

Abstract
Transbilayer movement, or flip-flop, of lipids across the endoplasmic reticulum (ER) is required for membrane biogenesis, protein glycosylation, and GPI anchoring. Specific ER membrane proteins, flippases, are proposed to facilitate lipid flip-flop, but no ER flippase has been biochemically identified. The glycolipid Glc3Man9GlcNAc2-PP-dolichol is the oligosaccharide donor for protein N-glycosylation reactions in the ER lumen. Synthesis of Glc3Man9GlcNAc2-PP-dolichol is initiated on the cytoplasmic side of the ER and completed on the lumenal side, requiring flipping of the intermediate Man5GlcNAc2-PP-dolichol (M5-DLO) across the ER. Here we report the reconstitution of M5-DLO flipping in proteoliposomes generated from Triton X-100-extracted Saccharomyces cerevisiae microsomal proteins. Flipping was assayed by using the lectin Concanavalin A to capture M5-DLOs that had been translocated from the inner to the outer leaflet of the vesicles. M5-DLO flipping in the reconstituted system was ATP-independent and trypsin-sensitive and required a membrane protein(s) that sedimented at ∼4 S. Man7GlcNAc2-PP-dolichol, a higher-order lipid intermediate, was flipped >10-fold more slowly than M5-DLO at 25 °C. Chromatography on Cibacron Blue dye resin enriched M5-DLO flippase activity ∼5-fold and resolved it from both the ER glycerophospholipid flippase activity and the genetically identified flippase candidate Rft1 [Helenius, J., et al. (2002) Nature 415, 447−450]. The latter result indicates that Rft1 is not the M5-DLO flippase. Our data (i) demonstrate that the ER has at least two distinct flippase proteins, each specifically capable of translocating a class of phospholipid, and (ii) provide, for the first time, a biochemical means of identifying the M5-DLO flippase.
Protein N-glycosylation is catalyzed by oligosaccharyltransferase (OST)1 in the lumen of the endoplasmic reticulum (ER) (1–6). OST transfers a 14-sugar oligosaccharide from the glycolipid Glc3Man9GlcNAc2-PP-dolichol to select asparagine residues in the nascent protein. The newly formed N-glycoprotein is typically packaged into secretory vesicles for export from the ER while the lipid product of the OST reaction (dolichol-PP) is recycled (6,7).
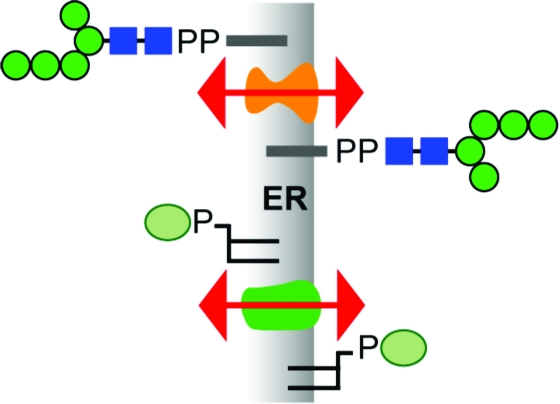
The biosynthesis of Glc3Man9GlcNAc2-PP-dolichol is a multistep process that is topologically split across the ER membrane (Figure 1)3,4. The first seven biosynthetic reactions, i.e., conversion of dolichol-P to Man5GlcNAc2-PP-dolichol (M5-DLO), occur on the cytoplasmic face of the ER, whereas the last seven reactions leading to the synthesis of Glc3Man9GlcNAc2-PP-dolichol occur on the lumenal face. This implies that M5-DLO must be translocated from the cytoplasmic face to the lumenal face of the ER membrane (8) (Figure 1). The lumenally oriented, membrane-bound glycosyltransferases responsible for elaborating M5-DLO to Glc3Man9GlcNAc2-PP-dolichol use dolichol-P-mannose and dolichol-P-glucose as sugar donors (3–6). Since these glycolipids are synthesized from dolichol-P and the corresponding nucleotide sugar on the cytoplasmic face of the ER, they too must be translocated across the ER membrane.
Figure 1.

Topology model for the dolichol pathway. The assembly of the dolichol-linked oligosaccharide (DLO) precursor of protein N-glycans is initiated on the cytoplasmic face of the ER when GlcNAc-P is transferred from UDP-GlcNAc to dolichol phosphate. Synthesis continues with the addition of another GlcNAc residue (from UDP-GlcNAc) to generate GlcNAc2-PP-dolichol and five mannose residues (from GDP-mannose) to generate the branched structure Man5GlcNAc2-PP-dolichol (M5-DLO). M5-DLO is flipped to the lumenal face of the ER where it is extended by four mannose residues (from dolichol-P-mannose) and three glucose residues (from dolichol-P-glucose) to yield mature DLO (Glc3Man9GlcNAc2-PP-dolichol). This lipid is the oligosaccharide donor for the protein N-glycosylation reaction catalyzed by oligosaccharyltransferase (OST) in the ER lumen. Flipping of M5-DLO across the ER is depicted as bidirectional, consistent with data presented in this paper.
Translocation of glycerophospholipids, M5-DLO, dolichol-P-mannose, and dolichol-P-glucose across the ER is required for ER membrane biogenesis, protein N-, O-, and C-glycosylation, and GPI anchoring (6,9,10). Flipping of polar lipids is energetically unfavorable and does not occur at a physiologically sufficient rate in protein-free liposomes (11–13); the energy barrier to flipping in liposomes is estimated to be 20−50 kcal/mol for glycerophospholipids (11) and considerably greater for M5-DLO [130−260 kcal/mol (4)]. However, phosphatidylcholine and other glycerophospholipids flip-flop rapidly across the ER membrane with an equilibration half-time of tens of seconds (14,15), and dolichol-based glycolipids must similarly translocate rapidly to satisfy the requirement for glycoprotein biosynthesis in vivo (13). To account for this, it has been proposed that lipid flip-flop in the ER is facilitated by specific membrane proteins or flippases. ER lipid translocation events are typically ATP-independent (14–25), indicating that ER flippases must be facilitators of transverse diffusion rather than pumps that translocate lipids in only one direction (9,26).2 This key feature distinguishes their activity from that of ABC transporters and P-type ATPases that have been proposed to translocate lipids at the plasma membrane9,26. Although ER flippases have yet to be biochemically identified, biochemical reconstitution data point to a role for specific proteins in flipping glycerophospholipids (21–24), GPIs (20), and dolichol-P-mannose (25). These precedents suggest the hypothesis that M5-DLO is flipped across the ER by a specific protein in an ATP-independent manner.
A genetic screen for analyzing the role of the unfolded protein response (UPR) pathway in the yeast Saccharomyces cerevisiae identified a mutant (per5-1) with a glycosylation defect; the mutant cells accumulated M5-DLO and hypoglycosylated the N-glycoprotein carboxypeptidase Y (CPY) (27). Additional genetic experiments concluded that the phenotype of per5-1 was due to a defect in M5-DLO flipping and that the essential PER5/RFT1 gene (28) encoded a polytopic membrane protein, Rft1, that was directly required for flipping M5-DLO (29). Rft1’s ability to flip M5-DLO was not biochemically tested in the latter study.
We now report the functional reconstitution of M5-DLO flipping in large unilamellar proteoliposomes generated from Triton X-100-solubilized S. cerevisiae ER membrane proteins. Using two distinct fractionation criteria, we resolve M5-DLO flippase activity from that of the flippase responsible for translocating glycerophospholipids in the ER. We also show that Rft1 plays no role in M5-DLO flipping in the reconstituted system. Our data (i) demonstrate that the ER has at least two distinct flippase proteins, each specifically capable of translocating a class of phospholipid, and (ii) provide, for the first time, a biochemical means of identifying the M5-DLO flippase.
