

Abstract
Spinal muscular atrophy (SMA) is caused by deletion or mutation of both copies of the SMN1 gene which produces an essential protein known as SMN. The severity of SMA is modified by variable copy number of a second gene, SMN2 that produces an mRNA that is incorrectly spliced with deletion of the last exon. We described previously the discovery of potent C5-substituted quinazolines that increase SMN2 gene expression by two-fold. Discovery of potent SMN2 promoter inducers relied on a cellular assay without knowledge of the molecular target. Using protein microarray scanning with a radiolabeled C5-quinazoline probe, we identified the scavenger decapping enzyme, DcpS as a potential binder. We show that the C5-quinazolines potently inhibit DcpS decapping activity, and that the potency of inhibition correlates with potency for SMN2 promoter induction. Binding of C5-quinazolines to DcpS holds the enzyme in an open, catalytically incompetent conformation. DcpS is a nuclear shuttling protein that binds and hydrolyzes the m7GpppN mRNA cap structure and a modulator of RNA metabolism. Therefore DcpS represents a novel therapeutic target for modulating gene expression by a small molecule.
Introduction
Selective modulation of gene expression by small molecule therapeutics has proved elusive except for those classes of drugs targeting transcription factors that are modulated by endogenous ligands. The steroid receptors comprise the largest family of such ‘drugable’ targets. These change conformation on binding ligand, and translocate from the cytoplasm to the nucleus where they interact with chromatin to alter gene transcription. Depending upon the ligand bound, steroid receptors adopt alternate conformations and may act either as activators or repressors of gene transcription. For example, binding of the endogenous ligand 17β-estradiol to the estrogen receptor induces a compact, active conformation of the receptor, while tamoxifen, a clinically important drug for treating estrogen-dependent breast cancer, is a synthetic antagonist of the estrogen receptor that prevents an activation loop from folding into the compact conformation, thereby holding the receptor in an inactive, open conformation 1. The peroxisome proliferator-actived receptor-α (PPAR-α), the molecular target of the fibrates for lowering plasma cholesterol and triglycerides, and PPAR-γ (the molecular target of the thiazolidinediones for treatment of insulin resistant diabetes), are further examples of molecular targets for important classes of therapeutics 2. PPARs form heterodimers with the retinoid X-receptor (RXR), which also is required for the function of the vitamin D and thyroid hormone receptors. Other nuclear receptors play key roles in the transcriptional regulation of xenobiotic and endobiotic metabolism, although these have not been exploited as therapeutic targets. Beyond the nuclear receptors, however, few therapeutic targets for modulating gene transcription have emerged. Global alteration of chromatin function can be achieved by inhibition of DNA- or nucleosome-modifying enzymes, for example, through inhibition of histone deacetylation. Valproic acid, a histone deactylase inhibitor, is marketed for the treatment of epilepsy, but the precise mechanism of its therapeutic action is unclear.
Spinal muscular atrophy is an autosomal recessive disease caused by deletion or mutational inactivation of the SMN1 gene. The disease affects 1 in 6000 live births and is the leading cause of hereditary infant death 3,4. Unusually, there is a second, duplicate copy of the SMN1 gene in humans that is located immediately centromeric to the functional gene.5,6 The second gene, designated SMN2, is transcriptionally active, however, it splices incorrectly due to a single nucleotide mutation, such that 90% of SMN2 mRNA lack exon 7, the last coding exon in the spliced transcript. Histone deacetylase inhibitors, including valproic acid 7,8 have been shown in cells to increase transcription of the SMN2 gene and to increase levels of the essential protein product of the gene, the survival motor neuron protein (SMN). These also have therapeutic benefit in mouse genetic models of spinal muscular atrophy and may be fruitful to explore in human clinical trials 9.
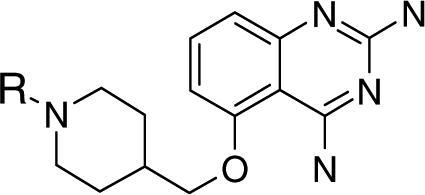
To explore additional molecular targets for transcriptional activation of the SMN2 gene, we used a cell based reporter assay to screen 550,000+ compounds for up-regulation of the SMN2 gene promoter. The reporter assay was constructed using a mouse neuroblastoma x motor neuron hybrid cell line in which was inserted a synthetic gene containing a fragment of the human SMN2 gene promoter linked functionally to a bacterial β-lactamase gene. The ultra-high throughput screen yielded two series of chemically amenable hits 10. These included C-5 substituted quinazolines which in confirmatory assays increased SMN mRNA levels in SMA patient-derived fibroblasts and also increased SMN protein levels and nuclear GEM/Cajal body numbers (a functional read-out of SMN protein levels), in patient-derived skin cells 10,11. Further optimization of the C-5 substituted quinazolines through directed medicinal chemistry has resulted in a clinical lead for SMA therapeutic trials (D156844) 11 (Figure 1). Clinical leads from the C-5 quinazoline series generally are well tolerated, distribute into brain, have good oral bioavailablity, and an adequate margin of safety for exploratory human clinical trials.
Figure 1. Structure of the C5-quinazoline D156844 and its effects on cellular SMN mRNA levels.

The structure of D156844, a C5 substituted ether-based 2,4-diaminoquinazoline is shown with its 2-fluoro-benzyl piperidine substituent. Atoms of the quinazoline scaffold are numbered in italics. (A) Fold increase of β-lactamase in NSC34 cells treated with D156844 (mean ± SD, triplicate wells), EC50 = 9.1 nM (B) Fold increase of mouse SMN mRNA in NSC-34 cells treated with D156844, 100 nM trichostatin A (TSA) or DMSO (mean ± SD, n=9) (C) Synthesis of 125I labeled C5-quinazoline. D156469, a tin derivative was synthesized from D156156 as described in the Supplemental Text. An 125I sodium iodide /chloramine T procedure was used to displace the SnBu3 of D156469, yielding the desired radiolabeled C5-quinazoline.
