

Abstract
Sphingosine-1-phosphate (S1P), produced by sphingosine kinase 1 (SphK1) or kinase 2 (SphK2), mediates biological effects through intracellular and/or extracellular mechanisms. Here we determined a role for these kinases in kidney injury of wild-type mice following ischemia-reperfusion. SphK1 but not SphK2 mRNA expression and activity increased in the kidney following injury relative to sham-operated animals. Although SphK1-/- mice had no alteration in renal function following injury, mice with a disrupted SphK2 gene (SphK2tr/tr) had histological damage and impaired function. The immune-modulating pro-drug, FTY720, an S1P agonist failed to provide protection in SphK2tr/tr mice. Injured kidneys of these mice showed increased neutrophil infiltration and neutrophil chemokine expression along with a 3- to 5-fold increase in expression of the G-protein-coupled receptor S1P3 compared to heterozygous SphK2+/tr mice. Kidney function and reduced vascular permeability were preserved in compared to mice after ischemia-reperfusion injury, suggesting increased S1P3 mRNA may play a role in the injury of SphK2tr/tr mice. Our study suggests that constitutive expression of SphK2 may contribute to reduced ischemia-reperfusion injury of the kidney, and its absence may enhance injury due to increased neutrophil infiltration and S1P3 activation. We also confirm that SphK2 is necessary to mediate the protective effects of FTY720.
Keywords: SphK, acute kidney injury, S1P, FTY720
Ischemic acute kidney injury is a major clinical problem with high morbidity and mortality and also accounts for delayed graft function of transplanted kidneys.1,2 Treatment of ischemic acute kidney injury still remains largely supportive and many drugs that reduced injury in animal models have failed in humans.3 Therefore, a refined understanding of the mechanisms of kidney ischemia-reperfusion injury (IRI) and development and testing of novel compounds is necessary.
Sphingosine-1-phosphate (S1P), a pleiotrophic lipid mediator is produced by phosphorylation of sphingosine by sphingosine kinases (SphKs) in response to a variety of stimuli. S1P is exported out of the cell (inside-out signaling) and serves as a ligand for five different G-protein-coupled receptors (S1P1-5).4 In addition to extracellular function, S1P also has an intracellular role and acts as a second messenger.5,6 S1P regulates diverse biological processes and functions, including cell growth, survival, and proliferation and angiogenesis; S1P inhibits apoptosis and effects lymphocyte trafficking.7-12 Furthermore, S1P1 receptor activation has been shown to protect kidneys from IRI.13,14
SphK1 and SphK2 are important rate-limiting steps in the formation of S1P. Despite sharing two large conserved regions, these kinases have different kinetics of expression during development as well as different subcellular localization,15,16 suggesting that these two isoforms may serve different functions. SphK1 promotes cell-survival, proliferation, and regulates cell transformation.17 In contrast, less is known about SphK2, although several reports suggest that SphK2 serves proapoptotic functions.18,19 More importantly, SphK2 phosphorylates FTY720,20 an immune modulatory prodrug currently in clinical trials for multiple sclerosis. Mice deficient in SphK1 or SphK2 do not exhibit conspicuous abnormalities, whereas double knockouts are lethal.21 The purpose of the current study was to investigate the role of SphK1 and SphK2 in kidney IRI and to determine the role of SphK2 in mediating the kidney-protective effect of FTY720.
RESULTS
SphK gene expression and enzyme activities after kidney IRI
We first examined SphK1 and SphK2 gene expression and enzyme activities at various time points after kidney IR in wild-type (WT) mice. SphK1 mRNA expression showed a robust increase as early as 2 h after reperfusion, peaking at 4-6 h with a 30-fold higher level compared to sham-operated mice and remaining elevated until 96 h. In contrast to the SphK1 mRNA, SphK2 mRNA did not increase significantly after IRI (Figure 1a). As expected from changes in cognate mRNA levels, SphK1 enzyme activity increased by threefold but there was no significant change in SphK2 enzyme activity (Figure 1b). These findings demonstrate that kidney IRI induces an increase in steady-state kidney SphK1 mRNA but not SphK2 mRNA, suggesting that SphK1 may be important in injury following kidney IR.
Figure 1. Time course of SphK mRNA expression and enzyme activities in kidney IRI.

Mouse kidneys were subjected to 32 min of ischemia and at various time points after reperfusion, kidneys were harvested for RNA extraction and measurement of SphK enzyme activities. Values are mean±s.e.m.; n=4-5 for each group; *P<0.05 compared with sham. (a) Time course of SphK mRNA expression following kidney IRI; (b) Time course of SphK enzyme activities following kidney IRI.
The effects of SphK1 and SphK2 on renal injury
To determine the importance of the SphK1 and SphK2 gene products in renal injury, we first evaluated functional and histological changes in SphK1-/- and SphK2tr/tr mice. We assumed that the marked increase in SphK1 activity following kidney IRI indicated an important role of this enzyme in the kidney. Unexpectedly, the increase in plasma creatinine in SphK1-/- mice following kidney IR was not significantly different from that in SphK1+/+ mice (Figure 2a). Surprisingly, although SphK2 gene expression and enzyme activity were unchanged after kidney IR, kidney injury at 24 h was worse in SphK2tr/tr as indicated by elevated plasma creatinine levels that were significantly greater in SphK2tr/tr than in SphK2+/tr or WT mice (Figure 2b). Histological examination with hematoxylin and eosin staining showed extensive tubular injury characterized by an increase in tubular cell necrosis, dilation of tubules, and cast formation in the outer medulla of SphK2tr/tr mice. Kidneys from SphK2+/tr and/or WT showed less tubular injury compared to SphK2tr/tr (Figure 3a). A semiquantitative assessment demonstrated histological damage was greater in SphK2tr/tr than in SphK2+/tr mice (Figure 3b).
Figure 2. Effect of the absence of SphK1 and SphK2 on plasma creatinine in kidney IRI.

Kidneys from (a) SphK1 (SphK1-/- and SphK1+/+) or (b) SphK2 (SphK2tr/tr SphK2+/tr and WT) mice were subjected to 32 min of ischemia and plasma creatinine concentrations were measured after 24 h of reperfusion. Values are mean±s.e.m.; n=4-5 for each group; *P<0.05 compared with SphK2+/tr in (b).