Experimental Procedures
Materials
Egg phosphatidylcholine (ePC) and 1-acyl-2-[6-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]hexanoyl-sn-glycero-3-phosphocholine (NBD-PC) were from Avanti Polar Lipids. Bovine serum albumin (BSA), Concanavalin A (Con A), PMSF, DTT, Arthrobacter luteus lyticase, Fast-flow Cibacron blue 3GA resin, and yeast mannan were from Sigma Chemical Co. ULTROL-grade Triton X-100 (TX-100) was from Roche Molecular Biochemicals. The micro BCA protein assay kit and BSA protein standard were from Pierce. Protease Inhibitor cocktail (set 1) was from Calbiochem. [2-3H]Mannose and [3H]phosphatidylcholine were from American Radiolabeled Chemicals. Antibodies against S. cerevisiae Sec61, Vph1, and Gas1 were gifts from T. Rapoport (Harvard Medical School, Boston, MA), P. Kane (State University of New York-Upstate Medical Center, Syracuse, NY), and A. Conzelmann (University of Fribourg, Fribourg, Switzerland), respectively. Antibodies against S. cerevisiae Dpm1 and Pep12 were from Molecular Probes. Horseradish peroxidase-coupled secondary antibodies were from Promega Corp. Standard laboratory chemicals were from Sigma Chemical Co.
Plasmids, Strains, and Growth Conditions
Plasmid pJH19, a multicopy YEp352-based URA3 plasmid (30) with RFT1* (encoding Rft1*, a fusion protein consisting of Rft1 fused at its C-terminus to Protein A) under control of the STT3 promoter, was a gift from J. Helenius and M. Aebi (ETH Zürich, Zurich, Switzerland). Strain YCF40 was obtained by sporulation and tetrad dissection of YG1128 (diploid strain heterozygous for RFT1) carrying plasmid pJH19. YCF40 was cultivated on selective medium lacking uracil at room temperature or 30 °C; for microsome preparation, large-scale cultures of YCF40 were inoculated in YPD medium at 30 °C. The yeast strains used in this paper are listed in Table 1. Standard yeast media and genetic techniques were used.
Table 1. Strains Used in This Work.
| strain | genotype | ref |
|---|---|---|
| SS328 | MATα ade2-101 his3Δ200 ura3-52 lys2-801 | 50 |
| YG248 | MATa ade2-101 ura3-52 his3Δ200 lys2-801 Δalg3::HIS3 | 51 |
| YG1056 | MATα ade2-101 ura3-52 his3Δ200 Δalg12::kanMX4 Δalg5::HIS3; SS328/SS330 strain background | Aebi strain collection |
| YG1057 | MATa ade2-201 ura3-52 his3Δ200 lys2-801 Δalg3::HIS3 Δalg5::HIS3; SS328/SS330 strain background | Aebi strain collection |
| YG1128 | MATa/α ade2-201/ade2-201 ura3-52/ura3-52 his3Δ200/his3Δ200 tyr1/+ lys2/+ Δrft1::HIS3/+; SS328/SS330 strain background | Aebi strain collection |
| YCF40a | MATα ade2-201 ura3-52 his3Δ200 tyr1 Δrft1::HIS3 pJH19 | this study |
| BJ2168 | MATa leu2 trp1 ura3-52 prb1-1122 pep4-3 prc1-407 gal2 | 52 |
The genomic copy of RFT1 is disrupted in strain YCF40; YCF40 contains URA3 plasmid pJH19 that encodes Rft1 tagged at its C-terminus with Protein A. We refer to this chimeric protein as Rft1*. YCF40 cells do not grow on minimal medium containing 5-fluoroorotic acid and uracil, indicating that the plasmid-encoded Rft1* protein is functional and required to complement the lack of RFT1 in the genome.
Radiolabeled Dolichol-Linked Oligosaccharides (DLOs)
Radiolabeled DLOs were isolated from [3H]mannose-labeled yeast as described previously (31). Strains YG248 and YG1057 were used to prepare [3H]M5-DLO, and YG1056 was used to prepare [3H]M7-DLO. [3H]DLOs were stored in a chloroform/methanol/water mixture (10:10:3, v/v/v) at −20 °C. The radiochemical purity of [3H]DLOs was assessed by thin layer chromatography (TLC) using Silica-60 plates (Merck) developed in a chloroform/methanol/water mixture (10:10:3, v/v/v). The chromatograms were visualized with a Berthold LB2842 TLC linear analyzer. The identity of [3H]DLOs was further verified by analyzing the [3H]oligosaccharide headgroup released by mild acid hydrolysis (32); these analyses were performed in H. Freeze’s laboratory (Burnham Institute, La Jolla, CA), using an HPLC method as described previously (33).
Yeast Microsomes and Preparation of the Triton X-100 Extract (TE)
Yeast microsomes were prepared from spheroplasts essentially as described previously (34). The spheroplasts were disrupted, and the resulting homogenate was clarified by centrifugation at low speed. The low-speed supernatant was further clarified (15000 rpm, Beckman JA-20 rotor, 10 min, 4 °C) and then centrifuged on a two-step sucrose gradient to generate “ER-enriched” microsomes. Alternatively, the low-speed supernatant was subjected to differential centrifugation in a Beckman JA-20 rotor (9000 rpm, 10 min, 4 °C, followed by centrifugation of the resulting supernatant at 20000 rpm for 30 min at 4 °C) to pellet microsomes. Microsomes prepared by either method were resuspended at ∼5 mg/mL in 20 mM Hepes-NaOH (pH 7.4) and 100 mM NaCl. The two types of microsome preparation were used interchangeably. A Triton extract (TE) was prepared by mixing the microsome suspension (on ice) with an equal volume of ice-cold buffer [20 mM Hepes-NaOH (pH 7.4) and 100 mM NaCl] containing 2% (w/v) Triton X-100. The mixture was left on ice for 45−60 min with occasional mixing before being centrifuged in a Beckman TLA 100.3 rotor at 70000 rpm (∼200000gav) for 1 h. The resulting supernatant (TE; protein concentration of typically ∼1 mg/mL) was either used directly for fractionation and/or reconstitution into proteoliposomes or snap-frozen and stored at −80 °C for future use. Snap-freezing and storage at −80 °C for several months did not cause any detectable loss of activity.
Reconstitution of Liposomes and Proteoliposomes
Proteoliposomes and liposomes were reconstituted as previously described (21–23,35,36). Briefly, 1 mL mixtures of TE, egg phosphatidylcholine (4.1 μmol), and ∼10000−20000 cpm of [3H]DLO in 20 mM Hepes-NaOH (pH 7.4), 100 mM NaCl, and 1% (w/v) Triton X-100 were treated with washed SM2 Bio-Beads for ∼18 h. The resulting vesicles were harvested by centrifugation in a Beckman TLA 100.3 rotor (70000 rpm, 4 °C, 60 min), resuspended in ∼250 μL of buffer [20 mM Hepes-NaOH (pH 7.4), 100 mM NaCl, 3 mM MgCl2, 1 mM CaCl2, and 1 mM MnCl2] and kept on ice until they were used. The amount of TE used was varied as required to generate proteoliposomes with different protein:phospholipid ratios; TE was omitted when preparing protein-free liposomes. All the reconstitution experiments reported in this paper were conducted with ePC since the results we obtained were indistinguishable from those obtained using mixtures of lipids, specifically, mixtures of ePC and egg phosphatidylethanolamine (molar ratio of 4:1 or 3:2) or ePC, egg phosphatidylethanolamine, phosphatidylinositol, and phosphatidylserine (molar ratio of 50:35:8:7).
Reconstitution of membrane proteins into proteoliposomes was verified by flotation in a sucrose step gradient (37). The protein:phospholipid ratio (PPR) of proteoliposome samples was determined as described previously (38–40). Proteoliposomes and liposomes were visualized by cryo-electron microscopy. For this, samples were deposited on a Cu 300 mesh (Quantifoil R2/4), blotted, and rapidly frozen by plunging the grid into liquid ethane in a humidity-controlled atmosphere. The frozen grid was visualized using a Tecnai G2 F20 electron microscope equipped with a 200 kV field emission gun.