The clinical lead in the C5-quinazoline series was optimized in the absence of knowledge of its molecular target based solely on data from the NSC34 cell-based, human SMN2 gene reporter assay. We therefore set out to identify the molecular target of the lead series. We did this by developing a highly potent, radioactively labeled C5-quinazoline analog which could be used to probe high-density human protein microarrays for potential binders. To develop molecular probes of the C5-quinazoline therapeutic target, we explored how to introduce a radioactive 125I label into the compounds while maintaining potency in the cell-based assay. Subsequently, microarrays containing thousands of recombinant human proteins immobilized on a solid substrate were probed to identify specific interactors. The scavenger decapping enzyme DcpS was identified as an interactor and modeling studies demonstrated a plausible binding mode for the lead series based on docking to the previously determined human and mouse DcpS protein structures 12,13. In vitro biochemical studies confirmed that the C5-quinazolines are potent DcpS inhibitors and furthermore, that their rank order of potency correlates with potency in the cellular SMN2 promoter activation assay. Finally, we confirmed the binding mode of the C5-quinazolines by X-ray crystal structure determination of co-crystals of several C5-quinazolines bound to DcpS. The structures reveal that the compounds trap the DcpS dimer in a catalytically inactive conformation with one monomer “closed” by binding inhibitor while the other monomer is held in an “open” catalytically incompetent conformation. Herein we report a “forward” chemical genetics approach to the identification of DcpS as a therapeutic target for treatment of SMA. The general utility of DcpS inhibition will be worthwhile to explore in other diseases where transcriptional activation may provide therapeutic benefit.
Results
C5-quinazolines increase endogenous SMN mRNA in NSC34 cells
Optimization of the C5-quinazoline series for human SMN2 promoter inducing activity utilized an NSC34 derived cell line expressing an intronless β-lactamase gene under control of the human SMN2 promoter 10. Since NSC34 cells also contain the mouse SMN gene in its normal chromosomal context, we therefore sought to learn if the C5-quinazolines would act on the NSC34 cells to increase endogenous mouse SMN mRNA. This would be important information for planned studies in mouse models of SMA, as well as demonstrate that the compounds were acting at the level of mRNA transcription or processing, rather than stabilizing the β-lactamase reporter protein. To that end, we carried out real-time PCR studies measuring mRNA levels in cells treated with D156844, a potent C5-quinzaoline, and compared that to the induction of β-lactamase activity driven by the human SMN2 promoter (Figure 1A & B). D156844 increased SMN mRNA by two-fold, a level of induction similar to that of β-lactamase reporter protein from the SMN2 reporter gene in the cell line. A similar two-fold increase in SMN mRNA is elicited by trichostatin A, a histone deacetylase (HDAC) inhibitor, although the mechanisms of SMN mRNA induction are clearly different in this system. Representative C5-quinazolines fail to generically up-regulate expression of other promoters unlike HDAC inhibitors 10. That induction of mouse SMN mRNA from the endogenous gene occurs in this cellular context implies that C5-quinazolines such as D156844 affect processes related to SMN mRNA synthesis, processing, or degradation.
Probing Immobilized Protein Target Arrays for Potential C5-Quinazoline Binders
In order to identify candidate molecular targets of the C5-quinazolines, we sought to produce radiolabeled analogs that retained potent SMN2 promoter activity. Our intent was to use these as radioactive probes against human protein arrays 14. We optimally wanted to derive high specific activity radioactive probes from the C5-quinazoline lead series with a preference for a 125I label. The SAR for the quinazoline series suggested that the C5-substituent binds in a hydrophobic pocket which is relatively narrow in the immediate vicinity of the C5 position, but which becomes progressively larger, and is probably relatively deep distal to the C5 position 11. Therefore it was hypothesized that a pendant iodo-substitutent might be tolerated at the C5 position, and indeed, the meta-iodo derivative (D156156) was found to be the most active piperidine amide-based analog 11 with an EC50 of 36 nM (Table 1).
Table 1.
C5-substituted quinazolines described in the text. The SMN2 β-lactamase NSC-34 cell reporter gene assay was performed multiple times. Average and low-high values are given.
 | ||||
|---|---|---|---|---|
| Cmpd_ID | R group | SMN2 NSC34 Cellular Assay EC50 nM Ave. (Low-Hi) | SMN2 NSC34 Cellular Assay Max Fold Induction Ave. (Low-Hi) | In Vitro DcpS Assay IC50 nM |
| D156844 | 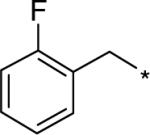 |
4.0 (1.4−6.7) | 2.29 (2.06−2.50) | 7.62 |
| D156676 | 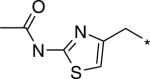 |
2540 | 1.95 | 339.45 |
| D157493 | 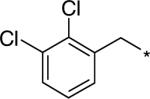 |
67 (17−109) | 2.20 (2.10−2.40) | 43.94 |
| D158963 | 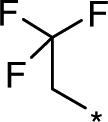 |
223 (81−364) | 1.7 (1.67−1.74) | 23.35 |
| D158885 | 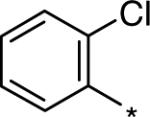 |
795 (791−800) | 1.78 (1.66−1.91) | 101.34 |
| D157555 | 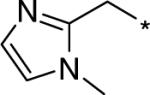 |
108 | 1.5 | 9.38 |
| D156095 | 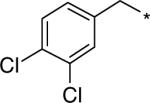 |
104 (51−142) | 1.9 (1.7−2.0) | n.d. |
| D157554 |  |
921 (370−2239) | 2.19 (1.90−2.40) | n.d. |
| D153215 |  |
243 (160−390) | 2.00 (1.89−2.14) | nd |
Having identified an appropriate iodinated C5-quinazoline for binding studies (D156156), we then prepared its tin derivative (D156469) which supports radioiodination using the iododestannylation reaction (Figure 1C). This reaction with 100 mole percent incorporation of iodine under carrier-free conditions would provide a theoretical specific activity of 2200 Ci/mmol. We selected additional quinazolines with potent SMN2 promoter activity to use as unlabeled competitors of 125I-D156156 in protein microarray binding experiments.
High density human protein microarrays were used as the test bed for probing with 125I-D156156 tracer. Represented on the protein microarrays are soluble proteins of potential therapeutic interest including kinases, phosphatases, nuclear receptors, and enzymes of intermediary metabolism. Proteins are purified under non-denaturing conditions and printed as adjacent duplicate spots on chemically modified glass slides. Additionally, the arrays contain control elements suitable for positional mapping of the data acquisition grid as well as positive and negative assay controls. The Human ProtoArray® Protein Microarray v3.0 containing more than 5,000 human proteins was probed with 100 nM of the 125I-D156156 tracer in the presence of 125I-streptavidin to facilitate accurate positional mapping (Figure 2).
Figure 2. Protein microarray identification of DcpS as a target for C5-substituted quinazolines.

