-E7 to see if the E7 mutant that cannot be phosphorylated has (or has not) a similar nuclear localization as the E7 wild type. Transient transfection experiments in HeLa cells revealed that the EGFP-E7ΔEDE variant is mostly nuclear, similar to the E7 wild type (Fig. 2, panels A and C), suggesting that CKII phosphorylation of HPV16 E7 is not required for E7 nuclear import and localization in vivo.
Fig. 2. An HPV16 E7 variant deficient in CKII phosphorylation is nuclear like the wild type E7.

HeLa cells were transfected with either EGFP-E7ΔEDE (panels A and B) or EGFP-E7 (panels C and D) plasmids and examined by fluorescence microscopy at 24 hours post transfection. Panels A and C represent the fluorescence of the EGFP and panels B and D the DAPI staining of the nuclei.
The N-terminal domain of HPV16 E7 contains a novel NLS
To map the NLS of HPV16 E7 we made EGFP fusion plasmids with the domains of E7 and performed transient transfection assays in HeLa cells. Fluorescence microscopy analysis revealed that the EGFP-E71-37 is localized mostly to the nucleus like the E7 wild type (Fig. 3, panels B and F), whereas the EGFP-E71-15 is throughout the cell like the EGFP itself (Fig. 3, panels D and H). The EGFP-E738-98 is mostly cytoplasmic, although there is also some nuclear localization (Fig. 4, panel B). These data suggest that the N terminal domain (aa 1-37, containing both CR1 and CR2) of HPV16 E7 may contain an NLS. However, as the EGFP-E71-37 is below the limit of passive diffusion through the nuclear pore complex (around 40 KDa), its nuclear accumulation could be due either to the presence of an NLS or to passive diffusion coupled with nuclear retention.
Fig. 3. EGFP-E71-37 localizes mostly to the nucleus in transfected cells.

HeLa cells were transfected with either EGFP-E7 (panels A and B), or EGFP-E71-15 (panels C and D), or EGFP-E71-37 (panels E and F), or EGPF (panels G and H) plasmids and examined by fluorescence microscopy at 24 hours post transfection. Panels A, C, E and G represent the DAPI staining of the nuclei and panels B, D, F and H the fluorescence of the EGFP.
Fig. 4. EGFP-E738-98 is mostly cytoplasmic in transfected cells.

HeLa cells were transfected with either EGFP-E738-98 (panels A and B), or EGFP-E7 (panels C and D), and examined by fluorescence microscopy at 24 hours post transfection. Panels A and C represent the DAPI staining of the nuclei and panels B and D, the fluorescence of the EGFP.
Therefore to finely map the NLS of HPV16 E7 we generated a plasmid containing a tandem copy of the EGFP fused to E7, because the 2xEGFP dimer is above the limit of passive diffusion through the nuclear pore complex. After transfection of HeLa cells the 2xEGFP-E7 fusion protein was localized mostly into the nucleus in the majority of transfected cells (Fig. 5A and 5B). We then made 2xEGFP fusions with the domains of E7 and carried out transfection assays with the 2xEGFP-E7, 2xEGFP-E71-15, 2xEGFP-E71-37, 2xEGFP-E738-98, and 2xEGFP plasmids in HeLa cells. Analysis of the localization of the translated fusion proteins showed mostly nuclear localization for the 2xEGFP-E7 and the 2xEGFP-E71-37 (Fig. 5A, panels A and E). This is similar with the nuclear localization of the EGFP-E7 and EGFP-E71-37 (Fig. 3, panels B and F). In contrast, both the 2xEGFP-E71-15 and 2xEGFP-E738-98 were cytoplasmic (Fig. 5A, panels C and G) as was the 2xEGFP itself (data not shown). The difference in the localization between EGFP-E71-15 (throughout the cell, Fig. 3, panel D) and 2xEGFP-E71-15 (cytoplasmic, Fig. 5A, panel G) is due to the fact that the EGFP-E71-15 can enter the nucleus via passive diffusion whereas the 2xEGFP-E71-15 cannot. Immunoblot analysis with an anti-GFP antibody of the translated 2xEGFP-E7 fusion proteins indicated that they were intact (data not shown), excluding the possibility that the cytoplasmic localization of 2xEGFP-E71-15 and 2xEGFP-E738-98 is due to protein degradation. To make sure that we are not missing additional amino acids that may be part of the nNLS we also made a 2xEGFP construct containing the E7 sequence aa 1-49. We then transfected HeLa cells with either 2xEGFP-E7, or 2xEGFP-E71-37, or 2xEGFP-E71-49 plasmids and analyzed the intracellular localization via fluorescence microscopy. Quantitation analysis of the number of cells with either nuclear, cytoplasmic, or throughout the cell fluorescent staining in three experiments revealed that there is no increase in the percent of cells with mostly nuclear fluorescence between 2xEGFP-E71-37 (70%) and 2xEGFP-E71-49 (46.67 +/- 1.9%) (Fig. 5B); the decrease noticed for the nuclear localization of 2xEGFP-E71-49 could be due to some potential folding problems of the E71-49 fragment. Overall these data indicate that the N-terminal domain of E7 (aa 1-37) contains an NLS (called nNLS) which can mediate the nuclear localization of the EGFP and 2xEGFP reporter proteins in vivo.
Fig. 5.