Assay for [3H]DLO Flipping in Proteoliposomes
Three 20 μL aliquots were taken from each reconstituted sample and mixed with 40 μL of either ice-cold buffer [20 mM Hepes-NaOH (pH 7.4), 100 mM NaCl, 3 mM MgCl2, 1 mM CaCl2, and 1 mM MnCl2], buffer containing Con A (3.6 mg/mL), or buffer containing Con A and 1.5% (w/v) Triton X-100. An additional control sample containing a 10 μL aliquot of vesicles was mixed with buffer containing 3.6 mg/mL Con A and 1.5% (w/v) Triton X-100; results obtained with this sample were compared with those obtained with the sample containing 20 μL of vesicles to verify that the amount of Con A used was sufficient to bind all the [3H]DLO present. The samples were incubated for 30 min on ice before addition of 140 μL of ice-cold blocking solution (20 mg/mL BSA and 2 mg/mL yeast mannan) and 1.4 mL of an ice-cold chloroform/methanol mixture (1:1, v/v) to produce a final chloroform:methanol:water ratio of 10:10:3 (v/v/v). After being vigorously vortexed, the samples were centrifuged (10 min, 4 °C, 16000gmax), and the supernatant in each case was transferred to a scintillation vial, dried, and dissolved in 200 μL of 1% (w/v) SDS. The pellets were air-dried and then solubilized with 200 μL of 1% (w/v) SDS.
Radioactivity in the supernatant and pellet of each of the three samples (“buffer only”, “Con A”, and “Con A + Triton X-100”) was determined by liquid scintillation counting. The fraction of total radioactivity in the pellet [pellet/(pellet + supernatant)] was calculated, and the value for the buffer only sample was subtracted from the values obtained for the Con A and Con A + Triton X-100 samples. The percent of [3H]DLO accessible to Con A in intact vesicles was calculated by dividing the corrected value for the Con A sample by the corrected value for the Con A + Triton X-100 sample.
Flippase activity was calculated as follows. Specific activity = [(percent [3H]DLO accessible to Con A in proteoliposomes) − (percent [3H]DLO accessible to Con A in liposomes)]/(PPR of proteoliposomes); specific activity measurements were conducted with proteoliposomes prepared in a PPR range such that the percent of [3H]DLO accessible to Con A in intact vesicles was 60−70%. Total activity in a protein fraction = (specific activity)(protein content of the fraction).
Assay for Glycerophospholipid Flipping in Proteoliposomes
1-Acyl-2-C6NBD-PC (NBD-PC) was used as a reporter for glycerophospholipid flipping. Trace amounts (∼0.3 mol %) of NBD-PC were included during vesicle reconstitution, and flip-flop was assayed by a fluorescence reduction method as described previously (23,24).
Glycerol Gradient Analysis of TE
TE (500 μL) was loaded onto a 3.8 mL glycerol gradient [5 to 25% (w/v)] prepared in 1% (w/v) Triton X-100, 100 mM NaCl, and 20 mM Hepes-NaOH (pH 7.4). Sedimentation standards [ovalbumin (3.6 S), BSA (4.2 S), β-amylase (8.9 S), and catalase (11.4 S)] were analyzed in a parallel gradient. The gradients were centrifuged in a Beckman MLS 50 rotor at 35000 rpm (∼165000gav) for 20 h at 4 °C, and fractions (300 μL) were collected from the top. Aliquots of each fraction were taken for refractive index measurement and protein determination using the Kaplan-Pedersen method (38). The fractions were desalted on Bio-Gel P6 spin columns to remove glycerol, and 150 μL aliquots were reconstituted into proteoliposomes and assayed for M5-DLO and NBD-PC flippase activity.
Chromatography of TE on Cibacron Blue Dye Resin
TE (500 μL) was adjusted to 20 mM Hepes-NaOH (pH 7.4), 50 mM NaCl, and 1% (w/v) Triton X-100 and incubated with Fast-flow Cibacron blue 3GA resin (300 μL bed volume) that had been pre-equilibrated with the same buffer. The sample was rotated end over end for 1 h at 4 °C, after which unbound material was collected and the resin was washed with the equilibration buffer. Bound proteins were eluted with salt in six sequential steps corresponding to 100 mM, 250 mM, 400 mM, 500 mM, 750 mM, and 1 M NaCl [all prepared in 20 mM Hepes-NaOH (pH 7.4) and 1% (w/v) Triton X-100]. Eluted material was adjusted to 20 mM Hepes-NaOH (pH 7.4), 100 mM NaCl, and 1% (w/v) Triton X-100 using Bio-Gel P6 spin columns. The protein content of the load, unbound, and salt-eluted fractions was determined by the Kaplan-Pedersen method (38). All fractions were reconstituted and tested for M5-DLO and NBD-PC flippase activity.
Results and Discussion
Assay for DLO Flipping in Proteoliposomes
We developed an assay for M5-DLO flipping based on the observation that the mannose-binding lectin Concanavalin A (Con A) has a high affinity for DLOs containing four or more mannose residues and that the M5-DLO−lectin complex thus formed is not soluble in organic solvents (8,41). We reasoned that if M5-DLO-containing liposomes are incubated with Con A, DLOs located in the outer leaflet of the vesicles will interact with the lectin whereas those in the inner leaflet will not. Upon being exposed to organic solvent, the DLO−lectin complexes formed on the outer leaflet will be precipitated whereas free M5-DLOs from the inner leaflet will be extracted (Figure 2A, top panel); the same result is predicted for proteoliposomes reconstituted with irrelevant membrane proteins, i.e., those that lack flippase activity. Thus, in intact, flippase-deficient vesicles with M5-DLO molecules symmetrically distributed between the two leaflets of the bilayer, ∼50% of the M5-DLO should be captured by Con A. However, in flippase-equipped proteoliposomes, M5-DLOs will flip back and forth across the membrane. Incubation of such vesicles with Con A will result in ∼100% of the M5-DLOs being captured by the lectin (Figure 2A, bottom panel; we note that since the binding reaction is essentially irreversible it is unlikely that M5-DLO molecules that are captured by Con A will return to the inner leaflet). For a mixed population of vesicles in which some vesicles contain a flippase and others do not, the percent capture will be intermediate between 50 and 100%, the exact value reflecting the proportion of flippase-containing vesicles in the ensemble. Thus, by exploiting the ability of Con A to capture M5-DLOs in the outer leaflet, the assay is able to distinguish between inactive vesicles and those that are equipped with DLO flippase.
Figure 2.

Strategy for assaying DLO translocation in reconstituted vesicles. (A) Assay for assessing DLO flipping in reconstituted vesicles. Unilamellar vesicles with [3H]DLO symmetrically distributed across the membrane are incubated with Con A. Con A captures DLO species that are initially located in the outer leaflet, as well as those that gain access to the outer leaflet after being translocated from the inner leaflet through the action of a flippase. After Con A binding is allowed to go to completion, the sample is extracted with organic solvent. DLOs that are bound to Con A precipitate with the protein, while free DLOs are extracted. In liposomes or inactive proteoliposomes (vesicles containing membrane proteins but not a flippase), 50% of the [3H]DLOs are expected to be captured by Con A, corresponding to the pool of DLO in the outer leaflet. [3H]DLOs in the inner leaflet of these vesicles cannot access Con A and are extracted by the solvent (top panel). For proteoliposomes containing a flippase (bottom panel), Con A binds all [3H]DLOs since those originally in the inner leaflet are flipped to the outer leaflet and captured by the lectin; thus, ∼100% of the DLOs are expected to be precipitated with Con A in this situation. In mixtures of vesicles in which some possess a flippase while others do not, the percent of [3H]DLO captured by Con A is predicted to be intermediate between 50 and 100%, reflecting the proportion of flippase-containing vesicles in the population. (B) Structures of M5-DLO and M7-DLO. The symbols used for dolichol, mannose, and GlcNAc are as described in the legend of Figure 1. The chemical structure of the oligosaccharide moiety of M5-DLO is Manα1−2Manα1−2Manα1−3(Manα1−6)Manβ1−4GlcNAcβ1−4GlcNAc; the oligosaccharide moiety of M7-DLO is Manα1−2Manα1−2Manα1−3(Manα1−2Manα1−3Manα1−6)Manβ1−4GlcNAcβ1−4GlcNAc. (C) Thin layer chromatography of [3H]M5-DLO and [3H]M7-DLO. Radiolabeled M5-DLO and M7-DLO were prepared from [3H]mannose-labeled yeast as described in . An aliquot of each preparation was analyzed by thin layer chromatography; chromatograms were visualized using a Berthold LB2842 radioactivity scanner. Each chromatogram contained a single peak of radioactivity; the relevant section of the chromatograms is shown.
