

Abstract
Enzymes capable of hydrolyzing N-acyl-l-homoserine lactones (AHLs) used in some bacterial quorum-sensing pathways are of considerable interest for their ability to block undesirable phenotypes. Most known AHL hydrolases that catalyze ring opening (AHL lactonases) are members of the metallo-β-lactamase enzyme superfamily and rely on a dinuclear zinc site for catalysis and stability. Here we report the three-dimensional structures of three product complexes formed with the AHL lactonase from Bacillus thuringiensis. Structures of the lactonase bound with two different concentrations of the ring-opened product of N-hexanoyl-l-homoserine lactone are determined at 0.95 and 1.4 Å resolution and exhibit different product configurations. A structure of the ring-opened product of the non-natural N-hexanoyl-l-homocysteine thiolactone at 1.3 Å resolution is also determined. On the basis of these product-bound structures, a substrate-binding model is presented that differs from previous proposals. Additionally, the proximity of the product to active-site residues and observed changes in protein conformation and metal coordination provide insight into the catalytic mechanism of this quorum-quenching metalloenzyme.
The metallo-β-lactamase enzyme superfamily is quite diverse in sequence, metal stoichiometry, metal identity, substrate specificity, reaction mechanism, and reaction type (1–3). Many of these enzymes are important to health and disease, so understanding how they work and how to control their activities is of significant interest. Despite numerous structural reports, there have not been many examples of substrate-bound or product-bound structures (4–9), making studies of mechanism and inhibition more difficult. One excellent counterexample is glyoxalase II, a dinuclear metal-dependent thioesterase involved in the detoxification pathway of methylglyoxal. Crystallization with a slow substrate revealed both substrate- and product-bound structures (4). The intact substrate is bound in an orientation that positions the carbonyl of the labile thioester over what is usually termed zinc 1 (Zn1) (Figure 1A), a site coordinated by three histidine residues. The sulfur of the intact thioester is poised over zinc 2 (Zn2), a site coordinated by two histidines and two aspartates. Although the metal ion sites in this superfamily are generally conserved, there are known deviations in the sequence of primary metal ion ligands at both sites (1–3). The substrate binding mode observed with glyoxalase II (4) orients the bridging hydroxide for attack, polarizes the substrate’s carbonyl bond, and stabilizes the developing negative charge on the substrate’s leaving group. The product-bound structure of a metallo-β-lactamase from Stenotrophomonas maltophilia has also been published and exhibits a similar orientation, with the lactam nitrogen leaving group ligated to Zn2 (Figure 1B) (6). These and other studies have led to a general acceptance of substrate orientation for the β-lactamases and thioesterases of the superfamily, which places the substrate’s carbonyl near Zn1 and the leaving group near Zn2.
Figure 1.

Comparison of selected substrate, product, and inhibitor binding orientations found in the metallo-β-lactamase superfamily. (A) Intact thioester substrate bound to human glyoxalase II (4). (B) Hydrolyzed β-lactam product bound to S. maltophilia metallo-β-lactamase (6). (C) Intact l-homoserine lactone inhibitor bound to Bacillus thuringiensis AHL lactonase (10). All line drawings are adapted from the structures referenced above.
Substrate placement is less well defined for the related family of N-acyl-l-homoserine lactone hydrolases (AHL1 lactonases), which catalyze a ring opening reaction that can block quorum sensing. One AHL lactonase structure has been reported in complex with an intact homoserine lactone (which lacks the N-acyl substituent of natural substrates) and exhibits an unexpected binding mode that places the carbonyl oxygen over Zn2 and the ring oxygen over Zn1 (Figure 1C) (10). This orientation is the opposite of that observed in the β-lactamase and thioesterase enzyme families and prompted us to make a more detailed investigation of the reaction mechanism. X-ray crystallography, molecular dynamics calculations, and a variety of functional studies are combined in investigating substrate binding, product binding, and the ring opening mechanism of AHL lactonase. These results are presented in two sequential papers. First, three product-bound structures of the quorum-quenching AHL lactonase (AiiA) from Bacillus thuringiensis are reported. Interactions of the bound products with metal ions and active-site residues along with an unusual change in the coordination of Zn2 support a substrate binding model different from that proposed elsewhere (10) providing specific mechanistic implications. Second, the substrate binding mode and the catalytic roles of active-site residues are investigated in more detail in the following paper (11), leading to the proposal of a catalytic mechanism for the quorum-quenching AHL lactonase from B. thuringiensis.
Materials and Methods
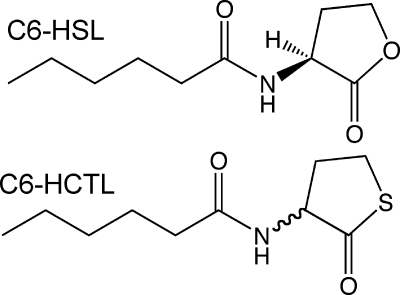
Unless otherwise noted, all chemicals were obtained from Sigma-Aldrich Chemical Co. (St. Louis, MO). Metal analysis of purified proteins was performed using inductively coupled plasma mass spectrometry (ICP-MS) as previously described (12). Wild-type and variant AHL lactonases were expressed in M9 minimal medium, supplemented with metal salts, as previously described (13). The substrates N-hexanoyl-l-homoserine lactone (C6-HSL, shown below) and N-hexanoyl-dl-homocysteine thiolactone (C6-HCTL, shown below) were synthesized according to the procedure reported previously (13).