(A) Array images identifying DcpS as a potential D156156 target protein. Arrays comprised of 5,000 immobilized human proteins were probed with 125I-D156156 in the presence and absence of unlabeled competitors. Specific displacement of 125I-labeled D156156 (at 100 nM concentration) by bioactive, but distinct C5-quinazolines (unlabeled at 10 μM concentration) was observed. Duplicate features boxed in white correspond to the positional mapping reagent 125I-streptavidin binding to a biotinylated control protein. Duplicate features boxed in red correspond to DcpS. (B) Quantified competition binding data with 10 μM competitor normalized against signals arising from 125I-streptavidin binding to the biotinylated control protein. Standard deviations across the replicate assays are indicated.
Evaluation of the resultant data set identified a single potential interacting protein that was consistently above background (Figure 2A), corresponding to the human scavenger decapping enzyme DcpS, a member of the histidine triad (HIT) superfamily of hydrolases 15. DcpS functions in the 3′−5′ exonucleolytic pathway for mRNA decay to hydrolyze the residual cap structure, m7GpppN, to m7GMP + pN 15,16 and in the 5′−3′ pathway to dephosphorylate the m7GDP decapping product to m7GMP 17. To address the specificity of DcpS for binding to the C5-quinazoline, ProtoArray® protein microarrays were probed with 125I-D156156 in the absence or presence of 10 μM unlabeled competitor molecules (Figure 2B). Unlabeled C5-quinazolines including D156156, D153215 and D156844 effectively competed with 125I-D156156 for binding to DcpS, while unrelated compounds such as staurosporine and tertbutylquinone did not.
Modeling of D156884 in the DcpS Substrate Binding Site
The role of DcpS in modulation of gene expression at the level of mRNA turnover 18 and pre-mRNA splicing 19 was suggestive that DcpS could be a plausible cellular target of compounds that could control gene expression. DcpS was also a plausible target from a chemical perspective as the 2,4 diamino quinazolines are isosteres of the guanine base in the m7GpppN substrate of DcpS. DcpS is known to have a symmetric dimer conformation in the absence of substrate 13,20. The N-terminal domains (residues Arg40-Arg145) of each DcpS protomer are intimately domain-swapped, while the C-terminal domains (residues Gln146-Ser337) which contain the conserved HIT motif active site residue are self-contained. Binding of m7GDP induces the movement of the domain-swapped N-terminal lobes of DcpS affecting closure of one active site allowing DcpS to adopt an asymmetric conformation in which the other, open DcpS monomer contains bound m7GMP and a phosphate 20. Comparison of the ligand-free and ligand-bound structures of DcpS indicates remarkable inherent flexibility with the open and closed forms corresponding to the substrate-bound complex (“closed” form) and the product bound complex (“open” form). Heterodimer mutations of DcpS prevent the overall hydrolytic activity of the protein by trapping one monomer of the protein in a closed substrate-bound configuration and restricting the second monomer in an open confirmation 21. Thus, the two active sites of the DcpS dimer appear to cycle dynamically between closed and open forms representing substrate binding and product release 12,13,20.
In both the open and closed conformations of DcpS, the m7G base occupies a narrow pocket staking between Leu206 and Trp175, and also forms hydrogen bonds to the Glu185 side chain and the carbonyl oxygen of Pro204. We assumed that the 2,4-diamino quinazoline core serves as an isostere of guanine. We therefore carried out a molecular docking study to assess computationally the binding mode of a potent C5-quinazoline (D156844) in the closed active site conformation of DcpS. The coordinate set of human DcpS containing an active site His277Asn mutation in complex with m7GpppG substrate was used as the docking model for these studies (PDB ID:1ST0) 12 (See Materials and Methods). Our docking studies demonstrated a plausible overlay of the quinazoline scaffold with m7G, such that a very similar hydrogen bond network is retained with the 2,4-diamino quinazoline core (Figure 3).
Figure 3. Computational model for D156844 binding to the closed active site conformation of DcpS.
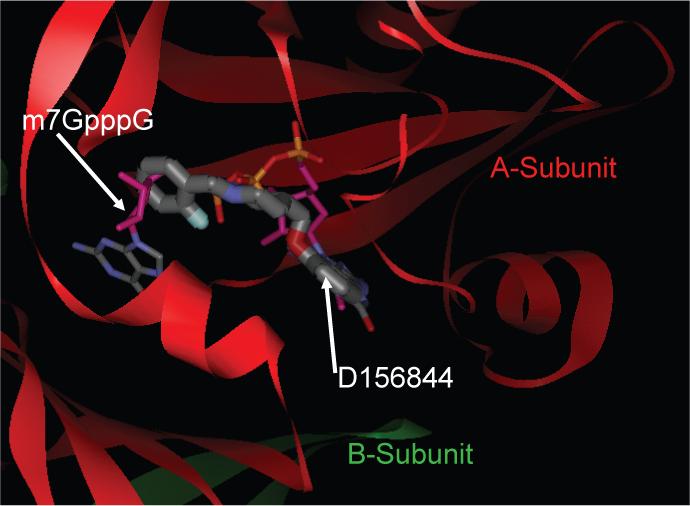
An energy minimized conformer of D156844 was computationally docked into the closed active site conformation of human DcpS (PDB ID:1ST0). The final pose for D156844 (thick stick) is shown superimposed on the structure of m7GpppG (thin stick), both bound into the asymmetric DcpS homodimer (red: A-subunit; green: B-subunit).
The docked pose for D156844 is consistent with our structure-activity relationship (SAR) data as we have previously found that any modification, derivatization or deletion of the 2 and/or 4-amino groups resulted in loss of activity in the β-lactamase promoter assay 11. In the docked pose, the quinazoline portion of D156844 nicely fills the m7G binding pocket with excellent shape, hydrophilic, and hydrophobic complementarity. The computationally reasonable binding pose thus explained why derivation at the 6, 7 and 8 positions of the quinazoline core were not acceptable. In the docked pose, the quinazoline ring displays a potential for π–π stacking with Trp175. The oxygen substituent at C5, by forming an intramolecular H-bond with the 4-amino group, pre-organizes the D156844 molecular scaffold to facilitate a preferred trajectory of the C5 substituent which follows an approximate space filling path occupied by the sugar and tri-phosphate groups of m7GpppG when it is bound in the closed active site conformation of DcpS. In turn, the trajectory of the C5 substitution allows the quinazoline group to maintain a good hydrogen bonding pattern in the relatively narrow portion of the cap binding pocket. Specifically, the C2-amino group makes a coordinated hydrogen bond network with the carbonyl oxygen of Pro204 and the side chain of Glu185. Also consistent with the SAR for the C5-quinazoline series is that the cap binding pocket becomes progressively larger distal to the m7G / quinazoline binding pocket. This explains in part how relatively large C5 substitutions were allowed within the SAR, some of which were found to be highly beneficial to potency. These presumably gain additional interactions to portions of the target outside of the tight cap binding pocket.