In the majority of the experiments described here, we analyzed the translocation of M5-DLO; however, we also tested flipping of the higher-order intermediate M7-DLO to determine transport specificity. Both lipids bind Con A (8); their structure is shown in Figure 2B, and thin layer chromatographic profiles of [3H]M5-DLO and [3H]M7-DLO are shown in Figure 2C.
Reconstitution of Proteoliposomes from Triton X-100-Solubilized Yeast ER Membrane Proteins
We used a Triton X-100 extract (TE) of yeast microsomes as a source of flippase. When microsomes were incubated with ice-cold Triton X-100, ER membrane proteins such as Sec61 and Dpm1 were solubilized, whereas membrane proteins such as Gas1, Vph1, and Pep12 that correspond to other organelles (plasma membrane, vacuole, and Golgi/endosome, respectively) found in the microsome preparation were not (Figure 3A). We prepared TE from YCF40 cells (Table 1) that express Rft1*, a Protein A-tagged version of Rft1. Like other ER membrane proteins, Rft1* was quantitatively extracted with ice-cold Triton X-100 (Figure 3A). Thus, TE is highly enriched in ER membrane proteins.
Figure 3.

M5-DLO flipping in proteoliposomes reconstituted from TE. (A) Characterization of TE. Microsomes from strain YCF40 were extracted with Triton X-100 as described in . Soluble (TE) and insoluble (Insol) fractions were separated by ultracentrifugation; sample equivalents were analyzed by SDS−PAGE and immunoblotting using antibodies against organelle marker proteins: ER, Sec61, Dpm1, and Rft1*; vacuole, Vph1; plasma membrane, Gas1; and Golgi/endosomes, Pep12. The immunoblotting profile indicates that ER membrane proteins are selectively extracted into the TE. (B) Protein reconstitution verified by flotation analysis. Proteoliposomes (prepared using TE from YCF40 cells, as well as trace amounts of [3H]phosphatidylcholine) were permeabilized, made up to 30% (w/w) sucrose, and layered under a sucrose step gradient. The gradient was centrifuged (SW41 rotor, 28000 rpm, 1.5 h), and fractions were collected as indicated and analyzed for protein and phospholipid content, as well as for Rft1*; the amount of phospholipid was determined by scintillation counting to detect [3H]phosphatidylcholine. The data show that the vesicles float to the 20%−10% and 10%−5% (w/w) sucrose interfaces, with the denser vesicles containing a higher protein:phospholipid ratio as well as the majority of Rft1*. (C) Cryo-electron micrographs of reconstituted vesicles. The scale bar is 100 nm. (D) M5-DLO flipping in proteoliposomes reconstituted from TE. Different amounts of TE were used during reconstitution to generate different proteoliposome samples with protein:phospholipid ratios ranging from 0 to 50 mg/mmol. The samples were incubated on ice with Con A for 30 min and processed as described in to determine the fraction of [3H]M5-DLO captured by Con A. The graph (representative of more than five experiments) shows a monoexponential increase in the percent of [3H]M5-DLO captured, from a value of ∼50% for protein-free vesicles to ∼80% for vesicles with a PPR of ∼50 mg/mmol. Identical results were obtained when the assay was carried out at 25 °C. The predicted maximum of 100% [3H]M5-DLO bound is not experimentally observed. The initial slope of the graph suggests that M5-DLO flippase represents ∼1% (w/w) of proteins in the TE (see the text for details of the calculation). (E) M5-DLO flipping is trypsin-sensitive. TE was treated with trypsin (30 μg/mL) for different amounts of time (from 0 to 90 min, as indicated) at 30 °C. Reactions were stopped with 30 μg/mL soybean trypsin inhibitor, and mixtures were reconstituted and assayed for [3H]M5-DLO flipping (◼). Samples incubated with trypsin in the presence of trypsin inhibitor were processed in parallel (◻). The percent of [3H]M5-DLO captured in the control sample was ∼67%, corresponding to a PPR of ∼15 mg/mmol. The data (mean and range) are derived from two independent experiments.
We combined TE with Triton X-100-solubilized egg phosphatidylcholine (ePC) and trace quantities of [3H]M5-DLO and reconstituted proteoliposomes by removing detergent with SM2 Bio-Beads; protein-free liposomes were generated similarly, except that TE was omitted. Detergent removal was nearly quantitative (>99.9%) as judged by absorption measurements of residual Triton X-100 (data not shown) (36), while protein and phospholipid recovery were typically >50 and >70%, respectively, as previously reported (21–24). Thin layer chromatography of samples obtained prior to and after reconstitution indicated that neither [3H]M5-DLO nor ePC was degraded or modified during the reconstitution process (data not shown). Cryo-electron microscopy established that the reconstituted vesicles were unilamellar, with an average diameter of ∼175 nm (Figure 3C).
To verify that TE proteins were reconstituted into vesicles, we floated the vesicles in a sucrose gradient (37). Reconstituted proteins would be expected to float with the vesicles, while nonreconstituted material would not float. Proteoliposomes were treated with 0.04% (w/v) Triton X-100 to increase their permeability to sucrose, adjusted to 30% (w/w) sucrose, layered under a series of sucrose steps [20, 10, 5, and 2.5% (w/w)], and centrifuged at ∼100000gav in a swing-out rotor. Figure 3B shows that ∼90% of the protein, including Rft1*, was recovered together with phospholipid at the 20%−10% and 10%−5% (w/w) sucrose interfaces, with vesicles with lower protein:phospholipid ratios banding at lower sucrose density as expected. These data suggest that TE proteins were successfully reconstituted into vesicles.
M5-DLO Flipping in Proteoliposomes
We next used our liposome and proteoliposome preparations to test the assay strategy outlined in Figure 2A. When protein-free liposomes were incubated with Con A for 30 min on ice [Con A binding to detergent-solubilized [3H]M5-DLO is complete in ∼5 min on ice (data not shown)], ∼50% of [3H]M5-DLO was captured. The percent of [3H]M5-DLO captured was not affected by increasing the ratio of Con A to vesicles, indicating that the reaction mixture contained sufficient Con A to capture all available [3H]M5-DLO (data not shown). Also, there was no increase in the amount captured when longer incubation times or higher incubation temperatures (25 °C) were used. Capture of M5-DLO was negligible if the blocking solution containing yeast mannan was added prior to the addition of Con A. These results indicate that 50% of the M5-DLO molecules are located in the outer leaflet of the liposomes where they are accessible to Con A; the remaining 50% are located in the inner leaflet and protected from the lectin.