Figure 3. Effect of the absence of SphK2 on histology in IR-induced acute kidney injury.

(a) Mouse kidneys from SphK2tr/tr or SphK2+/tr mice were subjected to 32 min of ischemia or sham surgery, and kidneys were harvested after 24 h of reperfusion and processed for histological examination of sections of kidney outer medulla under × 200 magnification. (b) Semiquantitative assessment of histological damage. Values are mean±s.e.m.; n=4-5 for each group. *P<0.05 compared with SphK2tr/tr.
Compensatory SphK mRNA expression in SphK-deficient mice
To examine the possibility that compensatory changes in expression of SphKs may occur in SphK null mice, we measured SphK1 mRNA expression in SphK2tr/tr and SphK2 mRNA expression in SphK1-/- after 6 h of reperfusion in sham and IR mice. Although the marked increase in expression of SphK1 and SphK2, as shown in Figure 1, was confirmed in this experiment, there were no significant differences in the magnitude of the increase in SphK1 mRNA in SphK2tr/tr compared to SphK2+/tr mice or in SphK2 mRNA in SphK1-/- compared to SphK1+/+ mice (Figure 4a and b). These results suggest that following kidney IRI, a compensatory mechanism between SphK1 and SphK2 does not occur when one enzyme is absent.
Figure 4. Changes in SphK1 mRNA expression in SphKtr/tr and SphK+/tr mice and SphK2 mRNA expression in SphK+/+ and SphK-/- mice.
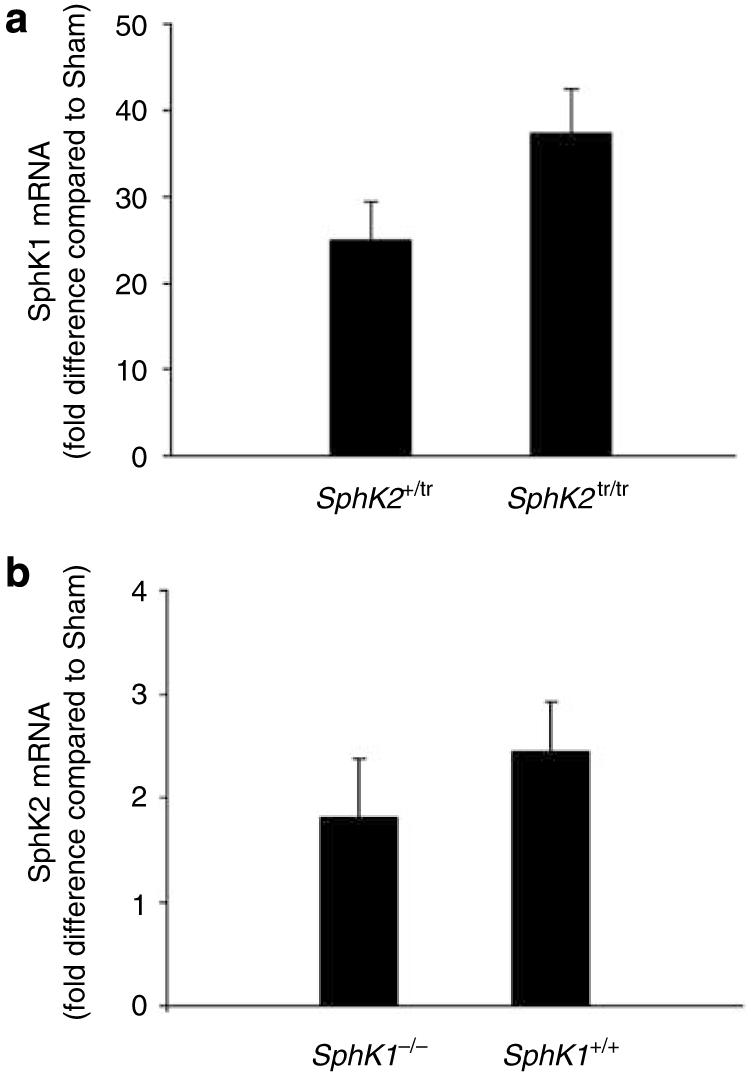
Mouse kidneys from SphK1-/-, SphK1+/+, SphK2tr/tr, or SphK2+/tr mice were subjected to 32 min of ischemia and 6 h of reperfusion. RNA was extracted and quantitative real-time RT-PCR was performed. (a) SphK1 mRNA in SphK2tr/tr and SphK2+/tr mice and (b) SphK2 mRNA in SphK1-/- and SphK1+/+ mice are shown as fold change compared to sham; n=4-5 for each group.
Enhanced cell death in SphK2 in kidney IRI
Our results with SphK2tr/tr mice, which suggest a role for SphK2 rather than SphK1 in kidney IRI, prompted further studies on SphK2. Together with necrosis, apoptosis is also an important mode of cell death following IR. We measured the number of terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL)-positive cells in kidneys following IRI and found that, unlike tubular cell necrosis, there was no significant difference in the increased number of apoptotic cells at 6 (Figure 5a and b) and 24 h (data not shown) post reperfusion in SphK2tr/tr compared to SphK2+/tr mice. Also, total kidney protein from sham or IR SphK2tr/tr and SphK2+/tr mice at 6 and 24 h reperfusion was used to assess apoptosis by western blot analysis using activated caspase 3 antibody. No significant changes in cleaved forms of caspase 3 were noted in SphK2tr/tr and SphK2+/tr mice; similarly no significant changes in mRNA levels of bax/bcl-2 ratio at 6 and 24 h were observed (data not shown).
Figure 5. Effect of SphK2 on apoptosis in kidney IRI.

Mouse kidneys from SphK2tr/tr or SphK2+/tr mice were subjected to 32 min of ischemia and kidneys were harvested after 6 h of reperfusion. (a, b) Sections of kidney (× 200 magnification) processed for detection of apoptosis using TUNEL staining (TUNEL-positive cells contain brown immunoperoxidase reaction product) and counterstained blue with hematoxylin. (c) Quantitative analysis (n=3-4) for each group of TUNEL-positive cells in kidneys from SphK2tr/tr or SphK2+/tr mice.