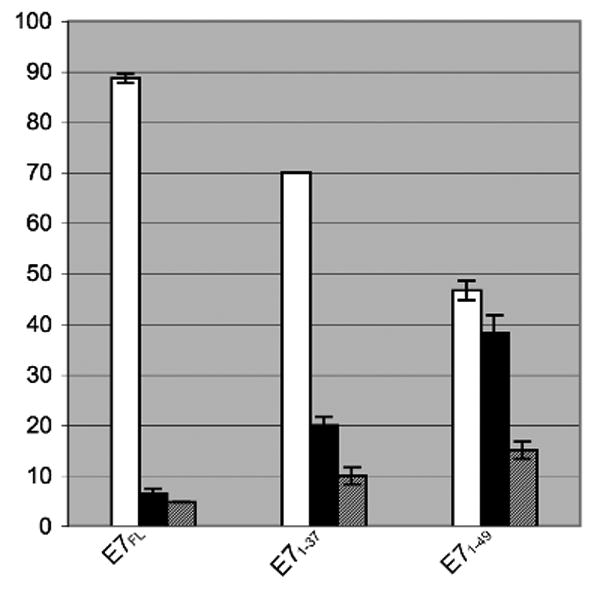
Fig. 5A. Both the 2xEGFP-E7 and 2xEGFP-E71-37 are localized mostly in the nucleus in vivo. HeLa cells were transfected with either 2xEGFP-E7 (panels A and B), or 2xEGFP-E71-15 (panels C and D), or 2xEGFP-E71-37 (panels E and F), or 2xEGPF-E738-98 (panels G and H) plasmids and examined by fluorescence microscopy at 24 hours post transfection. Panels A, C, E and G represent the fluorescence of the EGFP and panels B, D, F and H the DAPI staining of the nuclei.
Fig. 5B. Quantitative analysis of the intracellular localization of 2xEGFP fusions with E7FL, E71-37, and E71-49. The data from three experiments have been used for the graphic representation. White bars, mostly nuclear; black bars, mostly cytoplasmic; gray bars, throughout the cell.
The C-terminal domain of HPV16 E7 contains both a leucine-rich NES and a second NLS
Cytoplasmic localization of a protein which has a size above the limit of passive diffusion through the nuclear pore complex can be the result of either the absence of an active NLS, or the presence of both a leucine-rich NES and a weaker NLS. Leucine-rich NESs have been identified in both cellular and viral proteins and they interact with CRM1 nuclear export receptor. This nuclear export pathway can be inhibited by two specific export inhibitors, Leptomycin B (LMB) and ratjadone (RJA) which bind and inactivate CRM1 (Weis, 2003). We therefore examined the localization of the 2xEGFP-E738-98 in the absence of any drug, or in the presence of either LMB or RJA, as previously described (Strunze et al., 2005). Interestingly, both LMB and RJA treatment consistently changed the localization of the 2xEGFP-E738-98 protein from cytoplasmic to mostly nuclear (Fig. 6, compare panels C and E with panel A). Quantitation analysis of the percent of cells with nuclear versus cytoplasmic EGFP fluorescence showed a change from 84.1 +/- 4.8% cytoplasmic for 2xEGFP-E738-98 in the absence of a drug to 84.1 +/- 1.6% nuclear in the presence of LMB and 81.8 +/- 6.4% nuclear in the presence of RJA. Conversely, the LMB and RJA treatment had no significant effect on the localization of 2xEGFP-E71-15, 2xEGFP-E71-37, or 2xEGFP-E71-49 (data not shown). These data suggest the presence of both a putative leucine-rich NES and a weaker NLS in the C domain of E7 (aa 38-98). When the putative NES is inhibited by either LMB or RJA treatment, the NLS in the C domain is able to mediate the nuclear localization of the 2xEGFP-E738-98. As the 2xEGFP-E7 is localized mostly into the nucleus (Fig. 5, panel A) this suggests that the nNLS and cNLS together override the putative NES in the C domain in the context of the full length E7 protein in HeLa cells. If a second NES is added to the C-terminus of EGFP-E7 (the strong NES of HIV Rev), the resultant EGFP-16E7-NESRev fusion protein becomes cytoplasmic suggesting that the two NESs together can overpower the two NLSs of E7 (data not shown). Moreover, in the presence of the LMB inhibitor the EGFP-16E7-NESRev fusion protein is re-localized mostly to the nucleus (data not shown) suggesting that the cytoplasmic localization of EGFP-16E7-NESRev is due to CRM1-mediated nuclear export and not because of misfolding or masking the NLS of E7.
Fig. 6. LMB and RJA, two inhibitors of leucine rich NES-mediated nuclear export, change the localization of the 2xEGFP-E738-98 protein from cytoplasmic to mostly nuclear.

HeLa cells were transfected with 2xEGPF-E738-98 plasmid in the absence of any drug (panels A and B), or in the presence of either LMB (panels C and D), or RJA (panels E and F) and examined by fluorescence microscopy at 24 hours post transfection. Panels A, C and E represent the fluorescence of the EGFP and panels B, D and F the DAPI staining of the nuclei.