Upon incubation of proteoliposomes with Con A, the percent of [3H]M5-DLO captured was greater than that seen with liposomes. Capture increased monoexponentially as a function of the protein:phospholipid ratio (PPR) of the preparation, reaching a plateau of ∼80% in vesicle populations with a PPR of ∼50 mg/mmol (Figure 3D); experimentally, we never observed 100% capture in intact vesicles as depicted in Figure 2A (bottom panel), possibly because of steric effects that could weaken the ability of Con A to bind all the available M5-DLOs at the vesicle surface. These results are consistent with the interpretation (Figure 2A, bottom panel) that M5-DLO molecules located in the inner leaflet of flippase-equipped proteoliposomes are flipped to the outer leaflet where they are captured by Con A. The data imply that TE contains M5-DLO flippase activity that can be functionally reconstituted through the procedures we describe. ATP or other sources of metabolic energy were not included in the assay, nor did their inclusion have any effect. Thus, M5-DLO flippase activity, like other ER flippase activities (16–25), does not require metabolic energy and likely functions to facilitate bidirectional transbilayer diffusion of M5-DLO.
It was not possible to measure the rate of M5-DLO flipping in proteoliposomes since transport was complete within the time frame required for Con A binding. This was the case for measurements carried out on ice, as well as at 25 °C. However, as shown below, flipping of the higher-order intermediate M7-DLO was found to be substantially slower, allowing the flipping rate in that case to be measured. The observation that M5-DLO flipping is relatively rapid on ice is not surprising since a previous report of the temperature dependence of glycerophospholipid flipping indicated a half-time for flip-flop on the order of 100 s at 6 °C (42). On the basis of the limit imposed by the time required for Con A binding, we estimate the first-order rate constant for M5-DLO flipping on ice to be >2 × 10−3 s−1.
Trypsinization of TE prior to reconstitution caused a substantial reduction (>75%) in M5-DLO flippase activity (Figure 3E), as expected for a protein-mediated process. Similar results were obtained when papain, a more promiscuous protease, was used instead of trypsin (data not shown). Interestingly, reduction in activity was seen only after lengthy trypsinization, well after the stage when silver-stained SDS−PAGE analysis revealed that most (>80%) proteins in the TE had been digested. This suggests that the M5-DLO flippase may be a predominantly hydrophobic membrane protein with limited hydrophilicity and consequently few potential trypsin or papain cleavage sites. A similar relative resistance to proteolysis was previously reported for the flippase activities responsible for translocating glycerophospholipids and short chain analogues of dolichol-P-mannose and dolichol-P-glucose (18,19,21,22). Trypsinization of TE is expected to generate hydrophobic peptides corresponding to the transmembrane segments of membrane proteins. These peptides are likely to be incorporated into vesicles during the reconstitution process. However, it is clear from our demonstration of the trypsin sensitivity of flippase activity that the peptides themselves do not promote M5-DLO flipping. This result argues against proposals made elsewhere that hydrophobic membrane-spanning peptides alone, or the mere presence of membrane proteins in general, can mediate rapid translocation of phospholipids (43).
We conclude that TE contains a trypsin-sensitive protein(s) that when reconstituted into proteoliposomes endows the vesicles with the ability to translocate M5-DLO molecules from the inner to the outer leaflet rapidly and in an ATP-independent manner.
M5-DLO Flippase Represents ∼1% (w/w) of the Proteins in the TE
The percent of [3H]M5-DLO captured by Con A increases monoexponentially as a function of the PPR of the vesicle preparation (Figure 3D), reflecting the probability that a particular vesicle in the population is equipped with a M5-DLO flippase. The initial slope of the dose−response plot (Figure 3D) is ∼5% per PPR unit. Thus, at a PPR of ∼20 mg/mmol, every vesicle in the population would be predicted to be equipped with a M5-DLO flippase. This information can be used to estimate the abundance of the flippase in the mix of proteins found in TE. Taking the average external diameter of the vesicles to be 175 nm (Figure 3C) and assuming that the thickness of the membrane bilayer is 4 nm, the cross-sectional area of a phospholipid molecule is 0.7 nm2(44), and the average molecular mass of ER membrane proteins is ∼50 kDa (45), then at a PPR of 20 mg/mmol, each vesicle has ∼100 proteins, one of which is a M5-DLO flippase. Thus, functional M5-DLO flippases are predicted to represent ∼1% (w/w) of the proteins in TE.
M7-DLO Is Flipped More Slowly than M5-DLO
To test the specificity of the reconstituted flippase activity, we assayed flipping of the higher-order lipid-linked oligosaccharide, M7-DLO (Figure 2B), a biosynthetic intermediate synthesized from M5-DLO on the lumenal face of the ER (3–6). In preliminary experiments, we discovered that M7-DLO, unlike M5-DLO, was not detectably flipped in proteoliposomes that were incubated on ice, even if the incubation time was extended to 60 min. However, when proteoliposomes were incubated at 25 °C for >30 min, the percent of [3H]M7-DLO captured by Con A was identical to that seen for [3H]M5-DLO. Thus, M7-DLO is flipped at 25 °C but not on ice. TLC analyses of extracts of [3H]M7-DLO-containing proteoliposomes confirmed that [3H]M7-DLO was not degraded during the reconstitution procedure.
We determined the time course of M7-DLO and M5-DLO flipping at 25 °C (Figure 4A). Whereas M5-DLO flipping was complete by 5 min (the earliest time point taken because of the time required for Con A binding), [3H]M7-DLO flipping proceeded more slowly, reaching completion by ∼40 min. Using these data, we estimate a first-order rate constant of ∼2 × 10−4 s−1 for M7-DLO flipping at 25 °C. Although a precise comparison is difficult because of the time resolution imposed by Con A binding, M7-DLO flipping in the reconstituted system is likely to be more than 1 order of magnitude slower than that of M5-DLO. To verify that flipping of M7-DLO was mediated by the same protein that was responsible for the rapid transport of M5-DLO, we generated dose−response plots similar to Figure 3D. Proteoliposomes were reconstituted with different amounts of TE, and translocation of both [3H]M5-DLO and [3H]M7-DLO was assayed by incubating the vesicles with Con A for 40 min at 25 °C (Figure 4B). The identical protein dependence of both [3H]M5-DLO and [3H]M7-DLO flipping is consistent with the proposal that the same flippase mediates translocation of both precursors. We also chromatographed TE on a dye resin (described below) and assayed M5-DLO and M7-DLO flipping after reconstituting fractions eluted from the resin with salt; the profile of M5-DLO and M7-DLO flippase activity was identical (data not shown), confirming that the same protein is responsible for flipping both lipids. These data indicate that the M5-DLO flippase is able to discriminate sharply between lipids that differ by only two mannose residues.
Figure 4.

M7-DLO is flipped more slowly than M5-DLO. (A) Comparison of M7-DLO and M5-DLO flipping at 25 °C. Proteoliposomes with a PPR of ∼20 mg/mmol were reconstituted with [3H]M5-DLO (◻) or [3H]M7-DLO (◼) and assayed for DLO flipping at 25 °C. The kinetics of flipping are shown. The earliest time point analyzed was 5 min, corresponding to the time taken for completion of Con A binding to detergent-solubilized DLOs at 25 °C. The data (mean and range) are derived from two independent experiments. (B) Identical protein dependence of M7-DLO and M5-DLO flipping. M7-DLO and M5-DLO flipping were analyzed in vesicles prepared with different PPRs. Assays were carried out for 40 min at 25 °C.
M5-DLO Flippase Is 3-fold Less Abundant Than Glycerophospholipid Flippase: The Two Activities Can Be Resolved by Velocity Gradient Sedimentation
We considered whether the same protein was responsible for M5-DLO flippase and ER glycerophospholipid flippase activities. We previously reported the reconstitution of glycerophospholipid flippase activity using TE generated from yeast microsomes as a source of flippase (24) and the fluorescent glycerophospholipid analogue, NBD-PC, as a reporter. NBD-PC flipping was assayed as previously described (23,24), using the membrane impermeant reductant dithionite as a topological probe. We first determined the protein dependence of M5-DLO flipping and NBD-PC flipping in proteoliposomes. Inspection of the initial slope of the graphs shown in Figure 5A indicates that the protein responsible for glycerophospholipid flippase activity is ∼3-fold more abundant than the M5-DLO flippase, consistent with the two flippases being distinct proteins.
Figure 5.