The absence of SphK2 increases kidney neutrophil infiltration and neutrophil chemoattractants following kidney IRI
Kidney sections from SphK2tr/tr and SphK2+/tr were labeled immunohistochemically to assess neutrophil infiltration 24 h following kidney IRI. A marked increase in neutrophil infiltration following kidney IRI was seen primarily in the outer medulla (Figure 6a). Kidneys from SphK2tr/tr mice had significantly more neutrophil infiltration compared with SphK2+/tr mice (Figure 6b). We also examined neutrophil infiltration 24 h following kidney IR in SphK1-/- and SphK1+/+ mice. Consistent with the finding that there was no difference in plasma creatinine following kidney IRI in SphK1-/- and SphK1+/+ mice, we observed no difference in neutrophil infiltration between these two groups (Figure 6a and b). Similarly, the kidney sections from SphK1 and SphK2 were labeled for infiltrating monocytes (F4/80-positive cells). No difference in the number of infiltrating monocytes was observed between SphK1-/- and SphK2tr/tr IR (data not shown).
Figure 6. Effect of SphK on infiltrating leukocytes in IR kidney.

(a) Mouse kidneys from SphK2tr/tr SphK2+/tr SphK1+/+ and SphK1-/- mice were subjected to 32 min of ischemia and kidneys were embedded 24 h after reperfusion. Paraffin-embedded kidney sections of SphK mice from sham and IR labeled with 7/4 antibody show brown immunoreactivity and hematoxylin and eosin stain; × 200 magnification. (b) Quantification of DAB immunoperoxidase reaction product (brown pixels) indicative of neutrophil 7/4 labeling was carried out using Adobe Photoshop (see ‘Materials and Methods’). n=3-4 for each group; *P<0.05 compared to SphK2+/tr.
With our observation that kidney neutrophil infiltration was greater following IR in SphK2tr/tr compared with SphK2+/tr mice, we sought to determine whether neutrophil chemoattractant expression was increased in SphK2tr/tr compared with SphK2+/tr mice. mRNA of neutrophil chemoattractants, CXCL1, CXCL2, and CXCL5 was assessed in SphK2tr/tr and SphK2+/tr mice at 6 and 24 h post reperfusion (Figure 7). Kidney IRI led to an increase in CXCL1 and CXCL2 mRNA at both 6 and 24 h following kidney IRI in SphK2 mice; the increase in CXCL1 expression was significantly greater in SphK2tr/tr compared to SphK2+/tr mice (Figure 7a and b). CXCL5 mRNA expression, however, was reduced in the SphK2tr/tr mice compared to SphK2+/tr at 6 and 24 h following IRI (Figure 7c). In SphK1 mice no significant difference in neutrophil chemoattractant expression was observed between SphK1-/- and SphK1+/+ mice (data not shown). Furthermore, the expression of several proinflammatory cytokine and chemokine genes in SphK2tr/tr and SphK2+/tr mice was examined at 24 h following IRI. Tumor necrosis factor-α, interleukin-6 and monocyte chemoattractant protein-1 mRNA expression markedly increased at 24 h after IR in both SphK2tr/tr and SphK2+/tr mice compared to sham. However, there were no significant differences in mRNA levels between the SphK2tr/tr and SphK2+/tr mice (data not shown).
Figure 7. Effect of SphK2 on neutrophil chemokines in IR kidney.

Mouse kidneys from SphK2tr/tr and SphK2+/tr mice were subjected to 32 min of ischemia and were harvested for RNA at 6 and 24 h after reperfusion. Neutrophil chemoattractant chemokines CXCL1 (a), CXCL2 (b) and CXCL5 (c) mRNA were measured using real-time RT-PCR. CXCL1 and CXCL2 mRNA levels increased significantly in SphK2tr/tr mouse kidney compared to SphK2+/tr at 6 and 24 h. n=2-3 for each group; ***P<0.001 compared to SphK2+/tr.
S1P3 receptor role in mediating IRI injury in SphK2tr/tr
S1P1 receptor activation by selective agonists following kidney IR has been documented to be protective,13,14 whereas S1P3 activation may lead to injury.22 Thus, we sought to determine the effect of IRI on expression of S1P1 and S1P3 receptors in SphK2tr/tr and SphK2+/tr mice. S1P1 mRNA increased markedly at 6 and 24 h following IRI in SphK2 mice and the magnitude of the increase was greater in SphK2tr/tr than in SphK2+/tr (Figure 8a). S1P3 mRNA was not affected by IRI in SphK2+/tr but increased significantly in SphK2tr/tr mice (Figure 8b). Thus, although SphK2tr/tr and SphK2+/tr mice show increased expression of S1P1 mRNA following kidney IRI, a response that may be protective, only the SphK2tr/tr mice demonstrated heightened expression of S1P3 mRNA, a response that may be deleterious. To test this hypothesis we performed IR in , and mice and found that plasma creatinine levels 24 h following reperfusion were significantly lower in than and mice (Figure 8c). We next determined whether IRI in was associated with reduced vascular permeability by measuring kidney Evan’s blue dye content. mice had reduced Evan’s blue dye content following IRI consistent with a more intact vascular barrier than (Figure 8d). These results are consistent with results by Gon et al.22 who found that pulmonary permeability and injury was reduced in mice.
Figure 8. S1P3 receptor gene expression in IR kidney.