C5-Quinazolines are DcpS Inhibitors
We next tested if the C5-quinazolines could influence DcpS decapping activity in purified enzyme assays measuring the hydrolysis of m7GpppG to m7G. Decapping reactions were carried out with 32P-labeled m7Gp*ppG cap structure where the label was at the first phosphate following the methylated guanosine. Decapping reactions were carried out with a fixed amount of DcpS in the presence of increasing titration of D156844 and reaction products and substrate resolved by polyethylenimine (PEI) cellulose thin-layer chromatography (TLC) (Figure 4A). The decapping assays revealed that D156844, but not a related negative control D156676, was highly effective at inhibiting recombinant DcpS decapping activity. Surprisingly, the inhibition observed with D156844 (IC50 =∼15 nM) was more efficient than the natural cap analog m7GpppG (IC50 = ∼30 nM), Figure 4B. The differential inhibition of DcpS by D156844 and D156676 was also observed with endogenous DcpS in human cell extract. Endogenous DcpS decapping activity was selectively inhibited by D156844 added to cell extract (Figure 4C). To determine the extent of correlation between SMN2 promoter activation and DcpS decapping inhibition, we profiled a panel of compounds in the DcpS decapping assay with EC50 in the SMN2 promoter assay varying nearly 1,000 fold from 4 nM to 2.5 μM (Table 1). We observed a striking correlation between SMN2 promoter activation and DcpS decapping inhibition (r2 = 0.9857) over nearly three log orders of potency (Figure 5). These data support the thesis that DcpS is a molecular target of the C5-quinazolines.
Figure 4. Inhibition of DcpS decapping activity by C5-substituted quinazolines.

(A) Radiolabeled m7Gp*pppG was incubated with purified human DcpS (5nM) in the presence of increasing concentrations of C5-quinazolines (D156844 or D156676), a positive control for DcpS inhibition (Cap structure), or a negative vehicle (DMSO) control. The reaction products were separated by thin layer chromatography and visualized with a Phosphorimager. (B) Data for the relative decapping rate vs. concentration of compounds (nM). Structures of the compounds are shown in Table 1. (C) Inhibition of endogenous DcpS decapping activity in 20μg of human K562 erythroleukemia cellular extract with the D156844 or D156676 C5-quinazolines.
Figure 5.
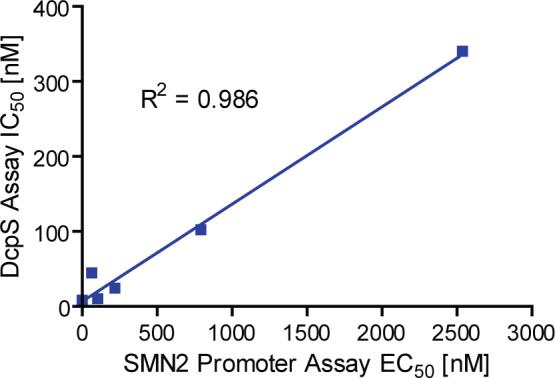
Linear correlation between SMN2 promoter inducing activity and DcpS decapping inhibition.
Co-crystal structures of DcpS with bound C5-quinazolines
To gain a better understanding of the mechanism of inhibition of the DcpS by the C5-quinazolines, we determined the X-ray crystal structures of human DcpS (residues Ala38 to terminal Ser337) together with D156844 (IC50 ∼4 nM, PDB ID:3BL7), D157493 (IC50 ∼66 nM, PDB ID:3BL9), and D153249 (IC50 ∼2 nM, PDB ID:3BLA) (Table 2, Supplemental Figures 1-6).
Table 2.
X-ray Crystal Structure Data and Statistics.
| (1). Numbers in parenthesis represent final shell of data. | |||
| Date | 6/23/2007 | 9/26/2007 | 9/27/2007 |
| Location/Beamline | Bainbridge/007HF | Bainbridge/007HF | Bainbridge/007HF |
| Ligand | D156844 | D157493 | D153249 |
| Data Collection | |||
| space group | P1 | P1 | P1 |
| A | 49.25 | 49.4 | 48.73 |
| B | 56.27 | 56.86 | 55.8 |
| C | 60.33 | 59.49 | 59.4 |
| A | 118.81 | 62.3 | 117.68 |
| B | 92.38 | 77.93 | 93.34 |
| Γ | 99.99 | 79.54 | 99.72 |
| # molecules/asymetric unit | 2 | 2 | 2 |
| Wavelength [Å] | 1.5418 | 1.5148 | 1.5418 |
| Resolution [Å] (1) | 50−2.31(2.39−2.31) | 50−1.8(1.86−1.80) | 50−2.6(2.69−2.60) |
| Reduncancy | 2.0(2.0) | 4.6(1.8) | 2.0(2.0) |
| Unique | 23462(2293) | 47420(4029) | 15959(1567) |
| Completeness [%] | 96.2(94.4) | 91.6(77.5) | 96.3(94.3) |
| Rsym [%] | 12.3(45.3) | 5.2(23.0) | 10.4(46.7) |
| I/sigI | 7.2(1.84) | 38.6(3.95) | 9.7(2.0) |
| Mosaicity | 0.64 | 0.89 | 1.4 |
| Refinement Statistics | |||
| No. of reflections | 22214(1577) | 44993(2811) | 15159(1024) |
| No. of non-hydrogen atoms | 4998 | 5341 | 4863 |
| Resolution range [Å] | 52.2−2.31(2.36−2.31) | 52.2−1.80(1.85−1.80) | 52−2.6(2.66−2.60) |