Examination of the E738-98 sequence shows the presence of two overlapping putative NES sequences (76IRTLEDLLM84 and 79LEDLLMGTLGI89) that matched the consensus of an NES (L-X2-3-L-X2-3-L-X-L; where L can be either L, I, V, F or M) (la Cour et al., 2004). We introduced the following separate substitutions in the context of 2xEGFP-E738-98: I76 to A, L79 to A, 82LLM84 to AAA, or 87LGI89 to AAA. After transient transfection in HeLa cells we analyzed the intracellular localization of the resultant 2xEGFP-E738-98 NES mutants in comparison with the wild type. As shown in Fig. 7A the 2xEGFP-E738-98l/a and 2xEGFP-E738-98i/a mutants are mostly nuclear (panels C and E) in contrast with the 2xEGFP-E738-98 wild type which is mostly cytoplasmic (panel A). The 2xEGFP-E7llm/aaa is also mostly nuclear (Fig. 7B, panel C) whereas for the 2xEGFP-E7lgi/aaa, the change in localization was less dramatic (Fig. 7B, panel E). Quantitation analysis of four experiments revealed that the most significant changes in the localization from cytoplasmic to nuclear are caused by the I76 to A, L79 to A, and 82LLM84 to AAA substitutions (Fig. 8). The 87LGI89 to AAA substitution led to a less significant change in localization (Fig. 8). These data suggest that the sequence 76IRTLEDLLM84 is the main functional NES for HPV16 E7 oncoprotein. However, the LGI amino acids downstream from the NES allow for better nuclear export mediated by this NES: when they are changed to alanines there is a decrease in the percent of cells with mostly cytoplasmic localization for 2xEGFP-E7lgi/aaa (Fig. 8).
Fig. 7. Mutations of critical amino acids in a putative NES of E738-98 change its localization from cytoplasmic to mostly nuclear.


A. HeLa cells were transfected with either 2xEGPF-E738-98wt (A and B), or 2xEGPF-E738-98l/a (C and D), or 2xEGFP-E738-98i/a (E and F) and examined by fluorescence microscopy at 24 hours post transfection. Panels A, C and E represent the fluorescence of the EGFP and panels B, D and F the DAPI staining of the nuclei. B. HeLa cells were transfected with either 2xEGPF-E738-98wt (A and B), or 2xEGPF-E738-98llm/aaa (C and D), or 2xEGFP- E738-98lgi/aaa (E and F) and examined by fluorescence microscopy at 24 hours post transfection. Panels A, C and E represent the fluorescence of the EGFP and panels B, D and F the DAPI staining of the nuclei.
Fig. 8. Quantitative analysis of the intracellular localization of 2xEGFP-E738-98 and corresponding NES mutants.

The data from four experiments have been used for the graphic representation. White bars, mostly nuclear; black bars, mostly cytoplasmic; gray bars, throughout the cell.
The data from the LMB and RJA treatments or from mutagenesis of critical amino acids in the NES suggest that there is also an NLS in the C-terminal domain able to mediate nuclear import when the export is blocked. To further analyze if the C domain of HPV16 E7 contains an active NLS we tested a GST fusion protein with the C terminal domain of HPV16 E7 in in vitro nuclear import assays. We used the GST-16cE744-98 which contains the zinc finger domain for these experiments. Digitonin-permeabilized HeLa cells were incubated with either GST-E7, or GST-16cE744-98, or GST-NLS16L1 (as a positive control), or GST (as a negative control) in the presence of HeLa cytosol. Both the full length E7 and the cE744-98 were able to mediate the nuclear import of the GST reporter, as was the positive control, the NLS16L1(Fig. 9). The negative control, GST itself, remained out of the nucleus (Fig. 9). These data indicate that the C terminal domain of E7 (aa 44-98) contains an active NLS. Previously we have shown that GST-16E7 enters the nuclei of digitonin-permeabilized cells in the presence of exogenous Ran and independent of karyopherins/importins (Angeline, Merle, and Moroianu, 2003). When we tested GST-16cE744-98 in import assays we found that it can also enter the nuclei of digitonin-permeabilized cells in the presence of only exogenous RanGDP (Fig. 10, panel B). As expected the GST-M9 (M9 is the NLS of hnRNP A1 and interacts with Kap β2 import receptor) entered the nuclei in the presence of RanGDP plus Kap β2 import receptor (Fig. 10, panel E). The negative control GST did not enter the nuclei in the presence of either RanGDP, or HeLa cytosol (Fig. 10, panels H and I).
Fig. 9. Nuclear import mediated by the 16E7 C-terminal domain.

Digitonin-permeabilized HeLa cells were incubated with either GST-E7 (panel A), GST-16cE750-98 (panel B), GST-NLS16L1 (panel C), or GST (panel D) in the presence of HeLa cytosol and energy mix. Note the nuclear import in panels A, B and C.
Fig. 10. Ran-mediated nuclear import of GST-16cE7.

Digitonin-permeabilized HeLa cells were incubated with either GST-16cE750-98 (panels A to C), GST-M9 (panels D to F), or GST (panels G to I) in the presence of transport buffer (panels A, D and G), RanGDP (panels B and H), RanGDP + Kap β2 (panel E), or HeLa cytosol (panels C, F and I). Note the nuclear import in panels B, C, E and F.
Discussion
In this study we investigated the requirements for the nuclear localization of HPV16 E7 oncoprotein in vivo and identified its nuclear transport signals. For this study we used EGFP and 2xEGFP fusions with E7 wild type, E7 mutants or E7 domains in transfection assays in HeLa cells and examined their intracellular localization. We used EGFP and 2xEGFP tags instead of a small tag like HA for two reasons: 1) to increase the molecular weight of the fusion proteins (containing E7 fragments) so that we could distinguish between active nuclear import or export mediated by an NLS or NES versus passive diffusion coupled with nuclear or cytoplasmic retention, and 2) to avoid the additional immunostaining step. Both the EGFP-E7 and 2xEGFP-E7 were mostly nuclear indicating that the EGFP tag at the N terminus of E7 does not interfere with the nuclear localization of 16E7. This is in agreement with previous reports showing that HA-E7 (the HA tag is at the N terminus of E7) is predominantly nuclear when expressed in both HaCaT and U2OS cells (Guccione et al., 2002). Interestingly, when the EGFP tag was placed at the C terminus of E7, the resultant E7-EGFP protein was mostly cytoplasmic, suggesting that it interferes with E7 nuclear localization (data not shown).