Mouse kidneys from SphK2tr/tr or SphK2+/tr mice were subjected to 32 min of ischemia and 24 h after reperfusion, RNA was extracted and quantitative real-time RT-PCR for S1P1 and S1P3 was carried out. (a) S1P1 mRNA and (b) S1P3 mRNA are expressed as fold change compared to sham. n=4 for each group. *P<0.05 compared with sham; #P<0.05 compared to SphK2tr/tr. (c) Plasma creatinine at 24 h following kidney IRI for (n=8), (n=6) and (n=7) mice. ***P<0.001 compared to ; #P<0.05 compared to . (d) Evan’s blue dye was measured in (n=7) and (n=7) mice after IR. Values are shown relative to respective sham mice; **P<0.02 compared to
The effect of absence of SphK2 gene on FTY720-mediated renal tissue protection
SphK2 is required for modulation of lymphocyte traffic by FTY720.13 To test the hypothesis that SphK2 is necessary in mediating the protective effect of FTY720, we administered FTY720 in SphK2tr/tr SphK2+/tr and WT mice. In contrast to WT mice, there was no protective effect of FTY720 in SphK2tr/tr or SphK2+/tr mice, indicating that SphK2 is required for mediating the protective effect of FTY720 (Figure 9a). However, administration of exogenous S1P reduced kidney IRI in the absence of SphK2 (Figure 9b).
Figure 9. Effect of FTY720 on renal function in SphK2tr/tr mice.
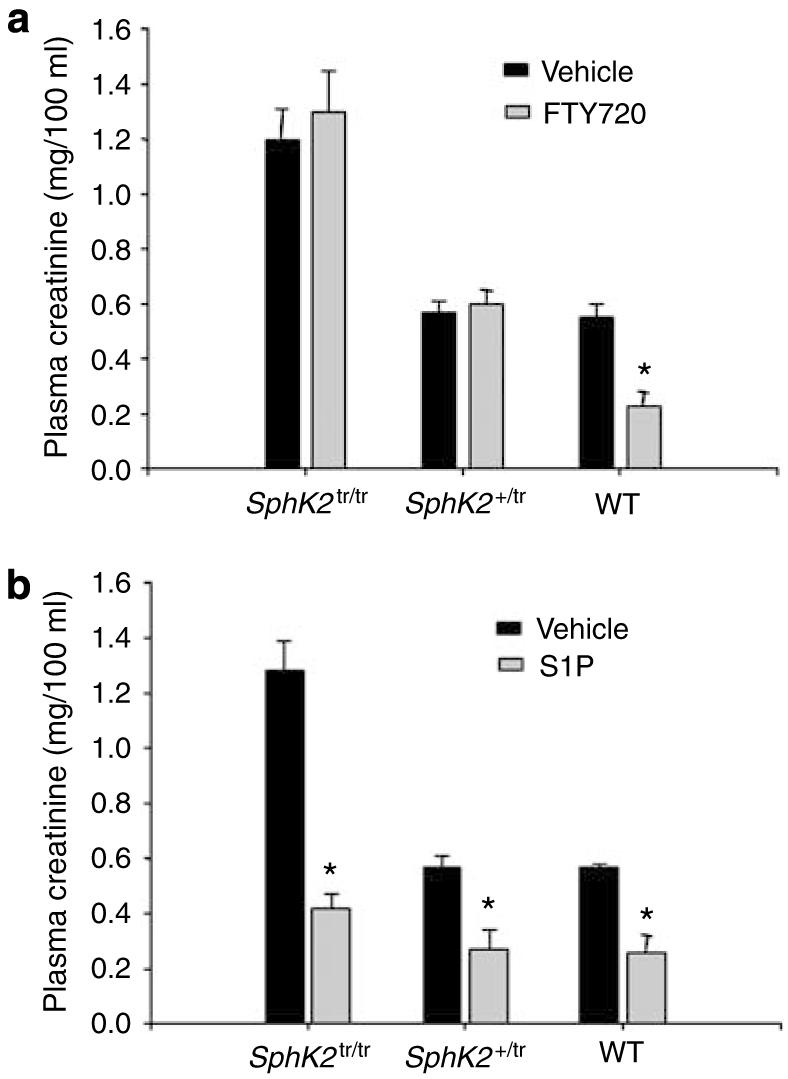
Mouse kidneys from SphK2tr/tr or SphK2+/tr mice were subjected to 32 min of ischemia and plasma creatinine was measured after 24 h of reperfusion. (a) FTY720 (48 mg/kg, i.p.) (b) S1P (50 ng/kg, i.v.) or vehicle (3% fatty acid-free bovine serum albumin/phosphate-buffered saline solution; i.p.) was administered (see ‘Materials and Methods’). n=4 for each group; *P<0.05 compared to vehicle.
DISCUSSION
In this study, we demonstrated that the absence of SphK2, but not SphK1, was associated with worse kidney damage after IRI. This result is counter to the observation that SphK1 but not SphK2 mRNA and activity increased in kidney subsequent to IRI. In WT mice SphK2 showed minimal regulation of mRNA and enzymatic activity after IRI. The more severe kidney IRI in SphK2tr/tr mice was associated with an increase in neutrophils and neutrophil chemoattractants and an increase in S1P3 mRNA expression. Heightened S1P3 expression may have had a deleterious effect in SphK2tr/tr mice, as kidney function following IR in mice was markedly protected and was associated with reduced vascular permeability. Our result suggests that constitutive expression of SphK2 is important in determining cell survival. However, in contrast to SphK2, the absence of SphK1 did not affect kidney damage. Lastly, we found that SphK2 is important in mediating the tissue-protective effect of FTY720 in IR-induced kidney injury, indicating that FTY720 phosphate is the active agent in protecting the kidney from IRI.
S1P is an emerging lipid signaling molecule that is important in diverse biological processes.7-9,11,12,23 SphK1 and SphK2 regulate levels of S1P, therefore we first examined SphK1 and SphK2 gene expressions and enzyme activities following kidney IR. Although SphK1 mRNA showed a marked increase shortly after reperfusion, SphK2 mRNA showed minimal changes. This finding suggested that SphK1 could be important in kidney IRI. To test this possibility, we induced kidney IRI in SphK1 and SphK2 null mice. Unexpectedly, the absence of SphK1 did not affect renal function 24 h after reperfusion despite a marked increase in SphK1 gene expression and activity in WT mice. In contrast, however, IRI led to worse injury in SphK2tr/tr mice compared with WT or SphK2+/tr mice. These findings were unexpected given that in vitro studies have demonstrated prosurvival and proapoptotic effects of SphK1 and SphK2, respectively.15,18,19,24 However, there is a possibility that the absence of any effect of SphK1 gene can be related to the fact that the function of SphK1 requires translocation to a specific membrane compartment. The majority of SphK1 activity is localized to the cytosol and on stimulation, it must travel to the plasma membrane to synthesize S1P. Kusner et al.25 showed SphK1 was co-localized with actin filaments at macrophage plasma membranes. Disruption of actin polymerization reduced plasma membrane SphK1 activity demonstrating that actin filaments are involved in SphK1 localization and regulation of activity. Following kidney IRI, proximal tubule epithelial and endothelial actin filaments are disrupted.,26,27 Thus, despite the marked increase in SphK1 mRNA expression and enzyme activity in kidneys of mice subjected to kidney IRI, translocation of SphK1 to critical intracellular sites in injured epithelial or endothelial cells may have been blocked, thereby preventing any change in kidney function and histology following kidney IRI. Finally, subcellular localization of SphKs may have an important role in production of S1P and its role in kidney IRI.