Crystallization
The crystallization of AHL lactonase (AiiA) was conducted via the hanging drop method using AiiA protein at 20 mg/mL. The well solution includes glycerol (20%, v/v), Tris-HCl (80 mM), polyethylene glycol (PEG) 4000 (24%, w/v), and MgCl2 (160 mM) and was adjusted to a final pH of 8.5 using NaOH. Two different stock solutions were prepared for the C6-HSL substrate. A lower-concentration stock solution (19 mM) was prepared by dissolving the substrate in water, and a higher-concentration stock solution (50 mM) was prepared in a 50% (v/v) aqueous methanol solution. A second substrate, C6-HCTL, was also dissolved in 50% aqueous methanol at 50 mM. The hanging drops were prepared by mixing a protein solution (2 μL), a substrate solution (1 μL), and a well solution (2 μL). During the processes of mixing and cocrystallization, the substrates were apparently converted to products either by the AHL lactonase or by nonenzymic hydrolysis. Considering the decrease in drop volume that occurs during cocrystallization, the resulting concentrations of substrates and products were in approximate ranges of 4−10 and 10−25 mM for C6-HSL, respectively, depending on whether low- or high-concentration stock solutions had been used, and were in a range of 10−25 mM for C6-HCTL. The hanging drops were kept at room temperature for 1−2 weeks during crystal growth. Crystals appeared in 2 days and were allowed to continue growing until they reached their maximum sizes. Crystals with good morphology and large sizes (approximately 1 mm × 0.4 mm × 0.4 mm) were picked directly from the hanging drops and cooled to liquid nitrogen temperature.
Data Collection and Processing
Monochromatic data were collected at the 23-IDD beamline at GM/CA-CAT at the Advanced Photon Source (APS), Argonne National Laboratory (ANL). The 23-IDD beamline uses dual-canted undulators to produce intense X-ray radiation and is equipped with a MarMosaic CCD detector for data collection. Data collection for crystals obtained from cocrystallizations with the higher concentrations of C6-HSL and C6-HCTL was conducted at a wavelength of 0.9793 Å with a 1° oscillation angle. The crystals obtained from cocrystallization with the lower concentration of C6-HSL diffracted to an ultrahigh (atomic) resolution at the 23-IDD beamline. Consequently, such data collection was conducted at a wavelength of 0.9184 Å with 0.4° oscillation angles to maximize the diffraction resolution collected and to resolve the crowded diffraction pattern. The atomic data set was collected via two passes. First, a high-resolution pass was collected to the maximum resolution (0.95 Å); later, a low-resolution pass was collected to a resolution of 2.0 Å to compensate for the overloads in lower-resolution bins during the high-resolution pass. During data collection, the crystal was translated along the longest dimension several times to minimize X-ray-induced radiation damage by exposing different parts of this single crystal.
Data sets were indexed and integrated by using the HKL2000 suite (14). The scaling process was also carried out in HKL2000 except for the atomic-resolution data set. The merging and scaling of the two passes of the atomic-resolution data set were done using SCALEPACKM16. A shell script written by T. Fenn (scale_mult.sh) was used to govern the merging and scaling of the two passes.
Model Building and Refinement
A previously published AHL lactonase structure (PDB entry 2A7M) (15) was used as the initial search model in molecular replacement conducted via PHASER (16) in the CCP4 suite (17). The solutions of the molecular replacement were then refined in REFMAC5 (18) through rigid body, isotropic, and anisotropic refinements. After each round of refinement, the model building and adjustment, including water picking, were conducted in COOT (19). The coordinates of the ligands were first generated in the Dundee PRODRG2 server (http://davapc1.bioch.dundee.ac.uk/programs/prodrg/) and then were used to generate the topology files using the MONOSKETCH program in the CCP4 suite. The ligand coordinates were then fitted into the difference electron density and included in the next round of refinement along with the topology files. In the final stage, the weighting factor for the chemical restraints in REFMAC5 was adjusted to produce the lowest possible R/Rfree values. The atomic-resolution data underwent further refinement in SHELXL (20). Conjugated gradient least-squares (CGLS) refinement against the square value of structure-factor amplitudes was used. The structural model went through isotropic and anisotropic restrained refinement to produce the lowest possible R/Rfree values before the hydrogen atoms were added to the model. The occupancy of the ligand was refined as a free variable in SHELXL after the structure model was well refined. The anisotropic refinement in SHELXL was validated via the PARVATI server (http://skuld.bmsc.washington.edu/parvati/).