Glycerophospholipid flippase is more abundant than M5-DLO flippase. Resolution of the two flippase activities by velocity gradient sedimentation. (A) Dose−response plots indicate that glycerophospholipid flippase is ∼3-fold more abundant than M5-DLO flippase. Glycerophospholipid flippase activity was assayed using NBD-PC as a reporter (○; the line represents a monoexponential fit); M5-DLO flippase activity was assayed in the same reconstituted samples (only the fit to the data is shown; data points are the same as those shown in Figure 3D). (B) TE was analyzed by velocity gradient sedimentation as described in . Fractions were collected from the top. The figure shows a silver-stained SDS−PAGE gel of fraction equivalents. (C) Resolution of NBD-PC and M5-DLO flippase activities. Gradient fractions were reconstituted and assayed for NBD-PC and M5-DLO flippase activities. The percent of activity (relative to the load) that was recovered in each fraction is indicated. Sedimentation standards analyzed in a parallel gradient are indicated at the top. Protein recovery in the fractions relative to the load was ∼80%; the enrichment of activity in the peak fractions was ∼4-fold in each case. The data are representative of five independent experiments.
We next attempted to separate the flippase activities. TE was fractionated by velocity sedimentation on a glycerol gradient. Gradient fractions were analyzed by SDS−PAGE and silver staining (Figure 5B) and also reconstituted into proteoliposomes to assay NBD-PC and M5-DLO flipping (Figure 5C). Figure 5B shows clear differences in the protein profile of the fractions, indicating successful separation of TE proteins. Reconstitution of the fractions into proteoliposomes showed that the M5-DLO and glycerophospholipid flippase activities were resolved (Figure 5C). The majority of M5-DLO activity peaked in fraction 7 corresponding to the 4.2 S sedimentation standard, while glycerophospholipid flippase activity sedimented more slowly and was recovered mainly in fractions 4 and 5. These data indicate that M5-DLO flippase does not translocate glycerophospholipids and, conversely, that the glycerophospholipid flippase does not translocate M5-DLO. We note that a number of gradient fractions are replete with proteins but have no flippase activity; for example, the complex mixtures of rapidly sedimenting proteins recovered in fractions 9−14 of the velocity gradient (Figure 5B,C) do not facilitate flipping of either M5-DLO or NBD-PC, while fractions 4 and 7 support the specific flipping of one lipid type or the other. This indicates that lipid flipping is not a general property of membrane proteins (43) and that specific transporters are required. We conclude that there are at least two distinct flippases in the ER, each responsible for transporting a specific class of lipid.
Separation of M5-DLO Flippase, Glycerophospholipid Flippase, and the Flippase Candidate Rft1 by Dye Resin Chromatography
As an initial step toward purifying M5-DLO flippase, we fractionated TE on Cibacron blue dye resin. Preliminary experiments indicated that the majority of the M5-DLO flippase activity bound to the resin and could be eluted with salt. To refine this result, we prepared TE from Rft1*-containing YCF40 cells (Table 1), incubated it with the dye resin, and eluted bound proteins in steps using progressively increasing salt concentrations. All fractions, including the load and nonbound material, were desalted to uniform ionic strength, before being subjected to protein determination, SDS−PAGE analysis, and reconstitution into proteoliposomes. Coomassie-stained SDS−PAGE gels showed that the nonbound material (0.05 M NaCl) and the different salt-eluted fractions had different protein compositions, indicating successful separation of TE proteins (Figure 6A). Immunoblotting indicated that Rft1* bound to the column and was eluted with >0.75 M NaCl (Figure 6A, bottom panel). Proteoliposomes were assayed for NBD-PC and M5-DLO flippase activities, both of which bind quantitatively to the dye resin. The majority (∼60%) of NBD-PC flippase activity was eluted with 100−250 mM NaCl, whereas M5-DLO flippase activity was eluted sharply with 400 mM NaCl (Figure 6B). These data reaffirm that NBD-PC and M5-DLO flipping are independent activities due to distinct proteins. Neither of these flippase proteins is Rft1* which requires considerably higher salt concentrations to be eluted from the resin.
Figure 6.

Resolution of glycerophospholipid flippase activity, M5-DLO flippase activity, and the flippase candidate Rft1* by Cibacron dye resin chromatography. (A) Fractionation of TE on Cibacron Blue dye resin. TE from Rft1*-containing YCF40 cells was loaded onto Cibacron blue dye resin, and the bound material was eluted in six sequential steps with salt. The 50 mM step corresponds to unbound (flow-through) material. The top panel shows a Coomassie-stained SDS−PAGE gel of the eluted fractions. The bottom panel shows the same fractions taken for immunoblotting to locate the elution behavior of Rft1*. (B) Resolution of NBD-PC and M5-DLO flippase activities. Fractions eluted from the Cibacron blue dye resin were reconstituted and assayed for M5-DLO (top) and NBD-PC (bottom) flippase activities. The percent of activity recovered (relative to the load) is indicated. Less than 5% of activity was recovered in the flow-through fraction (50 mM NaCl). Protein recovery in the fractions relative to the load was ∼85%; the enrichment of M5-DLO and NBD-PC flippase activities was 4.6- and 6.6-fold, respectively. The data are representative of three independent experiments.
Concluding Discussion
We functionally reconstituted M5-DLO flippase activity in proteoliposomes generated from Triton X-100-solubilized yeast ER membrane proteins. The ATP independence of M5-DLO flipping implies that transport occurs bidirectionally, by a facilitated diffusion mechanism. We enriched the activity by velocity gradient sedimentation and dye resin chromatography and resolved it from the ER glycerophospholipid flippase as well as the genetically identified flippase candidate Rft1. The ability to separately enrich the M5-DLO and glycerophospholipid flippase activities by completely distinct criteria (velocity gradient sedimentation and dye resin chromatography) eliminates previous proposals that a particular subset of membrane proteins, or the mere presence of transmembrane helices of proteins, is sufficient to promote lipid translocation (43).
The separation of M5-DLO flippase activity from Rft1* by dye resin chromatography (Figure 6) clearly indicates that Rft1 is not directly responsible for M5-DLO flipping as previously proposed on the basis of genetic experiments (27,29). This conclusion is reinforced by recent experiments in which Rft1-depleted TE was tested for M5-DLO flippase activity. In these experiments, Rft1 was immunodepleted from TE or TE was prepared from microsomes derived from Rft1-depleted cells; Rft1 depletion by either method had no effect on M5-DLO flippase activity (46). The glycosylation phenotype of yeast and mammalian cells depleted of Rft1 (27,29,47) nevertheless indicates that Rft1 may play some role in N-glycosylation. We suggest that early steps of DLO synthesis are laterally segregated from the M5-DLO flippase in the ER membrane and that the role of Rft1 is to regulate DLO traffic between this “synthesis domain” and the flippase. Alternatively, Rft1 may be a DLO chaperone that controls the lateral distribution of DLOs in the ER and in so doing regulates their access to the flippase. In these roles, Rft1 would have a crucial in vivo function but would be unnecessary in the reconstituted system where lateral compartmentalization is lost and transbilayer translocation of DLOs is directly measured. Similar models have been proposed and discussed in detail for the enigmatic Lec35/MPDU1 protein that is suggested to be required in vivo, but not in vitro, for the translocation of dolichol-P-mannose and dolichol-P-glucose across the ER membrane (48,49).
Translocation of M5-DLO in reconstituted proteoliposomes was rapid, even on ice, whereas translocation of M7-DLO was at least 1 order of magnitude slower. This result is consistent with the original report of Snider and Rogers (8), who showed that M7-DLO is essentially inaccessible to Con A in microsomes that are incubated with the lectin for 30 min on ice; had the experiment been conducted for a similar time period at 25 °C, it is likely, on the basis of our results, that M7-DLO capture would have been similar to that seen for M5-DLO. Our data on flipping of M5-DLO and M7-DLO suggest that the flippase either specifically recognizes features of the oligosaccharide headgroup or gates translocation of the DLO class of molecules according to headgroup size. In the former case, rapid translocation would be predicted for M5-DLO alone, with rates falling off for smaller and larger DLOs. In the latter scenario, all DLOs from GlcNAc-PP-dolichol to M5-DLO in the biosynthetic pathway would be rapidly translocated, with slower translocation being observed for higher-order structures. Future work will distinguish between these possibilities. It will also be interesting to learn what core features of the DLO structure, such as the α-saturated isoprene residue and the pyrophosphate linkage, are recognized by the M5-DLO flippase.