Analysis of the in vivo localization of HPV16 E7 deletion mutants defective in either binding pRB or CKII phosphorylation revealed that they are localized mostly to the nucleus as it is the case for the wild type E7. These results indicate that neither binding of pRB nor CKII phosphorylation of E7 is required for the nuclear import and localization of HPV16 E7 oncoprotein.
Previously we discovered that HPV16 E7 oncoprotein enters the nucleus via a novel Ran-dependent pathway which is independent of the nuclear import receptors belonging to the karyopherin β/importin β family (Angeline, Merle, and Moroianu, 2003). This is in agreement with the fact that HPV16 E7 has no classical basic NLS or other characterized type of NLS motif known to interact with karyopherins/importins. To identify the NLS(s) of E7 we performed in vivo localization experiments of EGFP and 2xEGFP fusions with E7 domains. The results suggest that HPV16 E7 oncoprotein contains two NLSs: one nNLS in the N-terminal domain (aa 1-37, representing both CR1 and CR2 domains) and a second cNLS in the CR3 domain. Both NLSs were able to mediate independently the nuclear localization of the 2xEGFP reporter in HeLa cells. The cNLS also mediated the nuclear import of a GST reporter in nuclear import assays in digitonin-permeabilized cells in the presence of HeLa cytosol. The nuclear accumulation of GST-cE744-98 in import assays (suggesting lack of nuclear export) could be due to the depletion of CRM1 nuclear export receptor in the digitonin-permeabilized cells. Importantly, the cNLS also mediated nuclear import in the digitonin-permeabilized cells in a Ran-dependent manner and in the absence of Kap β import receptors as previously shown for the full length 16E7 (Angeline, Merle, and Moroianu, 2003). Surprisingly, GST-E71-37 was not imported into the nucleus of digitonin-permeabilized cells in the presence of either HeLa cytosol or RanGDP (data not shown). This lack of nuclear import could be due to either folding problems (causing aggregation) or masking the nNLS by the GST reporter in this fusion protein, or requirement for an exogenous factor/protein which has been depleted in the digitonin-permeabilized cells.
Interestingly, our data with LMB and RJA nuclear export inhibitors suggest that the CR3 domain of HPV16 E7 contains both a cNLS and a leucine-rich NES. Examination of the CR3 domain sequence shows the presence of two overlapping putative NES sequences (76IRTLEDLLM84 and 79LEDLLMGTLGI89) that matched the consensus of an NES (LX2-3LX2-3LXL; L can be either L, I, V, F or M). Moreover, these putative NES sequences of E7 contain glutamate and aspartate residues which have been shown to be prevalent in characterized NESs (la Cour et al., 2004). In addition, the structure of the CR3 domain of E7 (Liu et al., 2006) reveals that the N terminal part of these putative NESs is α-helical which is also predominant for the characterized NESs based on structural data available (la Cour et al., 2004).
Functional mutagenesis of critical amino acids in these putative NESs revealed that it is the 76IRTLEDLLM84 sequence that is the main functional NES involved in nuclear export of HPV16 E7. However, the 87LGI89 amino acids downstream the E7's NES allow for better nuclear export activity of this NES. Previous reports indicated that the 16E7Δ79-83 deletion mutant was deficient in inducing S phase in differentiated keratinocytes (Helt, Funk and Galloway, 2002), had reduced transforming activity and no longer interacted with TATA box binding protein (Massimi, Pim and Banks, 1997). The 79-83 deletion disrupts the identified NES (76IRTLEDLLM84) and the resultant sequence now becomes IRTMGTLGI which also matches the consensus of an NES (LX2-3LX2-3LXL; L can be either L, I, V, F or M). If this putative NES artificially created by deletion is not functional then the 16E7Δ79-83 mutant will be deficient in nuclear export activity which may contribute to its reduced activities.
Although HPV16 E7 contains only 98 amino acids and as a monomer could enter and exit the nucleus via passive diffusion it has been shown that E7 forms dimers (and perhaps oligomeric complexes) in vivo and that the C terminal zinc binding domain is critical for this dimerization activity (Clemens et al., 1995; Liu et al., 2006). The presence of both NLSs and a leucine-rich NES in 16E7 suggests that E7 actively shuttles between the nucleus and the cytoplasm. This is not surprising as HPV16 E7 is known to interact with both nuclear and cytoplasmic proteins and has been found to be localized both in the nucleus and in the cytoplasm. The different interactions of 16E7 with its nuclear and cytoplasmic targets could regulate 16E7 nuclear import and export (via cytoplasmic or nuclear retention) and consequently modulate the steady-state localization of E7 in different cells and physiological conditions. The oligomerization state of E7 could also influence the steady state localization of E7. In this study in HeLa cells, although both the EGFP-E7 and 2xEGFP-E7 are mostly nuclear, there is also some cytoplasmic localization (Figs. 1, 2, 3, 5A and 5B). Moreover, this cytoplasmic localization of 2xEGFP-E7 remains even in the presence of LMB and RJA export inhibitors, or NES mutations (data not shown), suggesting that this small fraction of E7 is retained in the cytoplasm via an interaction with a target protein(s). Interestingly, in invasive cervical carcinoma in situ, 16E7 was also detected predominantly into the nucleus of the tumor cells and to some extent in the cytoplasm (Fiedler et al., 2004) reflecting both nuclear and cytoplasmic functions. Moreover, in transfected U-2OS cells when the protein synthesis was blocked, some 10% of E7 remained cytoplasmic suggesting that this fraction is retained in the cytoplasm via interaction with an E7 target protein (Fiedler et al., 2004). In our system we did not notice differences in the intracellular localization of the E7 proteins depending on their levels of expression. However, one could speculate that because E7 is translated in the cytoplasm and interacts with cytoplasmic proteins like p600 (Huh et al., 2005), at low levels of E7 expression it is possible that the cytoplasmic localization of E7 may be more predominant than nuclear localization. The levels of CRM1 nuclear export receptor in different cells or conditions may also influence the steady-state localization of 16E7 (and 16cE7) in those cells with high levels of CRM1 enhancing E7 nuclear export and cytoplasmic localization.