| Rcryst | 20.8(25.6) | 17.3(21.0) | 20.9(29.7) |
| Rfree | 27.3(33.4) | 22.1(28.8) | 27.8(37.4) |
| FreeR, # of reflections | 5%, 1185(92) | 5%, 2418(154) | 5%, 799(49) |
| average Bfactor [Å2] | 22.3 | 17.25 | 27.1 |
| Model Geometry | |||
| Bond length deviation [Å] | 0.009 | 0.012 | 0.008 |
| Bond angle deviation [o] | 1.196 | 1.394 | 1.121 |
1. Numbers in parenthesis represent high resolution bin.
As discussed above, the structures of human DcpS with bound substrate m7GDP, m7GpppA or m7GpppG have previously been shown to adopt an asymmetric dimer conformation 12,20. Similarly, DcpS with bound C5-quinazolines also forms an asymmetric dimer similar in conformation to the closed active site conformations observed when active site mutant (His277Arg) DcpS is bound to m7GpppG (PDB ID:1ST0) 12 (Figure 6A), or when wild-type DcpS is bound to m7GDP (PDB ID: 1XMM) 20. The overall rmsd between the Cα-carbons of DcpS with D156844 bound compared to that of m7GpppG substrate bound DcpS is 1.1 Å, excluding the residues Ala71-Gly77 which appear to be disordered in our C5-quinazoline bound crystal structures (the chemical structures of these residues can not be effectively modeled from our X-ray diffraction data). These disordered residues are relatively far removed from the active site regions and are likely to be disordered due to the nature of crystal packing in our crystal forms. The C5-quinazolines D156844 and D157493 could each be visualized bound to DcpS in the closed active site conformation of the asymmetric dimer, contacting residues from both the N-terminal and C-terminal domains of the two polypeptide chains. Unlike the structures with bound cap structure, where m7GpppG is observed to bind at the open active site, the DcpS protomer in the open conformation shows little or no clear binding of D156844 or D157493. However, D153249 was observed to bind within both the open and closed conformations, though in the open conformation, only the quinazoline group could be readily visualized in electron density maps.
Figure 6. Crystal Structures of DcpS Bound to C5-quinazolines.

(A) Superposition of DcpS with bound D156844 and m7GpppG. The structure of DcpS bound to D156844 (cyan CPK space filling model) to human DcpS (pink Subunit A, and light green Subunit B) has been superimposed onto the previously reported structure of human DcpS (dark red Subunit A, and dark green Subunit B) with m7GpppG (stick model) bound to both the closed (left side) and open (right side) active site regions (PDB ID:1ST0) 29. The superposition illustrates that D156844 is bound only in closed active site conformation state, while m7GpppG is capable of binding into both the open and closed active site pockets. The DcpS structures with D156844 and m7GpppG bound superimpose with and RMSD on all atoms of 1.1 Å indicating that the conformations of the two are very similar.
(B) Superposition of D156844 and m7GpppG in the DcpS active site. The structure of DcpS bound to D156844 (labeled stick model) to human DcpS (pink Subunit A, and light green Subunit B) has been superimposed onto the previously reported structure of human DcpS (dark red Subunit A, and dark green Subunit B) with m7GpppG (labeled stick model) bound active site regions (PDB ID:1ST0) 29. The “closed” active-site conformation of DcpS is shown. Amino acid side chains are numbered and the ligands are identified. The 2−4 diamino quinazoline of for D156844 ligand adopts an identical binding mode as the m7G group of m7GpppG in DcpS. The C5-substituent of D156844 takes on a different conformation in binding to DcpS as compared to the pppG portion of the m7GpppG. Side chains which take on significantly different conformations between the two structures are Tyr273 and Lys142.
(C) Comparison of the binding pose of D156844 to m7GpppG cap substrate. The DcpS protein structures of our C5-quinazoline bound structure and that of the m7GpppG bound structure (PDB ID:1ST0) 12 were superimposed with COOT 29. The superposition the DcpS protein structures revealed that the quinazoline moiety D156844 (thick ball and stick) overlays nicely with that of the m7G moiety of m7GpppG (thin ball and stick) of the 1ST0 structure. In contrast, the C5 substituent of the quinazoline follows a different trajectory in the binding pocket compared to the pppG appendage of m7GpppG. The two images are rotated 90 degrees with respect to each other and the protein has been removed.
(D) Superposition of DcpS with bound D156844 and D153249. The “closed” active-site conformation of DcpS is shown in superposition mode for the DcpS co-crystal structures with D156844 (light pink for A subunit residues and light green for B subunit residues) and D153249 (dark pink for A subunit residues and dark green for B subunit residues). Amino acid side chains are numbered and the ligands are identified. The 2−4 diamino quinazoline of for each ligand adopts an identical binding mode in DcpS. The C5-substituent of each ligand takes on a different conformation in binding to DcpS.
(E) Superposition of DcpS with bound D156844 and D157493. The “closed” active-site conformation of DcpS is shown in superposition mode for the DcpS co-crystal structures with D156844 (light pink for A subunit residues and light green for B subunit residues) and D157493 (dark pink for A subunit residues and dark green for B subunit residues). Amino acid side chains are numbered and the ligands are identified. The 2−4 diamino quinazoline of for each ligand adopts an identical binding mode in DcpS. The C5-substituent of each ligand takes on a different conformation in binding to DcpS.
Inspection of the binding mode for D156844 reveals that the quinazoline-2−4-diamine excellent mimic of the binding mode observed for the m7G group m7GpppG or m7Gpp bound in the closed conformation of DcpS. Specifically, the quinazoline-2−4-diamine makes hydrogen bond contacts to the “A” subunit (nomenclature of PDB ID:1ST0) side chains of residues Glu185 and Asp205, as well as a main chain carbonyl from Pro204 (Figures 6B and 7). On one side, the quinazoline ring shows π–π stacking interaction with Trp175, and on the other side it is in contact with the hydrophobic Leu206 side chain. The quinazoline ring is also perpendicular to Tyr113 of the “B” subunit which forms one side of the binding cavity. Overall, the m7G moiety and the quinazoline-2−4-diamine share similar interactions with A subunit side chains Trp175, Glu185, Pro204, Leu206, and Ile219; and with B subunit side chains Phe108, Asn110, and Tyr113 (Figure 6B). In addition, the role of side chain of Asp205 in forming hydrogen bonds to the m7G-ribose of m7GpppG in the closed active site conformation is mimicked (without significant movement of the Asp205 side chain) by a hydrogen bond with the 4-amino substituent of D156844 (Figure 6B). Moreover, the observed crystallographic binding mode for the quinazoline-2−4-diamine of both D153249 and D157493 in the closed active site conformation of DcpS are essentially identical to that of D156844 (Figure 6C and 6D), which provides further evidence of consistency in our understanding of SAR for binding and inhibition of DcpS.
Figure 7. Ligand Interaction Figure of D156844 bound to Human DcpS “Closed” Active Site.