The need for transbilayer movement of lipid intermediates during DLO synthesis in the ER has been recognized for more than 20 years (3,4,8). With the exception of a single previous report describing the reconstitution of protein-dependent transport of a water-soluble analogue of dolichol-P-mannose (25), and the genetic identification of the Rft1 and Lec35/MPDU1 proteins (29,48,49) which may serve an accessory role in glycolipid flipping as we discuss above, there has been little progress toward identifying the lipid flippases required for DLO synthesis. The work described in this paper provides, for the first time, a biochemical means of identifying the M5-DLO flippase using a robust assay and a radiolabeled natural lipid as a transport reporter. We have recently successfully extended the reconstitution and fractionation studies described here to detergent-extracted ER membrane proteins from rat liver, enabling purification from a more abundant source of starting material.
Acknowledgments
We thank Patrick Barré and the facilities at the New York Structural Biology Center for cryo-EM images of reconstituted vesicles, Christian Kranz, Bobby Ng, and Hudson Freeze for headgroup analysis of [3H]DLOs, Olga Boudker, Enrique Rodriguez-Boulan, Fred Maxfield, and Beate Schwer for the use of equipment, Jonne Helenius for plasmid pJH19, Andreas Conzelmann, Patty Kane, and Tom Rapoport for antibodies, and Markus Aebi for providing yeast strains as well as financial support for C.G.F.ʼs initial visits to the Menon laboratory.
Funding Statement
National Institutes of Health, United States
Footnotes
Abbreviations: BSA, bovine serum albumin; Con A, Concanavalin A; DLO, dolichol-linked oligosaccharide; ePC, egg phosphatidylcholine; ER, endoplasmic reticulum; M5-DLO, Man5GlcNAc2-PP-dolichol; M7-DLO, Man7GlcNAc2-PP-dolichol; NBD-PC, 1-acyl-2-[6-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]hexanoyl-sn-glycero-3-phosphocholine; OST, oligosaccharyltransferase; PPR, protein/phospholipid ratio of reconstituted vesicles (milligrams per millimole); Rft1*, Protein A-tagged Rft1; TE, Triton extract of yeast microsomes.
We refer to these “bidirectional” transporters as flippases and the process they facilitate as flipping (or translocation), even though these terms suggest vectorial transport.
References
- Hanover J. A.; Lennarz W. J. (1980) N-Linked glycoprotein assembly. Evidence that oligosaccharide attachment occurs within the lumen of the endoplasmic reticulum. J. Biol. Chem. 255, 3600–3604. [PubMed] [Google Scholar]
- Snider M. D.; Robbins P. W. (1982) Transmembrane organization of protein glycosylation: Mature oligosaccharide-lipid is located on the luminal side of microsomes from Chinese hamster ovary cells. J. Biol. Chem. 257, 6796–6801. [PubMed] [Google Scholar]
- Hirschberg C. B.; Snider M. D. (1987) Topography of glycosylation in the rough endoplasmic reticulum and Golgi apparatus. Annu. Rev. Biochem. 56, 63–87. [DOI] [PubMed] [Google Scholar]
- Lennarz W. J. (1987) Protein glycosylation in the endoplasmic reticulum: Current topological issues. Biochemistry 26, 7205–7210. [DOI] [PubMed] [Google Scholar]
- Helenius J.; Aebi M. (2002) Transmembrane movement of dolichol-linked carbohydrates during N-glycoprotein biosynthesis in the endoplasmic reticulum. Semin. Cell Dev. Biol. 13, 171–178. [DOI] [PubMed] [Google Scholar]
- Schenk B.; Fernandez F.; Waechter C. J. (2001) The ins(ide) and outs(ide) of dolichyl phosphate biosynthesis and recycling in the endoplasmic reticulum. Glycobiology 11, 61R–70R. [DOI] [PubMed] [Google Scholar]
- Rush J. S.; Gao N.; Lehrman M. A.; Waechter C. J. (2008) Recycling of dolichyl monophosphate to the cytoplasmic leaflet of the endoplasmic reticulum after the cleavage of dolichyl pyrophosphate on the lumenal monolayer. J. Biol. Chem. 283, 4087–4093. [DOI] [PubMed] [Google Scholar]
- Snider M. D.; Rogers O. C. (1984) Transmembrane movement of oligosaccharide-lipids during glycoprotein synthesis. Cell 36, 753–761. [DOI] [PubMed] [Google Scholar]
- Pomorski T.; Menon A. K. (2006) Lipid flippases and their biological functions. Cell. Mol. Life Sci. 63, 2908–2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlean P.; Menon A. K. (2007) GPI anchoring of protein in yeast and mammalian cells, or how we learned to stop worrying and love glycophospholipids. J. Lipid Res. 48, 993–1011. [DOI] [PubMed] [Google Scholar]
- Kornberg R. D.; McConnell H. M. (1971) Inside-outside transitions of phospholipids in vesicle membranes. Biochemistry 10, 1111–1120. [DOI] [PubMed] [Google Scholar]
- Hanover J. A.; Lennarz W. J. (1979) The topological orientation of N,N′-diacetylchitobiosylpyrophosphoryldolichol in artificial and natural membranes. J. Biol. Chem. 254, 9237–9246. [PubMed] [Google Scholar]
- McCloskey M. A.; Troy F. A. (1980) Paramagnetic isoprenoid carrier lipids. 2. Dispersion and dynamics in lipid membranes. Biochemistry 19, 2061–2066. [DOI] [PubMed] [Google Scholar]
- Marx U.; Lassmann G.; Holzhütter H.-G.; Wüstner D.; Müller P.; Höhlig A.; Kubelt J.; Herrmann A. (2000) Rapid flip-flop of phospholipids in endoplasmic reticulum membranes studied by a stopped-flow approach. Biophys. J. 78, 2628–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishwakarma R. A.; Vehring S.; Mehta A.; Sinha A.; Pomorski T.; Herrmann A.; Menon A. K. (2005) New fluorescent probes reveal that flippase-mediated flip-flop of phosphatidylinositol across the endoplasmic reticulum membrane does not depend on the stereochemistry of the lipid. Org. Biomol. Chem. 3, 1275–1283. [DOI] [PubMed] [Google Scholar]
- Bishop W. R.; Bell R. M. (1985) Assembly of the endoplasmic reticulum phospholipid bilayer: The phosphatidylcholine transporter. Cell 42, 51–60. [DOI] [PubMed] [Google Scholar]
- Backer J. M.; Dawidowicz E. A. (1987) Reconstitution of a phospholipid flippase from rat liver microsomes. Nature 327, 341–343. [DOI] [PubMed] [Google Scholar]
- Rush J. S.; Waechter C. J. (1995) Transmembrane movement of a water-soluble analogue of mannosylphosphoryldolichol is mediated by an endoplasmic reticulum protein. J. Cell Biol. 130, 529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush J. S.; Waechter C. J. (1998) Transbilayer movement of Glc-P-dolichol and its function as a glucosyl donor: Protein-mediated transport of a water-soluble analog into sealed ER vesicles from pig brain. Glycobiology 8, 1195–1205. [DOI] [PubMed] [Google Scholar]
- Vishwakarma R. A.; Menon A. K. (2005) Flip-flop of glycosylphosphatidylinositols (GPIs) across the ER. Chem. Commun., 453–455. [DOI] [PubMed] [Google Scholar]
- Menon A. K.; Watkins W. E.; Hrafnsdóttir S. (2000) Specific proteins are required to translocate phosphatidylcholine bidirectionally across the endoplasmic reticulum. Curr. Biol. 10, 241–252. [DOI] [PubMed] [Google Scholar]