Recently, it has been shown that HPV16 E7 is able to change the localization of steroid receptor coactivator 1 (SRC-1) from the nucleus to the cytoplasm (Baldwin, Huh, and Munger, 2006). Future studies could explore if the NES and nuclear export of E7 is required for this re-localization of SRC1 from the nucleus to the cytoplasm.
Previous studies have identified and characterized the NLSs of other HPV proteins. The L1 major and L2 minor capsid proteins of both high risk and low risk HPVs have classical basic monopartite and bipartite NLSs (Bordeaux et al., 2006; Darshan et al., 2004; Klucevsek et al., 2006; Merle et al., 1999; Nelson, Rose, and Moroianu, 2002; Sun et al., 1995). Low risk HPV11 E2 has a classical monopartite NLS in the hinge region whereas high risk HPV16 E2 has a novel alpha-helix NLS in the C domain (Klucevsek et al., 2007; Zou et al., 2000). The HPV11 E1 DNA helicase has a codominant bipartite NLS (Yu et al., 2007). In this study we discovered that HPV16 E7 has two NLSs, one nNLS in the CR1 and CR2 domains and a second cNLS in the CR3 domain. The cNLS mediates nuclear import via a Ran-dependent pathway which is independent of the Kap β nuclear import receptors as we have previously shown for the full length HPV16 E7 oncoprotein (Angeline, Merle, and Moroianu, 2003). It remains to be determined if the nNLS mediates nuclear localization in a similar manner as the cNLS or differently. It will be interesting to determine the critical amino acids in these two novel NLSs functioning in nuclear import of HPV16 E7 and correlate the lack of nuclear import and localization of E7 mutants with deficiencies in E7 functions. However, it may be difficult to find specific E7 mutations deficient in nuclear import that do not interfere with one or more of the E7 interactions as E7 has so many cellular targets. Moreover, as E7 has two NLSs, if one NLS is impaired the other one can compensate for the nuclear localization of E7.
One possible mechanism for E7 nuclear entry is via direct low affinity hydrophobic interactions between 16E7 and the FG (Phe-Gly) repeats in nucleoporins, in a similar manner as the nuclear import receptors (Weis, 2003). Preliminary experiments in support of this model showed that HPV16 E7 can interact with a Phenyl-Sepharose matrix (Knapp and Moroianu, observations unpublished) which mimics the selectivity of the hydrophobic interactions of the FG nucleoporins and binds all the nuclear import receptors (Ribbeck and Gorlich, 2002).
The cNLS of HPV16 E7 is in the CR3 domain that contains the zinc-finger domain (aa 58-94) consisting of two copies of a Cys-X-X-Cys sequence motif separated by 29 amino acids. Interestingly, it has been reported recently that the Tax protein of human T Lymphotropic Virus Type 1 enters the nucleus via direct interactions with nucleoporins containing phenylalanine-glycine (FG) repeats, and independent of the nuclear import receptors. Functional studies revealed that the zinc-finger domain of Tax functions as an NLS mediating the nuclear import of Tax via direct interactions with FG nucleoporins (Tsuji et al., 2007). In the future it will be interesting to analyze the interactions of the cNLS of E7 with the FG-containing nucleoporins at the nuclear pore complex.
Materials and Methods
Generation of EGFP fusion plasmids with E7 wild type, E7 mutants and different E7 domains
The enhanced green fluorescent protein (EGFP) expression plasmid pEGFP-C1 (Clontech, Inc.) was used to construct the EGFP fusion protein expression plasmids (EGFP-E7, EGFP-E71-15, EGFP-E71-37, EGFP-E738-98) as follows. The pEGFP-C1 plasmid was double digested with EcoR1 and BamH1, run on a 0.7% agarose gel and the digested vector was extracted using the protocol from the QIAquick Gel Extraction Kit (Qiagen). DNA fragments spanning the full length HPV16 E7 or the C domain (aa 38-98) were amplified using PCR oligonucleotides that added EcoR1 and BamH1 restriction endonuclease sites and the PCR products were digested with EcoR1 and BamH1 restriction enzymes and then ligated using T4 DNA ligase into the EcoR1 and BamH1 cloning sites of pEGFP-C1. The resulting plasmids were used to transform XL1-Blue cells and the purified plasmids were confirmed by sequencing (MGH DNA Sequencing Department). In all these EGFP fusion proteins the EGFP is at the N-terminus of E7 or E7 fragments. It should be noted that we also made an E7-EGFP construct where the EGFP was at the C terminus of E7.