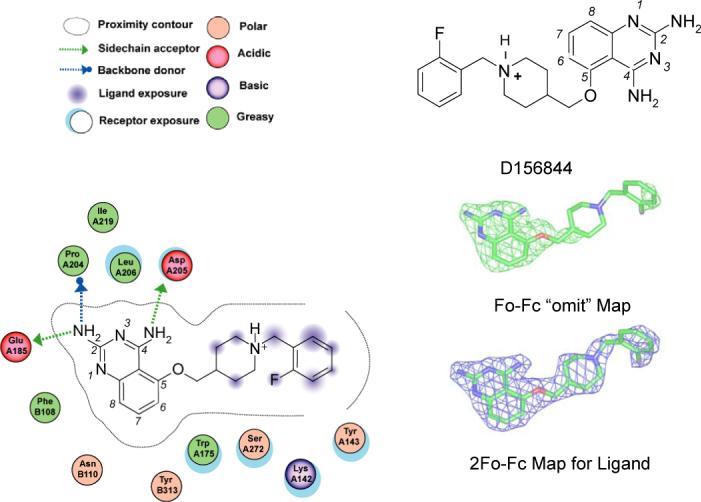
Left Panel. DcpS amino acid interactions with D156844 are depicted schematically for the closed active site. The “A” and “B” subunit residues are listed with “A” or “B” prefixing the residue number. The figure was generated by the MOE software package (Chemical Computing Group, Montreal, Canada), with legend shown. Upper Right Panel. The chemical structure of D156844. Middle Right Panel. Green wire, 3.0 sigma positive electron density(|Fo|-|Fc|) of omit map of D156844 bound in the closed active site of human DcpS. Lower Right Panel. Blue wire, final 2|Fo|-|Fc| electron density map of D156844 bound in the closed active site of human DcpS.
The C5-quinazoline substituent of D156844 connects to the piperidine moiety which spans the sugar-phosphate binding pocket of DcpS, 4.0 Å above Tyr273 which adopts an alternative rotated conformation to accommodate D156844 binding. Notably, Tyr273 has previously been shown to adopt alternative rotomer conformations in the open versus closed substrate binding conformations of DcpS 12,20. The sugar-phosphate backbone of m7GpppG turns deep into the active site pocket, whereas the C5 substituent of D156844 crosses above the active site, blocking potential access to the catalytic residues. Consequently, the piperidine ring of D156844 occupies the same space as the phosphate group of m7GMP in the closed active site conformation of DcpS observed in PDB ID: 1XMM 20 (Figure 6B). In this regard, it is possible that the C5 quinazolines may stabilize at least one catalytic intermediate transition state conformation of DcpS. The terminal fluorophenyl moiety of D156844 is partially solvent exposed forming a perpendicular T-stacking interaction with Tyr143. In addition, Lys142 is found to adopt an alternative conformation (compared to the m7GpppG bound structure) which accommodates positioning of the fluorphenyl moiety. In this way the aliphatic portion of the Lys142 side chain fits nicely between the fluorophenyl group of D156844 and Tyr143.
Together, the computational modeling (Figure 3) and crystal structures of DcpS bound to different C5-quinazoline derivatives (Figure 6) demonstrate that functional binding to DcpS can be supported by promiscuous substitution at the C5-quinozaline position, an observation that is congruent with the SAR data generated during the development of SMN2 promoter activators which showed that diverse C5-quinazoline substituents maintain potent promoter activity. Furthermore, the computational docking and X-ray structure studies support the possibility that potency of the C5-quinazolines may be at least partially attributed to the possibility that various C5 substitutions may adopt alternative conformations within the conformationally dynamic active site pocket of DcpS.
Discussion
DcpS has not previously been considered a potential therapeutic target for the modulation of gene expression nor for the treatment of spinal muscular atrophy. Nevertheless, we have presented compelling chemical, enzymatic, and structural data to support a structure-activity hypothesis that DcpS is a molecular target of the C5-quinazolines. DcpS was identified as a binder of a potent C5-quinazoline based on probing the ∼5,000 proteins on the ProtoArray® Human Protein Microarray (Invitrogen) with an 125I radiolabeled tracer. We further have shown that the structure-activity relationship (SAR) data from cell-based assays of SMN2 promoter activity reflect the relative potency of such compounds with respect to their ability to inhibit DcpS in purified enzyme assays. One of the most potent C5-quinazolines, D156844 shows an IC50 of ∼10 nM for inhibiting DcpS, which is comparable to the inhibitory potency of an m7GpppG cap mimetic structure. Furthermore, across a panel of C5-substituted quinazolines that differ nearly 1,000 fold in SMN2 promoter inducing activity, there was a significant correlation with in vitro inhibition of DcpS decapping activity. Finally, we conducted ligand docking studies and co-crystal structure determination to define the binding mode of the C5-quinazolines to human DcpS, the results of which revealed that in the closed conformation of DcpS the quinazoline-2−4-diamine moiety of the C5-quinazolines occupies the same binding pocket as the m7G purine moiety of m7GpppG substrate. These structural observations closely corresponded with the SAR observed for the C5-quinazolines in cell-based SMN2 promoter assays and in measures of GEMs counts 11. Together, these data provide sound chemical, enzymatic, and structural evidence identifying DcpS as a candidate molecular target of the C5-quinazolines for the potential treatment of SMA.
DcpS as a Small Molecule Target for Modulating GeneExpression
How might inhibition of DcpS alter SMN2 promoter activity? We can imagine several indirect ways that C5-quinazolines may affect SMN gene activity by inhibiting DcpS.
First, DcpS is part of the RNA degradation machinery. Potentially inhibition of DcpS by locking it into an asymmetric conformation may help to reduce the efficiency of mRNA turnover. DcpS functions in the last step of mRNA decay to hydrolyze the cap structure following 3′to 5′ exonucleolytic decay 16 as well as the m7GDP following 5′ end decapping by the Dcp2 protein 17 converting both to m7GMP. As such, DcpS is a modulator of cap dinucleotide, m7GDP and m7GMP levels in cells and could indirectly affect downstream functions by controlling methylated nucleotide levels. Disruption of the yeast DcpS homolog, Dcs1, results in accumulation of cap dinucleotide and inhibition of 5′ to 3′ exonuclease activity 18. Therefore, scavenger decapping activity could feed back to earlier steps in mRNA decay. Similarly, inhibition of DcpS decapping by the C5-quinazolines is also expected to accumulate products of m7GDP and m7GpppN that could impact pre-mRNA splicing 19 or 5′ to 3′ exonucleolytic decay as observed in yeast 18.
Second, SMN2 production could be influenced by the aberrant accumulation of methylated nucleobases which increases their probability of being incorporated into nucleic acids.