Figures
All structural figures were made using PYMOL (http://www.pymol.org) or UCSF-CHIMERA (21). The secondary structure assignments in the cartoon style figures were determined via the STRIDE online server (http://bioweb.pasteur.fr/seqanal/interfaces/stride.html). The electron density maps were generated with FFT in the CCP4 format or were generated using SHELXPRO in O format and then converted to CCP4 format using MAPMAN (22).
Results
Crystallization, Data Collection, and Model Building
AHL lactonase can be inhibited by ring-opened products (Supporting Information, Figure 1), so cocrystallization studies were pursued to gain information about ligand binding. In comparison with previous crystallographic studies (15), addition of the N-hexanoyl-l-homoserine lactone (C6-HSL) substrate significantly improved the crystallization of AHL lactonase, in both size and morphology. As a result, the crystal obtained with 4−10 mM C6-HSL diffracts to an ultrahigh resolution of 0.95 Å. The statistics of the atomic-resolution data set and its resulting model are presented in Table 1. The addition of higher concentrations of C6-HSL or N-hexanoyl-l-homocysteine thiolactone (C6-HCTL) also improves crystallization. It is expected that the amount of methanol cosolvent remaining in the hanging drop is negligible due to diffusion into the well solution during crystallization. The temporary presence of this cosolvent did not prevent formation of the crystals, nor did it change the packing in the crystal lattice. Crystals from the cocrystallization mixtures containing higher concentrations (10−25 mM) of C6-HSL and C6-HCTL diffract to resolutions of 1.4 and 1.3 Å, respectively. The statistics of these two data sets and their resulting structural models are listed in Table 2. All three structural models presented here are structures of the AHL lactonase protein complexed with ring-opened products derived from the added lactone or thiolactone substrates.
Table 1. Crystallographic Statistics for Complex I.
| substrate | C6-HSL (4−10 mM) |
| resolution range (Å) | 50−0.95 |
| space group | P212121 |
| cell dimensions | |
| a (Å) | 54.9 |
| b (Å) | 55.6 |
| c (Å) | 78.7 |
| total no. of reflections | 882914 |
| no. of unique reflections | 141032 |
| completeness (%) | 93.9 (61.9)a |
| linear Rmerge (%)b | 9.1 (37.1)a |
| I/σ(I) | 12.5 (2.0)a |
| Rcrys/Rfree (%)c | 13.3/16.9 |
| Rcrys/Rfree (%) for FO > 4σ(FO) | 12.3/16.2 |
| mean protein Ueq (Å2)d | 0.25 |
| mean solvent Ueq (Å2) | 0.40 |
| mean protein anisotropy | 0.55 (σ = 0.17) |
| mean solvent anisotropy | 0.47 (σ = 0.15) |
| rmsd for bond lengths (Å) | 0.02 |
| rmsd for angle distances (Å) | 0.03 |
| rmsd for chiral volumes (Å3) | 0.01 |
The values in parentheses are for the highest-resolution shell (0.98−0.95 Å).
Linear Rmerg = ∑|Iobs − Iavg|/∑Iavg.
Rfree = ∑|Fobs − Fcalc|/∑Fobs. Five percent of the reflection data was selected at random as a test set, and only the test set was used to calculate Rfree. Rcrys was calculated with the same equation as Rfree, but both test and working sets were used in the calculation.
Ueq and anisotropy values were generated via the Parvati sever.
Table 2. Crystallographic Statistics for Complexes II and III.
| substrate | C6-HSL (10−25 mM) | C6-HCTL (10−25 mM) |
| resolution range (Å) | 19−1.4 | 50−1.30 |
| space group | P212121 | P212121 |
| cell dimensions | ||
| a (Å) | 55.4 | 54.8 |
| b (Å) | 55.6 | 55.5 |
| c (Å) | 79.9 | 79.2 |
| total no. of reflections (no. of unique reflections) | 332267 (48248) | 376140 (56793) |
| completeness (%) | 97.0 (90.6)a | 94.4 (67.5)a |
| linear Rmerge (%)b | 5.3 (51.2)a | 5.1 (48.1)a |
| I/σ(I) [I/σ(I)] | 31.9 (2.4)a | 31.3 (2.1)a |
| Rcrys/Rfree (%)c | 14.0/18.3 | 14.5/18.3 |
| rmsd for bonds (Å) | 0.02 | 0.02 |
| rsmd for angles (deg) | 1.9 | 2.0 |
| average B (Å2) | 20.5 | 22.4 |
The values in parentheses are for the highest-resolution shell (1.45−1.4 and 1.35−1.3 Å, respectively).
Linear Rmerg = ∑|Iobs − Iavg|/∑Iavg.
Rfree = ∑|Fobs − Fcalc|/∑Fobs. Five percent of the reflection data was selected at random as a test set, and only test set was used to calculate Rfree. Rcrys was calculated with the same equation as Rfree, but both test and working sets were used in the calculation.