The 2xEGFP-E7 and 2xEGFP-E738-98 were generated by first using PCR on the EGFP-E7 and EGFP-E738-98 with PCR oligonucleotides that added SacII and XmaI restriction endonuclease sites, then digesting the PCR products with SacII and XmaI restriction enzymes, and finally ligating them into the SacII and XmaI cloning sites of pEGFP-C1 using the T4 DNA ligase.
The EGFP-E71-15, EGFP-E71-37 and 2xEGFP-E71-15, 2xEGFP-E71-37 were generated using the QuikChangeTM Site-Directed Mutagenesis Kit from Stratagene with EGFP-E7 or 2XEGFP-E7 as a template together with mutated primers containing a stop codon at aa 16 or 38. The EGFP-E7ΔDLYC and EGFP-ΔEDE were generated using the QuikChange™ Site-Directed Mutagenesis Kit with EGFP-E7 as a template together with mutated primers containing the corresponding deletion. All the constructs were used to transform XL1 blue bacteria and the purified plasmids were confirmed by sequence analysis (MGH DNA Sequencing Department).
Transient expressions and fluorescence microscopy
HeLa cells (ATCC) were plated on 12 mm poly-L-lysine-coated glass coverslips to 50-70 % confluency 24 hours prior to transfection. Cells in each well were transfected with the corresponding plasmid (as indicated in Fig. legends) and FuGENE 6 reagent (Roche Applied Science, IN) in 500 μl DMEM prepared following product protocol. Media was changed to DMEM with 10% FBS and pen-strep after 5 hours and the cells were fixed 24 hours after the initial transfection with 10% formaldehyde in PBS (10 min).
For the experiments with Leptomycin B (LMB), the LMB reagent (Sigma) was added to the transfected cells at a final concentration of 0.01 ng/mL in DMEM and incubated for 4 hours before fixation and examination by fluorescence microscopy. For the experiments with Ratjadone A (RJA), the RJA reagent was added to the transfected cells at a final concentration of 10 ng/mL in DMEM and incubated for 4 hours before fixation and examination by fluorescence microscopy.
Coverslips were mounted using Vectashield-Dapi mounting medium (Vector Labs, CA) to identify the nuclei by DAPI staining and examined by fluorescence microscopy using a Nikon Eclipse TE 300 Microscope. The photos were taken at the same exposure time with a Sony DKC-5000 camera or a SPOT RT3 camera equipped with the Advanced SPOT computer software.
Preparation of GST fusion proteins with E7 wild type and E7 domains
The GST-16E7, GST-16E744-98 and GST-16E71-52 plasmids were kindly provided by Dr. Karl Munger (Harvard Medical School, Boston) and Dr. David Pim (ICGEB, Trieste, Italy). The GST-16E71-37 construct was generated using the QuikChange™ Site-Directed Mutagenesis Kit with the GST-E7 plasmid as a template together with mutated primers containing a stop codon at aa 38. The GST-E71-37 plasmid was transformed in XL1 blue bacteria and confirmed by automated sequence analysis (MGH DNA sequencing department). For protein expression, the different GST-E7 plasmids were used to transform E.coli BL21 CodonPlus. After induction of the bacteria with 1 mM IPTG for 2 hours at 30°, the GST-E7, GST-E71-37, and GST-E744-98 fusion proteins were purified in their native state on glutathione-Sepharose beads using a standard procedure. The GST-NLSHPV16L1 containing the monopartite NLS (AKRKKRKL) of HPV16 L1 major capsid protein was prepared as previously described (Nelson, Rose, and Moroianu, 2003). The proteins were checked for purity and lack of proteolytic degradation by SDS-PAGE and Coomassie Blue staining. All the purified proteins were dialyzed in transport buffer (20 mM HEPES-KOH, pH 7.3, 110 mM potassium acetate, 2 mM magnesium acetate, 1 mM EGTA, 2 mM DTT, plus protease inhibitors (Moroianu, Blobel, and Radu, 1995), and stored in aliquots at −80°C until use.
Nuclear import assays
The nuclear import assays were carried out as previously described (Nelson, Rose, and Moroianu, 2002). Briefly, subconfluent HeLa cells, grown on poly-L-lysine coated glass coverslips for 24 h, were permeabilized with 70 μg/μl digitonin for 5 min on ice, and washed with cold transport buffer. All import reactions contained an energy regenerating system (1 mM GTP, 1mM ATP, 5 mM phosphocreatine, and 0.4 U creatine phosphokinase), HeLa cytosol (5 μl) and the different GST-fusion proteins (as indicated in the Fig. legends). In the experiments with recombinant transport factors, 2μg RanGDP and 1μg Kap β2 were used per assay. The final import reaction volume was adjusted to 20 μl with transport buffer. For visualization of nuclear import the GST fusion proteins were detected by immunofluorescence with an anti-GST antibody, as previously described (Nelson, Rose, and Moroianu, 2002). Coverslips were mounted using Vectashield-DAPI mounting medium (Vector Labs, CA) to identify the nuclei by DAPI staining. Nuclear import was analyzed with a Nikon Eclipse TE 300 Microscope that has a fluorescence attachment and photos were taken with a Sony DKC-5000 camera or a SPOT RT3 camera with the Advanced SPOT computer software.