Third, SMN mRNA and protein levels could also be influenced by the interplay between DcpS and the cap binding proteins, Cbp20 and eIF4E 22 19. The ability of DcpS to clear potentially deleterious cap structure from cells is expected to be important in maintaining proper cellular mRNA homeostasis. An shRNA-directed knockdown of DcpS leads to a reduction of first intron splicing due to sequestration of the nuclear cap binding protein, Cbp20, by the aberrant accumulation of cap structure 19. As such, DcpS can modulate pre-mRNA splicing. Similarly, DcpS activity is expected to also modulate mRNA translation by sequestration of the cytoplasmic eIF4E cap binding protein. Therefore, disruption of DcpS activity by D156844 is expected to alter the steady state level of potentially deleterious cap structure in a cell and sequester Cbp20 and eIF4E resulting in the abrogation of normal mRNA processing and translation.
DcpS could also be directly involved in the stimulation of SMN2 production by a function independent of its decapping activity. A decapping-independent function has already been demonstrated for the yeast Dcs1 gene, where glycerol lethality in a dcs1Δ strain could be complemented by both a wild type as well as a decapping catalytically incompetent mutant 18. Moreover, there is ample evidence for proteins involved in mRNA metabolism to also have a dual function in other pathways including transcription 23-27. DcpS could therefore also have distinct direct roles in both mRNA decay as well as transcription. The ability of D156844 to trap DcpS in a decapping inactive form might result in shifting the equilibrium away from a role in decapping and towards an alternative nuclear function. The predominant nuclear localization of DcpS 22,28 would be consistent with this property. Determining whether DcpS can directly or indirectly function in SMN2 transcription, RNA-processing or mRNA translation will begin to address potential roles of this protein in mRNA biogenesis. Future efforts to generate DcpS gene knock-out mice and active site mutation knock-in mice will provide significant insights in the mechanism of action of DcpS in both wild type and SMA disease model mice.
Considering the Value of Protein Structure in Translational Research
It is worthwhile to discuss the value of protein arrays and protein structures with respect to leveraging information from chemical genomics research such as we have presented herein. The sequencing of the human and other genomes allows annotation and production of protein arrays such as the ∼5,000 human protein ProtoArray® as used in this study. Our interpretation of the binding data greatly benefited from the public availability of protein crystal structures of both human DcpS (published by researchers in the US and Singapore) and mouse DcpS (published by the Joint Center for Structural Genomics, an NIH funded Protein Structure Initiative Center). These available structures allowed us to apply computational methods to test hypotheses as to how the C5-quinazolines might bind to the target. Moreover, the reported methods for protein production and crystal structure determination for both the human and mouse DcpS enzymes provided an excellent road map for determining the crystal structure of DcpS bound to the C5-quinazolines. Publicly available databases enabled us to identify a potential molecular target of the C5-quinazolines. These are clinically relevant compounds. Knowledge of their probable mechanism of action through inhibition of DcpS might provides important insights into additional pharmacodynamic markers of drug action for use in human clinical trials, for example, accumulation of m7GpppG cap structures. Like histone deacetylase inhibitors, DcpS inhibitors appear to be well tolerated in pre-clinical animal studies, and therefore may provide a useful therapeutic class for the modulation of gene function.
Materials and Methods
Supplementary Material
Acknowledgements
We thank the crystallization core team at deCODE biostructures for their support in crystallization and X-ray diffraction data collection. We thank the Protein Structure Initiative for funding work in structural proteomics enabling our rapid structure determination of DcpS-ligand bound co-structures. We thank Jaclyn Bonin, Fang Zhou and Dee Chen for manufacturing the protein arrays.
Grant Support
FSMA supported the generation of the quinazoline series of compounds used in this project and their radiolabeling. The synthetic gene design work for this project was funded in part by the NIGMS-NCRR co-sponsored PSI-2 Specialized Center Grant U54 GM074961 which supports the Accelerated Technologies Center for Gene to 3D Structure. Funding for the DcpS in vitro studies was provided by NIH grant GM67005 and FSMA to M.K.
Footnotes
PDB Entries
The X-ray crystal structures of human DcpS in complex with C5-substituted quinazolines have been made publicly available through the Protein Data Bank (www.rcsb.org/pdb) entries for ligands D156844 (PDB ID:3BL7), D157493 (PDB ID:3BL9), and D153249 (PDB ID:3BLA).
Contributor Information
Jasbir Singh, deCODE chemistry , Inc. 2501 Davey Rd., Woodridge, IL 60517.
Michael Salcius, Yale University, 219 Prospect St, New Haven, CT, 06511.
Shin-Wu Liu, Rutgers University, Dept. of Cell Biology and Neuroscience 604 Allison Road, Piscataway, NJ 08854−8082.
Bart L. Staker, deCODE biostructures, Inc., 7869 NE Day Rd. West, Bainbridge Island, WA 98110.
Rama Mishra, deCODE chemistry , Inc. 2501 Davey Rd., Woodridge, IL 60517.
John Thurmond, deCODE chemistry , Inc. 2501 Davey Rd., Woodridge, IL 60517.
Gregory Michaud, Novartis, 250 Massachusetts Avenue, Cambridge, MA, 02139.
Dawn R. Mattoon, Invitrogen Corporation, 688 East Main Street, Branford, CT 06405
John Printen, Invitrogen Corporation, 1600 Faraday Avenue, Carlsbad, CA 92008.
Brian A. Pollok, Invitrogen Corporation, 1600 Faraday Avenue, Carlsbad, CA 92008
Megerditch Kiledjian, Rutgers University, Dept. of Cell Biology and Neuroscience 604 Allison Road, Piscataway, NJ 08854−8082.
Jeffery Christensen, deCODE biostructures, Inc., 7869 NE Day Rd. West, Bainbridge Island, WA 98110.
Lance Stewart, deCODE biostructures, Inc., 7869 NE Day Rd. West, Bainbridge Island, WA 98110.
Jill Jarecki, P. O. Box 196, Libertyville, IL 60048−0196.
Jon Mar Bjornsson, deCODE genetics, Inc. Sturlugata 8 , IS-101 Reykjavik, Iceland.
Mark E Gurney, deCODE genetics, Inc. Sturlugata 8 , IS-101 Reykjavik, Iceland.