Structural Model of Product Complex I
An atomic-resolution (0.95 Å) data set collected on the crystal formed from cocrystallization with the lower concentration of C6-HSL resulted in the first structural model (complex I). Although the overall structure is similar to previously reported structural models (10,15), it has four additional N-terminal residues (Gly-Arg-Ile-Ser) derived from cloning (12), resulting in 254 total amino acid residues. These additional residues are numbered (−4, −3, −2, and −1) so that the sequence numbering of complex I can be directly compared to the other AHL lactonase structural models.
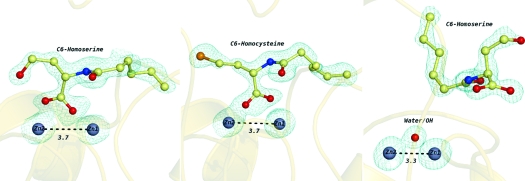
In comparison with the unliganded structure of AHL lactonase (PDB entry 2A7M) (15), the coordination of the dizinc metal center remains unchanged in complex I. The Zn1−Zn2 distance is 3.3 Å, which is also identical to that observed in the unliganded enzyme (Figure 2). The water/hydroxide species that bridges the two zinc ions is clearly present, as supported by the electron density map (Figure 2). The active-site cavity is large and Y-shaped, as reported previously (Figure 3A,B) (15). In one of the two smaller branches of the cavity, extra electron density was observed and was well fit by N-hexanoyl-l-homoserine, the ring-opened hydrolysis product of C6-HSL. Supporting this assignment, the electron density resulting from this high-resolution data set is only well fit by the ring-opened product (Figure 2). The occupancy of the bound product was refined as a free variable in SHELXL and indicates that this position is 70% occupied. Somewhat surprisingly, the product is not bound to the dizinc metal center, but rather to an adjacent branch of the active-site cavity (Figures 2 and 3B). The product interacts with the protein through a series of hydrogen bonds (Supporting Information, Figure 2) formed with two backbone atoms, Phe107 and Glu136, and with three ordered water molecules. These water molecules in turn recruit and interact with other residues and more water molecules to stabilize product binding. In unliganded AHL lactonase, the positions of the three ordered water molecules mentioned above were occupied by two molecules of glycerol (15), which is a component in all crystallization conditions used in these studies.
Figure 2.

Wall-eyed stereoview of the complex I active site. Two zinc ions are shown as silver-gray spheres. The bridging water/hydroxide is shown as a red sphere. Metal-coordinating residues and the N-acyl-l-homoserine product are shown in ball-and-stick representations. Oxygen atoms are colored red, and nitrogen atoms are colored blue. The carbon atoms in the homoserine molecule are colored pale yellow. The carbon atoms in the coordinating residues are colored white. Electron density maps (with 2Fo − Fc coefficients) are colored blue at 6σ (for the clarity of the figure) around the zincs and the bridging water/hydroxide and at 1σ around the homoserine product. The Zn1−Zn2 distance is given in angstroms and indicated with a dashed black line.
Figure 3.

Active-site cavity and ligand position. (A) Active-site cavity of unliganded AHL lactonase with glycerol molecules bound in the cavity (15). (B) Active-site cavity of complex I with the N-acyl-l-homoserine product bound distant from the dizinc metal center. (C) Active-site cavity of complex II with the N-acyl-l-homoserine product bound at the dizinc metal center. The zinc ions are shown as cyan spheres. All ligand molecules are shown in ball-and-stick form. Carbon, oxygen, and nitrogen atoms are colored white, red, and blue, respectively.
Structural Model of Product Complex II
A 1.4 Å resolution data set collected on the crystal formed from cocrystallization with the higher concentration of C6-HSL resulted in the second structural model (complex II). Although the protein used was the same as that used for complex I, the four additional N-terminal residues are not observed in complex II, likely the result of conformational disorder. Minor differences in the crystallization conditions such as the initial addition of methanol and different amounts of ligands may have slightly affected the pH, ionic strength, and crystal quality, resulting in this disorder. The structural model of complex II has 250 residues, starting with the first methionine encoded in the AHL lactonase gene (12).