By contrast, cell survival following kidney IRI appears to require constitutive expression of SphK2. Compared to WT or SphK2+/tr SphK2tr/tr mice had more severe kidney damage following IR. Insofar as innate mechanisms participate in kidney IRI,28,29 analogous results have been shown in bacterial infections. In a model of bacterial lung infection, the extent of disease was worse in SphK2 null mice in comparison to SphK1 null mice.30
Apoptosis does not explain the difference in extent of kidney IRI in SphK2tr/tr compared to SphK2+/tr mice, suggesting that SphK2 has different effects on cell death pathways. Change in SphK2 did not affect the increase in the levels of expression of the proinflammatory cytokines (tumor necrosis factor-α, interleukin-6 and monocyte chemoattractant protein-1), suggesting SphK2 does not affect key surrogate markers of inflammation. However, it should be noted that our assays were limited to whole kidney mRNA expression. It is possible that selective S1P deficiency from immune cells isolated from SphK2tr/tr mice may have been altered although the overall effect on inflammation in IRI kidneys was not.
Kidney IRI in SphK2tr/tr mice was associated with an increase in infiltration of neutrophils and neutrophil chemoattractants such as CXCL1 and CXCL2. In SphK1-/- that had a similar level of injury compared to SphK+/+ mice, there was no change in number of neutrophils infiltrating the kidney or in neutrophil chemoattractants as compared to the SphK2tr/tr mice. Activation of SphK is likely to increase S1P levels and in its absence there may be an increase in ceramide/sphingosine levels. Because these sphingolipid metabolites have opposite actions, the concept of a ‘sphingolipid rheostat’ has been developed that addresses the importance of balance of these mediators and not the absolute amount of metabolites in determining the ultimate cell fate.31 S1P inhibits transendothelial migration of neutrophils and is thought to be an effective motility regulator of human neutrophils32 through a reduction in neutrophil chemoattractants, CXLC1 (homologue of human interleukin-8)33,34 and CXCL2.35 Thus, our findings that kidney IRI in SphK2tr/tr mice (and presumably a decrease in S1P in critical areas) was associated with an increase in kidney injury, neutrophil infiltration, and increased CXCL1 and CXCL2 expression are consistent with these reports.
S1P, produced by SphK, mainly functions by binding to S1P receptors. Activation of S1P1 has been recently shown to reduce kidney IRI.13,14 Activated S1P1-induced peripheral lymphopenia with a subsequent decrease in T-cell infiltration into kidney is thought to be important. In our study, we found S1P1 and also S1P3 gene expression increased markedly in SphK2tr/tr mice. Although the increase in S1P1 may serve to reduce injury, the increase in S1P3 may not. Although S1P activation of S1P1 mediates cortical actin reassembly by Rac activation and leads to endothelial barrier enhancement, S1P3 activation in pulmonary epithelial cells leads to disruption of tight junctions, possibly by activating Rho, resulting in increased lung vascular permeability.22,36,37 S1P3 KO mice however were protected.22 Thus, it may be possible that S1P3 activation after kidney IRI negatively regulates endothelial or epithelial barrier function and that marked increase of S1P3 in SphK2tr/tr mice might be responsible for worse renal injury. In support of this possibility we have observed that the protective effect of FTY720 reduces kidney IRI and vascular permeability through the activation of S1P1 receptors.13 However, the protective effect of FTY720 on kidney IRI and vascular permeability is lost with high doses of FTY720 that may in part be due to effects on S1P3 receptors (M.D. Okusa and A.E. Awad). To directly test this possibility we performed kidney IRI in mice and found that these mice were protected from injury and the ischemic kidneys demonstrated reduced vascular permeability compared to mice. Thus, the increase in S1P3 expression following kidney IRI in SphK2tr/tr mice could have led to enhanced injury.
FTY720 is phosphorylated in vivo by SphK2 to form the active principle, FTY720-P.20 Although necessary for FTY720 induced lymphopenia,38-40 the importance of FTY720 phosphorylation in reducing kidney IRI has not previously been tested. We found that FTY720 did not have a protective effect in SphK2tr/tr or in SphK2+tr confirming the necessity of SphK2 in mediating FTY720-induced protection in kidney IRI. This further confirms the possibility that S1P receptor activation is necessary for the protective effects of S1P and/or its receptor agonist, FTY720.
In summary, we showed that the absence of SphK2 was associated with more severe kidney damage after IR. This finding suggests that despite minimal regulation of SphK2 after IR, constitutive expression of SphK2 is essential for cell-survival pathways after injury. However, the absence of SphK2 was not associated with increased apoptotic cell death (measured by TUNEL and western blot for cleaved caspase 3), but appeared to be important in neutrophil infiltration into inflamed kidneys through an increase in neutrophil chemoattractants. Furthermore, the absence of SphK2 enhanced expression of kidney S1P3 following IRI and increased vascular permeability following IRI, an observation that could be in part responsible for the increase in injury. In addition, SphK2 is thought to be important in mediating the kidney-protective effects of FTY720, a novel immune modulator. Sphingolipids are important constituents in injury associated with kidney IRI necessitating further studies to demonstrate the precise role of SphK, S1P, and S1P receptors in kidney IRI. More refined knowledge on the role of sphingolipids in kidney will likely lead to development of new therapeutic drugs for treatment of ischemic acute kidney injury in humans.