- Gummadi S. N.; Menon A. K. (2002) Transbilayer movement of dipalmitoylphosphatidylcholine in proteoliposomes reconstituted from detergent extracts of endoplasmic reticulum: Kinetics of transbilayer transport mediated by a single flippase and identification of protein fractions enriched in flippase activity. J. Biol. Chem. 277, 25337–25343. [DOI] [PubMed] [Google Scholar]
- Chang Q.; Gummadi S. N.; Menon A. K. (2004) Chemical modification identifies two populations of glycerophospholipid flippase in rat liver ER. Biochemistry 43, 10710–10718. [DOI] [PubMed] [Google Scholar]
- Vehring S.; Pakkiri L.; Schröer A.; Alder-Baerens N.; Herrmann A.; Menon A. K.; Pomorski T. (2007) Flip-flop of fluorescently labeled phospholipids in proteoliposomes reconstituted with Saccharomyces cerevisiae microsomal proteins. Eukaryotic Cell 6, 1625–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush J. S.; Waechter C. J. (2004) Functional reconstitution into proteoliposomes and partial purification of a rat liver ER transport system for a water-soluble analogue of mannosylphosphoryldolichol. Biochemistry 43, 7643–7652. [DOI] [PubMed] [Google Scholar]
- van Meer G.; Voelker D. R.; Feigenson G. W. (2008) Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng D. T. W.; Spear E. D.; Walter P. (2000) The unfolded protein response regulates multiple aspects of secretory and membrane protein biogenesis and endoplasmic reticulum quality control. J. Cell Biol. 150, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koerte A.; Chong T.; Li X.; Wahane K.; Cai M. (1995) Suppression of the yeast mutation rft1-1 by human p53. J. Biol. Chem. 270, 22556–22564. [DOI] [PubMed] [Google Scholar]
- Helenius J.; Ng D. T.; Marolda C. L.; Walter P.; Valvano M. A.; Aebi M. (2002) Translocation of lipid-linked oligosaccharides acros the ER membrane requires Rft1 protein. Nature 415, 447–450. [DOI] [PubMed] [Google Scholar]
- Hill J. E.; Myers A. M.; Koerner T. J.; Tzagoloff A. (1986) Yeast/E. coli shuttle vectors with multiple unique restriction sites. Yeast 2, 163–167. [DOI] [PubMed] [Google Scholar]
- Zufferey R.; Knauer R.; Burda P.; Stagljar I.; te Heesen S.; Lehle L.; Aebi M. (1995) STT3, a highly conserved protein required for oligosaccharyl transferase activity in vivo. EMBO J. 14, 4949–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank C. G.; Aebi M. (2005) ALG9 mannosyltransferase is involved in two different steps of lipid-linked oligosaccharide biosynthesis. Glycobiology 15, 1156–1163. [DOI] [PubMed] [Google Scholar]
- Kim S.; Westphal V.; Srikrishna G.; Mehta D. P.; Peterson S.; Filiano J.; Karnes P. S.; Patterson M. C.; Freeze H. H. (2000) Dolichol phosphate mannose synthase (DPM1) mutations define congenital disorder of glycosylation Ie (CDG-Ie). J. Clin. Invest. 105, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoni Y.; Schekman R. (2002) Vesicle budding from endoplasmic reticulum. Methods Enzymol. 351, 258–278. [DOI] [PubMed] [Google Scholar]
- Lévy D.; Bluzat A.; Seigneuret M.; Rigaud J. L. (1990) A systematic study of liposome and proteoliposome reconstitution involving Bio-bead-mediated Triton X-100 removal. Biochim. Biophys. Acta 1025, 179–190. [DOI] [PubMed] [Google Scholar]
- Hrafnsdóttir S.; Menon A. K. (2000) Reconstitution and partial characterization of phospholipid flippase activity from detergent extracts of the Bacillus subtilis cell membrane. J. Bacteriol. 182, 4198–4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigaud J. L.; Paternostre M. T.; Bluzat A. (1988) Mechanisms of membrane protein insertion into liposomes during reconstitution procedures involving the use of detergents. 2. Incorporation of the light-driven proton pump bacteriorhodopsin. Biochemistry 27, 2677–2688. [DOI] [PubMed] [Google Scholar]
- Kaplan R. S.; Pedersen P. L. (1989) Sensitive protein assay in the presence of high levels of lipid. Methods Enzymol. 172, 393–399. [DOI] [PubMed] [Google Scholar]
- Bligh E. G.; Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Med. Sci. 37, 911–917. [DOI] [PubMed] [Google Scholar]
- Rouser G.; Fleischer S.; Yamamoto (1970) Two-dimensional thin layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids 5, 494–496. [DOI] [PubMed] [Google Scholar]
- Naismith J. H.; Field R. A. (1996) Structural basis of trimannoside recognition by concanavalin A. J. Biol. Chem. 271, 972–976. [DOI] [PubMed] [Google Scholar]
- Hrafnsdóttir S.; Nichols J. W.; Menon A. K. (1997) Transbilayer movement of fluorescent phospholipids in Bacillus megaterium membrane vesicles. Biochemistry 36, 4969–4978. [DOI] [PubMed] [Google Scholar]
- Kol M. A.; de Kroon A. I.; Killian J. A.; de Kruijff B. (2004) Transbilayer movement of phospholipids in biogenic membranes. Biochemistry 43, 2673–2681. [DOI] [PubMed] [Google Scholar]
- Huang C.; Mason J. T. (1978) Geometric packing constraints in egg phosphatidylcholine vesicles. Proc. Natl. Acad. Sci. U.S.A. 75, 308–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn P.; Griffiths G.; Warren G. (1984) Density of newly synthesized plasma membrane proteins in intracellular membranes II. Biochemical studies. J. Cell Biol. 98, 2142–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank C. G.; Sanyal S.; Rush J. S.; Waechter C. J.; Menon A. K. (2008) Is Rft1 a flippase for Man5GlcNAc2-PP-dol?. Nature XXX–XXX(in press). [Google Scholar]
- Haeuptle M. A.; Pujol F. M.; Neupert C.; Winchester B.; Kastaniotis A. J.; Aebi M.; Hennet T. (2008) Human RFT1 deficiency leads to a disorder of N-linked glycosylation. Am. J. Hum. Genet. 82, 600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand M.; Rush J. S.; Ray S.; Doucey M.-A.; Weik J.; Ware F. E.; Hofsteenge J.; Waechter C. J.; Lehrman M. A. (2001) Requirement of the Lec35 gene for all known classes of monosaccharide-P-dolichol-dependent glycosyltransferase reactions in mammals. Mol. Biol. Cell 12, 487–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk B.; Imbach T.; Frank C. G.; Grubenmann C. E.; Raymond G. V.; Hurvitz H.; Raas-Rotschild A.; Luder A. S.; Jaeken J.; Berger E. G.; Matthijs G.; Hennet T.; Aebi M. (2001) MPDU1 mutations underlie a novel congenital disorder of glycosylation, designated type If. J. Clin. Invest. 108, 1687–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayraghavan U.; Company M.; Abelson J. (1989) Isolation and characterization of pre-mRNA splicing mutants of Saccharomyces cerevisiae. Genes Dev. 3, 1206–1216. [DOI] [PubMed] [Google Scholar]
- Aebi M.; Gassenhuber J.; Domdey H.; te Heesen S. (1996) Cloning and characterization of the ALG3 gene of Saccharomyces cerevisiae. Glycobiology 6, 439–444. [DOI] [PubMed] [Google Scholar]
- Jones E. W. (1991) Tackling the protease problem in Saccharomyces cerevisiae. Methods Enzymol. 194, 428–453. [DOI] [PubMed] [Google Scholar]