The extra density observed in the active site of complex II is fit well by N-hexanoyl-l-homoserine, the ring-opened product derived from C6-HSL (Figure 4). Notably, the product’s position in complex II differs from that in complex I. In complex II, the carboxylate group of the product coordinates to both zinc ions in the dinculear site (Figure 4; Supporting Information, Figure 3). The resulting three-center bridge replaces the original bridging water/hydroxide and results in a Zn1−Zn2 distance (3.7 Å) much longer than that observed in both the unliganded structure (3.3 Å) (15) and complex I (3.3 Å). The coordination of the dizinc site is also altered (Figure 4; Supporting Information, Figure 3). The most striking difference occurs at Asp108, which now requires two alternative conformations (conformers a and b) to account for the observed electron density. In conformer a, the closest carboxylate oxygen of Asp108 is located farther from Zn2 (2.8 Å) than in both the unliganded protein structure and complex I (2.3 Å). In conformer b, the carboxyl group of Asp108 is completely dissociated from Zn2 with its two carboxyl oxygens located 3.9 and 4.7 Å from Zn2, respectively. Notably, in both conformers a and b, a side chain oxygen of Asp108 is within hydrogen bonding distance (2.6 and 2.7 Å, respectively) of the hydroxyl group of the ring-opened homoserine product (Figure 4; Supporting Information, Figure 3). In addition to the conformational changes observed in Asp108, another significant difference in metal coordination is observed with the bridging ligand Asp191. In both the unliganded structure (15) and complex I, Asp191 forms a monodentate bridge between both zinc ions, with the sole coordinating oxygen atom of Asp191 located 2.6 Å from Zn1 and 2.0 Å from Zn2, and the noncoordinated oxygen located 3.3 Å from Zn2. However, in complex II, the Asp191 side chain moves slightly toward Zn1, shortening its coordination distance to 2.4 Å even with the elongated Zn1−Zn2 distance. The coordination distance between Zn2 and the bridging oxygen atom of Asp191 remains at 2.0 Å, but the second carboxylate oxygen of Asp191 moves much closer (2.7 Å), indicating that Asp191 now forms a bidentate interaction with Zn2. A similar change in coordination is observed in the phosphate-bound structure of a related dizinc AHL lactonase from Agrobacterium tumefaciens(23). This type of aniso−bidentate interaction between zinc and carboxylate groups is well-established in coordination complexes (23). In fact, the flexibility of zinc to accommodate these additional partial bonds (typically with 2.4−2.8 Å Zn−O distances) has been suggested to be an important characteristic of zinc in enzyme mechanisms (24).
Figure 4.

Wall-eyed stereoview of the complex II active site. Two zinc ions are shown as silver-gray spheres. The bridging water/hydroxide is shown as a red sphere. Metal-coordinating residues and the homoserine product are shown as ball-and-stick representations. Two alternate conformations of the Asp108 side chain are labeled a and b. Oxygen atoms are colored red, and nitrogen atoms are colored blue. The carbon atoms in the product molecule are colored pale yellow. The carbon atoms in the coordinating residues are colored white. Electron density maps (with 2Fo − Fc coefficients) are shown in blue at 6σ around the zinc atoms and at 1σ around the product. Several key distances are given in angstroms and indicated with dashed black lines.
Another notable change in structure is observed in complex II. The large Y-shaped active-site cavity observed in the unliganded enzyme and in complex I is now collapsed, eliminating the smaller branch occupied by product in complex I and contouring nicely around the hydrolyzed lactone moiety of the bound product in complex II (Figure 3C). Although this change in cavity shape is dramatic, it results mostly from subtle side chain movements and is not the result of large changes in backbone positioning (Figure 5).
Figure 5.

Superimposed overall structures of complexes I and II. Complex I is colored green and complex II red. The N-acyl-l-homoserine products are shown in stick form. The zinc ions are shown as spheres: (A) side view and (B) top view. The most significant backbone variances between the two are indicated with arrows.
Structural Model of Product Complex III
A 1.3 Å resolution data set collected on the crystal formed from cocrystallization with C6-HCTL resulted in the third structural model (complex III). The overall structure and active-site configurations (Figure 6; Supporting Information, Figure 4) are very similar to complex II. In the active site, the ring-opened product derived from C6-HCTL, N-hexanoyl-l-homocysteine, is modeled with its carboxylate bridging the two zinc ions, although with a configuration slightly twisted from that in complex II. Although racemic substrate is used during crystallization, only the l-enantiomer is observed at the active site. Unlike that in complex II, the side chain of Asp108 in complex III only shows one configuration. The distance between the closest carboxyl oxygen of Asp108 and Zn2 is 3.3 Å. Notably, the side chain of Asp108 is not within hydrogen bonding distance of the leaving (thiol) group of the N-hexanoyl-l-homocysteine product because this thiol is pointing in a different direction than the corresponding hydroxyl group in complex II (Figure 6).
Figure 6.

Wall-eyed stereoview of the complex III active site. Two zinc ions are shown as silver-gray spheres, and the bridging water/hydroxide is shown as a red sphere. All metal-coordinating residues and the homoserine product are shown as ball-and-stick representations. All oxygen atoms are colored red and all nitrogen atoms blue. The carbon atoms in the N-acyl-l-homocysteine molecule are colored yellow and the carbon atoms in the coordinating residues white. Electron density maps (with 2Fo − Fc coefficients) are colored blue at 6σ around the zinc atoms and at 1σ around the N-acyl-l-homocysteine product. The Zn1−Zn2 distance is given in angstroms and indicated with a dashed black line.