MATERIALS AND METHODS
Animals and IRI experimental protocol
All experiments were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals. We used C57BL/6 mice (∼20 g, between 6 and 8 weeks of age; Charles River Laboratories, Wilmington, MA, USA), SphKtr/tr and SphK2+/tr mice (129/Sv X C57BL/6) were generated and provided by Dr Kevin R. Lynch (University of Virginia).20 SphK1-/- and mice (congenic on C57BL/6) were provided by Dr Richard L. Proia (National Institutes of Health). For SphK1-/- mice, C57BL/6 mice served as WT controls. Bilateral flank incisions were performed as previously described.41 Mice were anesthetized and were placed on a thermoregulated pad to maintain body temperature at 37 °C. Both renal pedicles were exposed and cross-clamped for 32 (SphK1/2 mice) and 26 min (S1P3 mice), and then clamps were released to allow for reperfusion for different times. In sham-operated mice, renal pedicles were exposed but not clamped. At various time points after reperfusion, blood was collected and tissues were processed for molecular and histological examination.
Drug treatment
Three doses (24, 20, and 1 h before ischemia) of FTY720 (48 μg/kg, i.p.; Novartis, Basel, Switzerland) or vehicle (3% fatty acid-free bovine serum albumin/phosphate-buffered saline solution) (Sigma, St Louis, MO) were administered. S1P (Biomol International, Plymouth Meeting, PA) from methanol stocks was air-dried and dissolved in phosphate-buffered saline/1% bovine serum albumin and administered (50 ng/kg, i.v.) 1 h before ischemia.
Biochemical and histological examination
Plasma creatinine was determined using a colorimetric assay according to the manufacturer’s protocol (Sigma). For histological examination, 4-μm sections of paraformaldehyde-fixed (4%) and paraffin-embedded kidney tissues were stained with hematoxylin and eosin. Briefly, semiquantitatively outer medulla tubular damage was estimated in 8-10 high-power fields (× 200 magnification) per section by using a scoring system based on the percentage of damaged tubules per field (1≤25%; 2=25-50%; 3=50-75%; and 4≥75%), and the mean scores of each mouse were compared. Apoptotic cells in the kidney were detected using ApopTag Plus (Intergen, Purchase, NY) on paraffin-embedded kidney tissue sections following the manufacturer’s protocol. The number of apoptotic cells in the outer medulla and cortex was measured semiquantitatively by counting 8-10 high-power fields (× 200) per section, and the mean number of TUNEL-positive cells was compared between groups.
Neutrophil labeling of kidney sections and quantification
Paraffin sections (5 μm) were labeled with a rat monoclonal antibody to murine neutrophils (clone 7/4; Caltag Laboratories, Carlsbad, CA, USA, catalog no. RM6500 at 1:100) as previously described.42 Photographs, taken and adjusted for brightness/contrast with a SPOT RT camera (software version 3.3; Diagnostic Instruments), were stored as TIF files and analyzed using Adobe Photoshop CS2. Use of Photoshop allows the color of any individual pixel to be sampled and set as foreground color. The select/color range tools allow all pixels in the image having a color similar to the foreground color to be selected and counted using the histogram tool. An average of 5-6 different areas and 3-4 different mice were analyzed and averaged.
Analysis of kidney vascular permeability
Changes in vascular permeability were assessed by measuring extravasation of Evan’s blue dye into the kidney tissue as previously described.13
Measurement of SphK activity
Kidney SphK activity was assessed as previously described.20 Briefly, kidney homogenates were prepared and to determine the fractional activity of SphK1 and SphK2, the buffer was supplemented with either 0.05% Triton X-100 or 1 m KCl, respectively. Radiolabeled enzyme products were detected by autoradiography and identified by migration relative to authentic standards. For quantification, the silica gel containing radiolabeled lipid was scraped into a scintillation vial and counted.
Real-time RT-PCR
Total RNA and cDNA synthesis was performed as previously described.13 Subsequently, real-time RT-PCR was run on the MyCycler system (Bio-Rad, Hercules, CA, USA) with gene-specific primer sets of mouse tumor necrosis factor-α (catalog no. PPM03113), interleukin-6 (catalog no. PPM03015), monocyte chemoattractant protein-1 (PPM03151), and glyceraldehyde-3-phosphate dehydrogenase (catalog no. PPM02946) developed by SuperArray Bioscience Corporation (Frederick, MD). Primer sequences for mouse SphK1, SphK2, S1P1, S1P3, CXCL1, CXCL2, and CXCL5 are shown in Table 1. The abundance of gene expression was normalized to that of GAPDH.
Table 1. Primer pairs for RT-PCR.
| Gene | Forward primer (5′→3′) | Reverse primer (5′→3′) |
|---|---|---|
| S1P1 | TTCTCATCTGCTGCTTCATCATCC | GGTCCGAGAGGGCTAGGTTG |
| S1P3 | GCGTGTTCCTTCTGATTGG | GCAAGATGGTAGAGCAGTC |
| SphK1 | GGAACTTGACTGTCCATACC | TACCATCAGCTCTCCATCC |
| SphK2 | GCACGGCGAGTTTGGTTC | GAGACCTCATCCAGAGAGACTAG |
| CXCL1 | TGGCTGGGATTCACCTCAAGAACA | TGTGGCTATGACTTCGGTTTGGGT |
| CXCL2 | AAAGTTTGCCTTGACCCTGAAGCC | TTTCCAGGTCAGTTAGCCTTGCCT |
| CXCL5 | AATGCACTCGCAGTGGAAAGAACG | TGAGCAGGAAGCTTCAGGGACAAT |
Statistical analysis
Data were analyzed by SigmaStat 2.03 (San Jose, CA, USA) and GraphPad InStat 3 presented as mean±s.e.m. Analysis was carried out by using unpaired t-test or analysis of variance, and a P-value less than 0.05 was considered statistically significant.
ACKNOWLEDGMENTS
We gratefully acknowledge Michael Rouse, MS (Dept of Medicine) for expert technical assistance and helpful discussions, Dr Diane Rosin (Dept of Pharmacology) for careful reading of the paper and helpful discussions, Dr Richard Prioa (NIH) for providing the SphK1-/- and mice, and Dr V. Brinkmann (Novartis) for providing FTY720. This work was supported by grants from the National Institutes of Health PO1 073361, RO1 DK62324, RO1 DK065957, RO1 HL070065, and R01 GM067958.