Acknowledgments
We thank Dr. Karl Munger and Dr. David Pim for their generous gifts of GST-16E7, GST-16E71-52 and GST-16E744-98 expression plasmids. We thank Zackary Piccioli for technical assistance in preparation of GST-16E7 proteins and for nuclear import assays with GST-16E71-37 and GST-16E71-52; and Jeremy Eberhard for the mutagenesis generating the GST-16E71-37 construct and transfection assays with 2XEGFP-E7 and NES mutants. This work was supported by a grant from the National Institutes of Health (R01 CA94898) to JM.
Abbreviations
- HPV
human papillomavirus
- NLS
nuclear localization signal
- NES
nuclear export signal
- EGFP
enhanced green fluorescent protein
- GST
glutathione-S-transferase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Angeline M, Merle E, Moroianu J. The E7 oncoprotein of high-risk human papillomavirus type 16 enters the nucleus via a nonclassical Ran-dependent pathway. Virology. 2003;317(1):13–23. doi: 10.1016/j.virol.2003.08.024. [DOI] [PubMed] [Google Scholar]
- Baldwin A, Huh KW, Munger K. Human papillomavirus E7 oncoprotein dysregulates steroid receptor coactivator 1 localization and function. J Virol. 2006;80(13):6669–77. doi: 10.1128/JVI.02497-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa MS, Edmonds C, Fisher C, Schiller JT, Lowy DR, Vousden KH. The region of the HPV E7 oncoprotein homologous to adenovirus E1a and Sv40 large T antigen contains separate domains for Rb binding and casein kinase II phosphorylation. Embo J. 1990;9(1):153–60. doi: 10.1002/j.1460-2075.1990.tb08091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordeaux J, Forte S, Harding E, Darshan MS, Klucevsek K, Moroianu J. The L2 minor capsid protein of low-risk human papillomavirus type 11 interacts with host nuclear import receptors and viral DNA. J Virol. 2006;80(16):8259–62. doi: 10.1128/JVI.00776-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens KE, Brent R, Gyuris J, Munger K. Dimerization of the Human Papillomavirus E7 Oncoprotein in Vivo. Virology. 1995;214:289–293. doi: 10.1006/viro.1995.9926. [DOI] [PubMed] [Google Scholar]
- Darshan MS, Lucchi J, Harding E, Moroianu J. The l2 minor capsid protein of human papillomavirus type 16 interacts with a network of nuclear import receptors. J Virol. 2004;78(22):12179–88. doi: 10.1128/JVI.78.22.12179-12188.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duensing S, Munger K. Mechanisms of genomic instability in human cancer: insights from studies with human papillomavirus oncoproteins. Int J Cancer. 2004;109(2):157–62. doi: 10.1002/ijc.11691. [DOI] [PubMed] [Google Scholar]
- Dyson N, Guida P, Munger K, Harlow E. Homologous sequences in adenovirus E1A and human papillomavirus E7 proteins mediate interaction with the same set of cellular proteins. J Virol. 1992;66(12):6893–902. doi: 10.1128/jvi.66.12.6893-6902.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson N, Howley PM, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243(4893):934–7. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- Fiedler M, Muller-Holzner E, Viertler HP, Widschwendter A, Laich A, Pfister G, Spoden GA, Jansen-Durr P, Zwerschke W. High level HPV-16 E7 oncoprotein expression correlates with reduced pRb-levels in cervical biopsies. Faseb J. 2004;18(10):1120–2. doi: 10.1096/fj.03-1332fje. [DOI] [PubMed] [Google Scholar]
- Fujikawa K, Furuse M, Uwabe K, Maki H, Yoshie O. Nuclear localization and transforming activity of human papillomavirus type 16 E7-beta-galactosidase fusion protein: characterization of the nuclear localization sequence. Virology. 1994;204(2):789–93. doi: 10.1006/viro.1994.1594. [DOI] [PubMed] [Google Scholar]
- Guccione E, Massimi P, Bernat A, Banks L. Comparative analysis of the intracellular location of the high- and low- risk human papillomavirus oncoproteins. Virology. 2002;293(1):20–5. doi: 10.1006/viro.2001.1290. [DOI] [PubMed] [Google Scholar]
- Helt AM, Funk JO, Galloway DA. Inactivation of both the Retinoblastoma Tumor Suppressor and p21 by the Human Papilomavirus Type 16 E7 Oncoprotein Is Necessary to Inhibit Cell Cycle Arrest in Human Epithelial Cells. J Virol. 2002;76(20):10559–10568. doi: 10.1128/JVI.76.20.10559-10568.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh KW, DeMasi J, Ogawa H, Nakatani Y, Howley PM, Munger K. Association of the human papillomavirus type 16 E7 oncoprotein with the 600-kDa retinoblastoma protein-associated factor, p600. Proc Natl Acad Sci U S A. 2005;102(32):11492–7. doi: 10.1073/pnas.0505337102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DL, Munger K. Interactions of the human papillomavirus E7 protein with cell cycle regulators. Semin Cancer Biol. 1996;7(6):327–37. doi: 10.1006/scbi.1996.0042. [DOI] [PubMed] [Google Scholar]