Discussion
High-resolution structures of substrate-bound or product-bound enzymes in the metallo-β-lactamase superfamily are not plentiful. For the AHL lactonase family in particular, there have only been a few protein structures reported (10,15,23), and none with substrate or product bound. Despite the potential importance of these lactonases in quorum-quenching applications, the basis for their substrate discrimination and catalytic rate enhancements remains unresolved. Toward these ends, our crystallographic studies of the dizinc AHL lactonase from B. thuringiensis cocrystallized with low and high concentrations of a typical substrate (C6-HSL), and with a non-natural thiolactone substrate (C6-HCTL), resulted in the three product-bound structures reported herein. In each case, the observed ring-opened products were presumably derived from the intact substrates by AHL lactonase activity or by nonenzymic hydrolysis during crystal growth. These structural models are the first reported product-bound complexes in the AHL lactonase enzyme family. Observed interactions of the products with the dizinc metal center and active-site residues provide insight into the substrate binding determinants and catalytic mechanisms of these enzymes.
Cocrystallization of AHL lactonase with a typical substrate, C6-HSL [kcat = 91 s−1; KM = 5.6 mM for dizinc AHL lactonase (13)], resulted in extra electron density in the active-site cavity. This density was fit well by the expected ring-opened hydrolysis product, N-hexanoyl-l-homoserine, and the resulting product-bound structure is named complex I (Figure 2). Surprisingly, the product is located a considerable distance from the dizinc metal center (the product’s carboxylate is approximately 6 Å from Zn1), with its hydrophilic moieties occupying one of the smaller branches of the Y-shaped active-site cavity (Figure 3B). The product’s hydrophobic N-hexanoyl chain makes nonspecific interactions along the surface of the largest branch of the active-site cavity, which points toward the enzyme’s surface. Because the product is distant from the mechanistically important dinuclear metal center (12,13), the importance of the interacting protein residues for catalysis or specificity is not immediately clear. It is possible that complex I may reflect interactions that are made as the product exits the enzyme active site, but further experiments will be required to determine whether this structure is found on the reaction pathway.
Interestingly, the mode of product binding shows concentration dependence. Cocrystallizations containing a higher concentration of C6-HSL result in a structural model, named complex II, that shows a different product configuration (Figure 4). Notably, the product’s carboxylate is directly coordinated to the catalytic metal center, bridging the dinuclear zinc site in a bidentate fashion with each oxygen coordinated to a different zinc atom. The product’s hydroxyl leaving group interacts with the side chain of Asp108 (described in more detail below), and the amide nitrogen and carbonyl make through-water hydrogen bonds to the phenol group of Tyr194 and the backbone nitrogen of Phe107, respectively. The product’s hydrophobic N-hexanoyl chain again makes relatively nonspecific interactions along the surface of the same large branch of the active-site cavity as observed in complex I, although the particular interactions differ in detail. The finding that, in both complexes, the products’ N-hexanoyl chains make nonspecific surface interactions along a wide unconstrained cavity is very consistent with functional data. Typical AHL substrates have millimolar KM values, and there is very little discrimination between substrates with varying chain lengths or substitutions (11, 12,25).
In contrast to the relatively unconstrained binding cavity for the N-acyl chain, the rest of the product binding site has a more tailored fit. Part of the Y-shaped active-site cavity observed in the unliganded enzyme constricts in complex II and forms a tighter binding pocket around the hydrophilic portion of the product (Figure 3C). In fact, the smaller branch that was occupied by product in complex I is collapsed and is no longer visible in complex II. Active-site plasticity in enzymes within this superfamily has been noted before and is typically mediated by flap movements (26–29). However, the active-site constriction observed here results mostly from side chain motions; the main chain of the protein shows very little difference in positioning except for a small stretch of flexible loops (Figure 5). The proximity of the product to the metal center and the ability of the enzyme to mold around the product are consistent with complex II representing a species found on the reaction pathway. The mechanistic implications are discussed below. Product inhibition studies (Supporting Information, Figure 1) indicate that C6-homoserine is present at subsaturating concentrations (at least in solution phase), so it is not entirely surprising that a change in concentration may favor different binding modes.
Cocrystallization of AHL lactonase with a non-natural thiolactone substrate, C6-HCTL [kcat = 4.1 s−1 and KM = 36 mM for dizinc AHL lactonase (13)], resulted in extra electron density at the active site. This density was fit well by the expected hydrolysis product, N-hexanoyl-l-homocysteine, and the resulting structural model was named complex III (Figure 6). Although racemic substrate was used during crystallization, only one enantiomer of the product was observed, consistent with the finding that AHL lactonase is a stereospecific catalyst that hydrolyzes only the l-enantiomer of C6-HSL (12). The product’s location in complex III is quite similar to that observed in complex II, although there are minor differences. The product’s carboxylate binds directly to the metal center, replacing the bridging water/hydroxide observed in unliganded enzyme with a three-center bridge. This bidentate interaction is similar to that observed in complex II, but the product’s carboxylate is now somewhat twisted, placing one of the oxygens closer to the original position of the bridging water/hydroxide. The interactions of the product’s amide nitrogen and carbonyl are the same as those observed in complex II. In contrast, the leaving group thiol is not found within hydrogen bonding distance of the side chain of Asp108, which was observed with the corresponding hydroxyl leaving group in complex II. Further experiments will be required to determine whether the differences observed between complexes II and III reflect different species along the same reaction coordinate or instead reflect a change in mechanism upon sulfur substitution. It is somewhat surprising that the product’s thiol group is not coordinated to an active-site zinc ion because in most [but not all (30)] other examples of thiol-inhibited enzymes in this superfamily, the sulfur atom either bridges both zinc ions or binds directly to Zn2 (4,31–34).