Footnotes
DISCLOSURE
All the authors declared no competing interests.
References
- 1.Star RA. Treatment of acute renal failure. Kidney Int. 1998;54:1817–1831. doi: 10.1046/j.1523-1755.1998.00210.x. [DOI] [PubMed] [Google Scholar]
- 2.Bonventre JV, Weinberg JM. Recent advances in the pathophysiology of ischemic acute renal failure. J Am Soc Nephrol. 2003;14:2199–2210. doi: 10.1097/01.asn.0000079785.13922.f6. [DOI] [PubMed] [Google Scholar]
- 3.Jo SK, Rosner MH, Okusa MD. Pharmacologic treatment of acute kidney injury. Why drugs haven’t: why drugs haven’t worked and what is on the horizon. Clin J Am Soc Nephrol. 2007;2:256–365. doi: 10.2215/CJN.03280906. [DOI] [PubMed] [Google Scholar]
- 4.Spiegel S, Milstien S. Functions of a new family of sphingosine-1-phosphate receptors. Biochim Biophys Acta. 2000;1484:107–116. doi: 10.1016/s1388-1981(00)00010-x. [DOI] [PubMed] [Google Scholar]
- 5.Morita Y, Perez GI, Paris F, et al. Oocyte apoptosis is suppressed by disruption of the acid sphingomyelinase gene or by sphingoine-1-phosphate therapy. Nat. Med. 2000;6:1109–1114. doi: 10.1038/80442. [DOI] [PubMed] [Google Scholar]
- 6.Rosenfeldt HM, Hobson JP, Maceyka M, et al. EDG-1 links the PDGF receptor to Src and focal adhesion kinase activation leading to lamellipodia formation and cell migration. FASEB J. 2001;15:2649–2659. doi: 10.1096/fj.01-0523com. [DOI] [PubMed] [Google Scholar]
- 7.English D, Welch Z, Kovala AT, et al. Sphingosine 1-phosphate released from platelets during clotting accounts for the potent endothelial cell chemotactic activity of blood serum and provides a novel link between hemostasis and angiogenesis. FASEB J. 2000;14:2255–2265. doi: 10.1096/fj.00-0134com. [DOI] [PubMed] [Google Scholar]
- 8.An S, Zheng Y, Bleu T. Sphingosine 1-phosphate-induced cell proliferation, survival, and related signaling events mediated by G protein-coupled receptors Edg3 and Edg5. J Biol Chem. 2000;275:288–296. doi: 10.1074/jbc.275.1.288. [DOI] [PubMed] [Google Scholar]
- 9.Donati C, Cencetti F, Nincheri P, et al. Sphingosine 1-phosphate mediates proliferation and survival of mesoangioblasts. Stem Cells. 2007;25:1713–1719. doi: 10.1634/stemcells.2006-0725. [DOI] [PubMed] [Google Scholar]
- 10.Goetzl EJ, Kong Y, Voice JK. Cutting edge: differential constitutive expression of functional receptors for lysophosphatidic acid by human blood lymphocytes. J Immunol. 2000;164:4996–4999. doi: 10.4049/jimmunol.164.10.4996. [DOI] [PubMed] [Google Scholar]
- 11.Karliner JS, Honbo N, Summers K, et al. The lysophospholipids sphingosine-1-phosphate and lysophosphatidic acid enhance survival during hypoxia in neonatal rat cardiac myocytes. J Mol Cell Cardiol. 2001;33:1713–1717. doi: 10.1006/jmcc.2001.1429. [DOI] [PubMed] [Google Scholar]
- 12.Maceyka M, Payne SG, Milstien S, et al. Sphingosine kinase, sphingosine-1-phosphate, and apoptosis. Biochim Biophys Acta. 2002;1585:193–201. doi: 10.1016/s1388-1981(02)00341-4. [DOI] [PubMed] [Google Scholar]
- 13.Awad AS, Ye H, Huang L, et al. Selective sphingosine 1-phosphate 1 (S1P1) receptor activation reduces ischemia-reperfusion injury in mouse kidney. Am J Physiol Renal Physiol. 2006;290:F1516–F1524. doi: 10.1152/ajprenal.00311.2005. [DOI] [PubMed] [Google Scholar]
- 14.Lien YH, Yong KC, Cho C, et al. S1P(1)-selective agonist, SEW2871, ameliorates ischemic acute renal failure. Kidney Int. 2006;69:1601–1608. doi: 10.1038/sj.ki.5000360. [DOI] [PubMed] [Google Scholar]
- 15.Olivera A, Kohama T, Edsall L, et al. Sphingosine kinase expression increases intracellular sphingosine-1-phosphate and promotes cell growth and survival. J Cell Biol. 1999;147:545–558. doi: 10.1083/jcb.147.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Igarashi N, Okada T, Hayashi S, et al. Sphingosine kinase 2 is a nuclear protein and inhibits DNA synthesis. J Biol Chem. 2003;278:46832–46839. doi: 10.1074/jbc.M306577200. [DOI] [PubMed] [Google Scholar]
- 17.Edsall LC, Cuvillier O, Twitty S, et al. Sphingosine kinase expression regulates apoptosis and caspase activation in PC12 cells. J Neurochem. 2001;76:1573–1584. doi: 10.1046/j.1471-4159.2001.00164.x. [DOI] [PubMed] [Google Scholar]
- 18.Maceyka M, Sankala H, Hait NC, et al. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J Biol Chem. 2005;280:37118–37129. doi: 10.1074/jbc.M502207200. [DOI] [PubMed] [Google Scholar]
- 19.Liu H, Toman RE, Goparaju SK, et al. Sphingosine kinase type 2 is a putative BH3-only protein that induces apoptosis. J Biol Chem. 2003;278:40330–40336. doi: 10.1074/jbc.M304455200. [DOI] [PubMed] [Google Scholar]
- 20.Kharel Y, Lee S, Snyder AH, et al. Sphingosine kinase 2 is required for modulation of lymphocyte traffic by FTY720. J Biol Chem. 2005;280:36865–36872. doi: 10.1074/jbc.M506293200. [DOI] [PubMed] [Google Scholar]