- Klucevsek K, Wertz M, Lucchi J, Leszczynski A, Moroianu J. Characterization of the nuclear localization signal of high risk HPV16 E2 protein. Virology. 2006 doi: 10.1016/j.virol.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klucevsek K, Wertz M, Lucchi J, Leszczynski A, Moroianu J. Characterization of the nuclear localization signal of high risk HPV16 E2 protein. Virology. 2007;360(1):191–8. doi: 10.1016/j.virol.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- la Cour T, Kiemer L, Molgaard A, Gupta R, Skriver K, Brunak S. Analysis and prediction of leucine-rich nuclear export signals. Protein Eng Des Sel. 2004;17(6):527–36. doi: 10.1093/protein/gzh062. [DOI] [PubMed] [Google Scholar]
- Liu X, Clements A, Zhao K, Marmorstein R. Structure of the human Papillomavirus E7 oncoprotein and its mechanism for inactivation of the retinoblastoma tumor suppressor. J Biol Chem. 2006;281(1):578–86. doi: 10.1074/jbc.M508455200. [DOI] [PubMed] [Google Scholar]
- Massimi P, Pim D, Banks L. Human papillomavirus type 16 E7 binds to the conserved carboxy-terminal region of the TATA box binding protein and this contributes to E7 transforming activity. 1997;78:2607–2613. doi: 10.1099/0022-1317-78-10-2607. [DOI] [PubMed] [Google Scholar]
- Merle E, Rose RC, LeRoux L, Moroianu J. Nuclear import of HPV11 L1 capsid protein is mediated by karyopherin alpha2beta1 heterodimers. J Cell Biochem. 1999;74(4):628–37. [PubMed] [Google Scholar]
- Moroianu J, Blobel G, Radu A. Previously identified protein of uncertain function is karyopherin alpha and together with karyopherin beta docks import substrate at nuclear pore complexes. Proc Natl Acad Sci U S A. 1995;92(6):2008–11. doi: 10.1073/pnas.92.6.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger K, Basile JR, Duensing S, Eichten A, Gonzalez SL, Grace M, Zacny VL. Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene. 2001;20(54):7888–98. doi: 10.1038/sj.onc.1204860. [DOI] [PubMed] [Google Scholar]
- Munger K, Phelps WC, Bubb V, Howley PM, Schlegel R. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J Virol. 1989a;63(10):4417–21. doi: 10.1128/jvi.63.10.4417-4421.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger K, Werness BA, Dyson N, Phelps WC, Harlow E, Howley PM. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. Embo J. 1989b;8(13):4099–105. doi: 10.1002/j.1460-2075.1989.tb08594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson LM, Rose RC, Moroianu J. Nuclear import strategies of high risk HPV16 L1 major capsid protein. J Biol Chem. 2002;23:23. doi: 10.1074/jbc.M200724200. [DOI] [PubMed] [Google Scholar]
- Rapp L, Chen JJ. The papillomavirus E6 proteins. Biochim Biophys Acta. 1998;1378(1):F1–19. doi: 10.1016/s0304-419x(98)00009-2. [DOI] [PubMed] [Google Scholar]
- Ribbeck K, Gorlich D. The permeability barrier of nuclear pore complexes appears to operate via hydrophobic exclusion. Embo J. 2002;21(11):2664–71. doi: 10.1093/emboj/21.11.2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S, Liem A, Miller JA, Lambert PF. Human papillomavirus types 16 E6 and E7 contribute differently to carcinogenesis. Virology. 2000;267(2):141–50. doi: 10.1006/viro.1999.0106. [DOI] [PubMed] [Google Scholar]
- Strunze S, Trotman LC, Boucke K, Greber UF. Nuclear targeting of adenovirus type 2 requires CRM1-mediated nuclear export. Mol Biol Cell. 2005;16(6):2999–3009. doi: 10.1091/mbc.E05-02-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun XY, Frazer I, Muller M, Gissmann L, Zhou J. Sequences required for the nuclear targeting and accumulation of human papillomavirus type 6B L2 protein. Virology. 1995;213:321–7. doi: 10.1006/viro.1995.0005. [DOI] [PubMed] [Google Scholar]
- Tsuji T, Sheehy N, Gautier VW, Hayakawa H, Sawa H, Hall WW. The nuclear import of the human T lymphotropic virus type I (HTLV-1) tax protein is carrier- and energy-independent. J Biol Chem. 2007;282(18):13875–83. doi: 10.1074/jbc.M611629200. [DOI] [PubMed] [Google Scholar]
- Weis K. Regulating access to the genome: nucleocytoplasmic transport throughout the cell cycle. Cell. 2003;112(4):441–51. doi: 10.1016/s0092-8674(03)00082-5. [DOI] [PubMed] [Google Scholar]
- Zou N, Lin BY, Duan F, Lee KY, Jin G, Guan R, Yao G, Lefkowitz EJ, Broker TR, Chow LT. The hinge of the human papillomavirus type 11 E2 protein contains major determinants for nuclear localization and nuclear matrix association. J Virol. 2000;74(8):3761–70. doi: 10.1128/jvi.74.8.3761-3770.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- zur Hausen H. Papillomaviruses causing cancer: evasion from host-cell control in early events in carcinogenesis. J Natl Cancer Inst. 2000;92(9):690–8. doi: 10.1093/jnci/92.9.690. [DOI] [PubMed] [Google Scholar]
- Zwerschke W, Jansen-Durr P. Cell transformation by the E7 oncoprotein of human papillomavirus type 16: interactions with nuclear and cytoplasmic target proteins. Adv Cancer Res. 2000;78:1–29. doi: 10.1016/s0065-230x(08)61022-2. [DOI] [PubMed] [Google Scholar]
- Yu JH, Lin BY, Deng W, Broker TR, Chow LT. Mitogen-activated protein kinases activate the nuclear localization sequence of human papillomavirus type 11 E1 DNA helicase to promote efficient nuclear import. J Virol. 2007;81(10):5066–78. doi: 10.1128/JVI.02480-06. [DOI] [PMC free article] [PubMed] [Google Scholar]