The product complexes described here are the first reported for the quorum-quenching AHL lactonases. They provide considerable insight into how substrates might bind and into the catalytic mechanism. A previously proposed mechanism places the substrate’s lactone carbonyl oxygen over Zn2 and the leaving group oxygen over Zn1 (10). If this mechanism were correct, then the substrate would have to flip almost 180° to form the product complex (complex II), necessitating a substantial reorganization during catalysis. Instead, it is more likely that the substrate’s lactone carbonyl is bound near Zn1 and the leaving group near Zn2, requiring only minor movements during catalysis to reach the product placement observed here. This proposed substrate placement is more similar to those of other hydrolytic enzymes found in this superfamily (Figure 1). It is likely that the previous mechanism (10) was based on a nonproductive inhibitory binding mode of l-homoserine rather than on an on-pathway species more relevant to the catalytic mechanism.
Two active-site residues, Tyr194 and Asp108, are positioned to participate in the catalytic mechanism and are absolutely conserved in all known AHL lactonase enzymes. The importance of Tyr194 is evidenced by qualitative and quantitative analysis of mutations at this position (10, 11, 35). A previous mechanism proposes that Tyr194 acts as a general acid to protonate the leaving group oxygen (10). However, phenols normally have high pKa values (ca. 10), and complexes II and III reported here clearly show that this residue is not located near the leaving group (Figures 4 and 6). It is more likely that this tyrosine provides stabilization for the substrate’s carbonyl and/or a tetrahedral intermediate formed subsequent to hydroxide attack (15). In further support of this hypothesis, the recent structure of a related AHL lactonase from A. tumefaciens (AiiB) complexed with phosphate shows that the homologous tyrosine residue is well placed to interact with a tetrahedral species bound at the active site (23). This proposed role of Tyr194 is further supported by results presented in the following paper (11).
The Asp108 residue is also well placed to participate in the reaction mechanism. In the unliganded enzyme (15), this residue ligates Zn2 and makes a hydrogen bond to the bridging water/hydroxide, the likely nucleophile in the reaction. In complex II, the side chain of Asp108 is fit by two alternative conformers, both of which are located farther from Zn2 and are within hydrogen bonding distance of the hydroxyl leaving group of the product (Figure 4). In fact, conformer b is quite distant from Zn2, showing a complete loss of metal coordination by Asp108. These alternative conformers immediately suggest that Asp108 may act as a proton shuttle during the reaction. In the unliganded enzyme, the carboxylate side chain is poised to abstract a proton from the bridging hydroxide (presumably after its attack on the substrate). After proton transfer, a protonated Asp108 side chain would be a poor Zn2 ligand and would be free to release the metal ion and reposition for proton transfer to the leaving group hydroxide. In support of this idea, structures of a related metallo-β-lactamase variant at low pH values show dissociation of a homologus aspartyl group from Zn2 upon protonation (36). This proposed role for Asp108 is consistent with the functional studies reported in the following paper (11).
In a related metallo-β-lactamase enzyme, the homologous aspartate is proposed to serve primarily as an anchor for the proper positioning of Zn2 during catalysis (37). In contrast, AHL lactonase appears to make compensatory changes in the dizinc center to allow for dissociation of Asp108 without causing deleterious Zn2 movement. In complexes II and III, the bridging Asp191 forms a new bidentate interaction with Zn2 (Figures 4 and 6), instead of the monodentate interaction observed in the unliganded enzyme. In addition, the Zn1−Zn2 distance becomes considerably longer (3.7Å) than when product is not bound (3.3 Å). This lengthening is reminiscent of the long Zn1−Zn2 distance (4.3 Å) observed in a related AHL lactonase with phosphate bound (23) and may be the result of replacing the bridging water/hydroxide with a three-center bridge. As noted with other dinuclear metalloenzymes (38), changes in the coordination bridges and lengths may represent the flexibility of the metal site to coordinate different species along the reaction coordinate.