- 21.Mizugishi K, Yamashita T, Olivera A, et al. Essential role for sphingosine kinases in neural and vascular development. Mol Cell Biol. 2005;25:11113–11121. doi: 10.1128/MCB.25.24.11113-11121.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gon Y, Wood MR, Kiosses WB, et al. S1P3 receptor-induced reorganization of epithelial tight junctions compromises lung barrier integrity and is potentiated by TNF. Proc Natl Acad Sci USA. 2005;102:9270–9275. doi: 10.1073/pnas.0501997102. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Goetzl EJ, Lee H, Dolezalova H, et al. Mechanisms of lysolipid phosphate effects on cellular survival and proliferation. Ann N Y Acad Sci. 2000;905:177–187. doi: 10.1111/j.1749-6632.2000.tb06549.x. [DOI] [PubMed] [Google Scholar]
- 24.Xia P, Gamble JR, Wang L, et al. An oncogenic role of sphingosine kinase. Curr Biol. 2000;10:1527–1530. doi: 10.1016/s0960-9822(00)00834-4. [DOI] [PubMed] [Google Scholar]
- 25.Kusner DJ, Thompson CR, Melrose NA, et al. The localization and activity of sphingosine kinase 1 are coordinately regulated with actin cytoskeletal dynamics in macrophages. J Biol Chem. 2007;282:23147–23162. doi: 10.1074/jbc.M700193200. [DOI] [PubMed] [Google Scholar]
- 26.Sutton TA, Mang HE, Campos SB, et al. Injury of the renal microvascular endothelium alters barrier function after ischemia. Am J Physiol Renal Physiol. 2003;285:F191–F198. doi: 10.1152/ajprenal.00042.2003. [DOI] [PubMed] [Google Scholar]
- 27.Schwartz N, Hosford M, Sandoval RM, et al. Ischemia activates actin depolymerizing factor: role in proximal tubule microvillar actin alterations. Am J Physiol. 1999;276:F544–F551. doi: 10.1152/ajprenal.1999.276.4.F544. [DOI] [PubMed] [Google Scholar]
- 28.Li L, Okusa MD. Blocking the Immune response in ischemic acute kidney injury: the role of adenosine 2A agonists. Nat Clin Pract Nephrol. 2006;2:432–444. doi: 10.1038/ncpneph0238. [DOI] [PubMed] [Google Scholar]
- 29.Rabb H. The T cell as a bridge between innate and adaptive immune systems: implications for the kidney. Kidney Int. 2002;61:1935–1946. doi: 10.1046/j.1523-1755.2002.00378.x. [DOI] [PubMed] [Google Scholar]
- 30.Zemann B, Urtz N, Reuschel R, et al. Normal neutrophil functions in sphingosine kinase type 1 and 2 knockout mice. Immunol Lett. 2007;109:56–63. doi: 10.1016/j.imlet.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 31.Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003;4:397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- 32.Kawa S, Kimura S, Hakomori S, et al. Inhibition of chemotactic motility and trans-endothelial migration of human neutrophils by sphingosine 1-phosphate. FEBS Lett. 1997;420:196–200. doi: 10.1016/s0014-5793(97)01516-0. [DOI] [PubMed] [Google Scholar]
- 33.Wang L, Cummings R, Usatyuk P, et al. Involvement of phospholipases D1 and D2 in sphingosine 1-phosphate-induced ERK (extracellular-signal-regulated kinase) activation and interleukin-8 secretion in human bronchial epithelial cells. Biochem J. 2002;367:751–760. doi: 10.1042/BJ20020586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cummings RJ, Parinandi NL, Zaiman A, et al. Phospholipase D activation by sphingosine 1-phosphate regulates interleukin-8 secretion in human bronchial epithelial cells. J Biol Chem. 2002;277:30227–30235. doi: 10.1074/jbc.M111078200. [DOI] [PubMed] [Google Scholar]
- 35.Man K, Ng KT, Lee TK, et al. FTY720 attenuates hepatic ischemia-reperfusion injury in normal and cirrhotic livers. Am J Transplant. 2005;5:40–49. doi: 10.1111/j.1600-6143.2004.00642.x. [DOI] [PubMed] [Google Scholar]
- 36.Garcia JG, Liu F, Verin AD, et al. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest. 2001;108:689–701. doi: 10.1172/JCI12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y, Wada R, Yamashita T, et al. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J Clin Invest. 2000;106:951–961. doi: 10.1172/JCI10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mandala S, Hajdu R, Bergstrom J, et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 2002;296:346–349. doi: 10.1126/science.1070238. [DOI] [PubMed] [Google Scholar]
- 39.Pinschewer DD, Ochsenbein AF, Odermatt B, et al. FTY720 immunosuppression impairs effector T cell peripheral homing without affecting induction, expansion, and memory. J Immunol. 2000;164:5761–5770. doi: 10.4049/jimmunol.164.11.5761. [DOI] [PubMed] [Google Scholar]
- 40.Yanagawa Y, Sugahara K, Kataoka H, et al. FTY720, a novel immunosuppressant, induces sequestration of circulating mature lymphocytes by acceleration of lymphocyte homing in rats. II. FTY720 prolongs skin allograft survival by decreasing T cell infiltration into grafts but not cytokine production in vivo. J Immunol. 1998;160:5493–5499. [PubMed] [Google Scholar]
- 41.Day Y-J, Huang L, McDuffie MJ, et al. Renal protection from ischemia mediated by A2A adenosine receptors on bone marrow-derived cells. J Clin Invest. 2003;112:883–891. doi: 10.1172/JCI15483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Day Y-J, Huang L, Ye H, et al. Renal ischemia-reperfusion and adenosine 2A receptor-mediated tissue protection: the role of CD4+ T cells and inteferon gamma. J Immunol. 2006;176:3108–3114. doi: 10.4049/jimmunol.176.5.3108. [DOI] [PubMed] [Google Scholar]