References
- 1.Shiau AK, et al. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–37. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 2.Kliewer SA, Lehmann JM, Milburn MV, Willson TM. The PPARs and PXRs: nuclear xenobiotic receptors that define novel hormone signaling pathways. Recent Prog Horm Res. 1999;54:345–67. discussion 367−8. [PubMed] [Google Scholar]
- 3.Pearn J. Incidence, prevalence, and gene frequency studies of chronic childhood spinal muscular atrophy. J Med Genet. 1978;15:409–13. doi: 10.1136/jmg.15.6.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roberts DF, Chavez J, Court SD. The genetic component in child mortality. Arch Dis Child. 1970;45:33–8. doi: 10.1136/adc.45.239.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lefebvre S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–65. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 6.Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. 1999;96:6307–11. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brichta L, et al. Valproic acid increases the SMN2 protein level: a well-known drug as a potential therapy for spinal muscular atrophy. Hum Mol Genet. 2003;12:2481–9. doi: 10.1093/hmg/ddg256. [DOI] [PubMed] [Google Scholar]
- 8.Sumner CJ, et al. Valproic acid increases SMN levels in spinal muscular atrophy patient cells. Ann Neurol. 2003;54:647–54. doi: 10.1002/ana.10743. [DOI] [PubMed] [Google Scholar]
- 9.Avila AM, et al. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J Clin Invest. 2007;117:659–71. doi: 10.1172/JCI29562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jarecki J, et al. Diverse small-molecule modulators of SMN expression found by high-throughput compound screening: early leads towards a therapeutic for spinal muscular atrophy. Hum Mol Genet. 2005;14:2003–18. doi: 10.1093/hmg/ddi205. [DOI] [PubMed] [Google Scholar]
- 11.Thurmond J, et al. Synthesis and biological evaluation of novel 2,4-diaminoquinazoline derivatives as SMN2 promoter activators for the potential treatment of spinal muscular atrophy. J Med Chem. 2008 doi: 10.1021/jm061475p. In Press. [DOI] [PubMed] [Google Scholar]
- 12.Gu M, et al. Insights into the structure, mechanism, and regulation of scavenger mRNA decapping activity. Mol Cell. 2004;14:67–80. doi: 10.1016/s1097-2765(04)00180-7. [DOI] [PubMed] [Google Scholar]
- 13.Han GW, et al. Crystal structure of an Apo mRNA decapping enzyme (DcpS) from Mouse at 1.83 A resolution. Proteins. 2005;60:797–802. doi: 10.1002/prot.20467. [DOI] [PubMed] [Google Scholar]
- 14.Schweitzer B, Predki P, Snyder M. Microarrays to characterize protein interactions on a whole-proteome scale. Proteomics. 2003;3:2190–9. doi: 10.1002/pmic.200300610. [DOI] [PubMed] [Google Scholar]
- 15.Liu H, Rodgers ND, Jiao X, Kiledjian M. The scavenger mRNA decapping enzyme DcpS is a member of the HIT family of pyrophosphatases. Embo J. 2002;21:4699–708. doi: 10.1093/emboj/cdf448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Z, Kiledjian M. Functional link between the mammalian exosome and mRNA decapping. Cell. 2001;107:751–62. doi: 10.1016/s0092-8674(01)00592-x. [DOI] [PubMed] [Google Scholar]
- 17.van Dijk E, Le Hir H, Seraphin B. DcpS can act in the 5'−3' mRNA decay pathway in addition to the 3'−5' pathway. Proc Natl Acad Sci U S A. 2003;100:12081–6. doi: 10.1073/pnas.1635192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu H, Kiledjian M. Scavenger decapping activity facilitates 5' to 3' mRNA decay. Mol Cell Biol. 2005;25:9764–72. doi: 10.1128/MCB.25.22.9764-9772.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen V, Liu H, Liu SW, Jiao X, Kiledjian M. DcpS scavenger decapping enzyme can modulate pre-mRNA splicing. Rna. 2008;14:1132–42. doi: 10.1261/rna.1008208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen N, Walsh MA, Liu Y, Parker R, Song H. Crystal structures of human DcpS in ligand-free and m7GDP-bound forms suggest a dynamic mechanism for scavenger mRNA decapping. J Mol Biol. 2005;347:707–18. doi: 10.1016/j.jmb.2005.01.062. [DOI] [PubMed] [Google Scholar]
- 21.Liu SW, Rajagopal V, Patel SS, Kiledjian M. Mechanistic and kinetic analysis of the DcpS scavenger decapping enzyme. J Biol Chem. 2008 doi: 10.1074/jbc.M800341200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu SW, et al. Functional analysis of mRNA scavenger decapping enzymes. Rna. 2004;10:1412–22. doi: 10.1261/rna.7660804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao C, Guo H, Mi Z, Wai PY, Kuo PC. Transcriptional regulatory functions of heterogeneous nuclear ribonucleoprotein-U and -A/B in endotoxin-mediated macrophage expression of osteopontin. J Immunol. 2005;175:523–30. doi: 10.4049/jimmunol.175.1.523. [DOI] [PubMed] [Google Scholar]
- 24.Leverrier S, et al. Purification and cloning of type A/B hnRNP proteins involved in transcriptional activation from the Rat spi 2 gene GAGA box. Biol Chem. 2000;381:1031–40. doi: 10.1515/BC.2000.127. [DOI] [PubMed] [Google Scholar]
- 25.Tomonaga T, Levens D. Heterogeneous nuclear ribonucleoprotein K is a DNA-binding transactivator. J Biol Chem. 1995;270:4875–81. doi: 10.1074/jbc.270.9.4875. [DOI] [PubMed] [Google Scholar]
- 26.Eggert H, Schulz M, Fackelmayer FO, Renkawitz R, Eggert M. Effects of the heterogeneous nuclear ribonucleoprotein U (hnRNP U/SAF-A) on glucocorticoid-dependent transcription in vivo. J Steroid Biochem Mol Biol. 2001;78:59–65. doi: 10.1016/s0960-0760(01)00074-7. [DOI] [PubMed] [Google Scholar]
- 27.Dempsey LA, Hanakahi LA, Maizels N. A specific isoform of hnRNP D interacts with DNA in the LR1 heterodimer: canonical RNA binding motifs in a sequence-specific duplex DNA binding protein. J Biol Chem. 1998;273:29224–9. doi: 10.1074/jbc.273.44.29224. [DOI] [PubMed] [Google Scholar]
- 28.Cougot N, Babajko S, Seraphin B. Cytoplasmic foci are sites of mRNA decay in human cells. J Cell Biol. 2004;165:31–40. doi: 10.1083/jcb.200309008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Emsley P, Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.