In summary, comparison of the unliganded enzyme structure with product-bound structures reveals various conformational changes that are not favored in the resting enzyme: a tightening of the active site around the hydrophilic moieties of the product, the release of Asp108 from Zn2, and a lengthening of the Zn1−Zn2 distance with a compensating bidentate interaction of Asp191 with Zn2. These changes have the potential to facilitate the ring opening hydrolysis catalyzed by AHL lactonase. The product complexes reported here are consistent with proposing a new substrate binding model which places the lactone carbonyl over Zn1 and the leaving group oxygen over Zn2, with the less hindered re face of the lactone facing the available coordination sites of the metal center (Figure 7). This substrate orientation is consistent with that seen in the other superfamily members, including metallo-β-lactamase, which place the less hindered exoface of the β-lactam ring toward the active-site metals (Figure 1). This orientation would allow facile attack of the bridging hydroxide at the lactone’s carbonyl carbon, polarization of this carbonyl bond by coordination of oxygen to Zn1, and stabilization of the leaving group heteroatom by coordination to Zn2. The phenol of Tyr194 can stabilize substrate binding and/or a tetrahedral species. Finally, Asp108 is positioned to function as a proton shuttle between the attacking hydroxide and the leaving group, as evidenced by its altered conformations upon product binding. These specific hypotheses are experimentally tested in the following paper (11) and, along with previous studies, lead to the proposal of a detailed catalytic mechanism for AHL lactonases.
Figure 7.

Proposed substrate binding model. On the basis of the observed product complexes, a substrate binding model is presented that is consistent with substrate and product complexes observed in AHL lactonase and in other metallo-β-lactamase superfamily members.
From a larger perspective, there are intriguing similarities between β-lactam and AHL processing systems. Some small-molecule antibiotics have the ability to trigger cell signaling pathways, and it has been suggested that they may serve as cell-to-cell signaling molecules when produced in native environments (39). The structural and chemical similarities between the AHL signaling agents and naturally occurring β-lactams, the structural similarities between the metallo-β-lactamases and the AHL lactonases, and the substrate binding and mechanistic similarities between these two enzyme families all raise intriguing possibilities about the intertwined natural evolutionary history of these catalysts and their role in the chemical ecology of interacting microbial systems. Comparing the structural and mechanistic features of these catalysts will help us to better understand the divergence and specialization of function in this enzyme superfamily.
Acknowledgments
We thank the whole GM/CA-CAT staff for help and support during data collection and Drs. Mark Wilson and Edwin Pozharski for their assistance in SHELX refinement. We also thank Noah Wolfson for help preparing protein for crystallization.
Supporting Information Available
Four figures depicting product inhibition and product binding interactions observed in complexes I, II, and III. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Footnotes
Abbreviations: AHL, N-acyl-l-homoserine lactone; HSL, homoserine lactone; HCTL, homocysteine thiolactone; MBP, maltose binding protein; ICP-MS, inductively coupled plasma mass spectrometry; aiiA and AiiA, gene and gene product for AHL lactonase from B. thuringiensis, respectively; FFT, fast Fourier transfer; rmsd, root-mean-square deviation.
Supplementary Material
References
- Aravind L. (1999) An evolutionary classification of the metallo-β-lactamase fold proteins. In Silico Biol. 1, 69–91. [PubMed] [Google Scholar]
- Daiyasu H.; Osaka K.; Ishino Y.; Toh H. (2001) Expansion of the zinc metallo-hydrolase family of the β-lactamase fold. FEBS Lett. 503, 1–6. [DOI] [PubMed] [Google Scholar]
- Crowder M. W.; Spencer J.; Vila A. J. (2006) Metallo-β-lactamases: Novel weaponry for antibiotic resistance in bacteria. Acc. Chem. Res. 39, 721–728. [DOI] [PubMed] [Google Scholar]
- Cameron A. D.; Ridderstrom M.; Olin B.; Mannervik B. (1999) Crystal structure of human glyoxalase II and its complex with a glutathione thiolester substrate analogue. Struct. Folding Des. 7, 1067–1078. [DOI] [PubMed] [Google Scholar]
- Hagelueken G.; Adams T. M.; Wiehlmann L.; Widow U.; Kolmar H.; Tummler B.; Heinz D. W.; Schubert W. D. (2006) The crystal structure of SdsA1, an alkylsulfatase from Pseudomonas aeruginosa, defines a third class of sulfatases. Proc. Natl. Acad. Sci. U.S.A. 103, 7631–7636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer J.; Read J.; Sessions R. B.; Howell S.; Blackburn G. M.; Gamblin S. J. (2005) Antibiotic recognition by binuclear metallo-β-lactamases revealed by X-ray crystallography. J. Am. Chem. Soc. 127, 14439–14444. [DOI] [PubMed] [Google Scholar]
- Garau G.; Bebrone C.; Anne C.; Galleni M.; Frere J. M.; Dideberg O. (2005) A metallo-β-lactamase enzyme in action: Crystal structures of the monozinc carbapenemase CphA and its complex with biapenem. J. Mol. Biol. 345, 785–795. [DOI] [PubMed] [Google Scholar]
- Garau G.; Lemaire D.; Vernet T.; Dideberg O.; Di Guilmi A. M. (2005) Crystal structure of phosphorylcholine esterase domain of the virulence factor choline-binding protein e from Streptococcus pneumoniae: New structural features among the metallo-β-lactamase superfamily. J. Biol. Chem. 280, 28591–28600. [DOI] [PubMed] [Google Scholar]
- Silva M. S.; Barata L.; Ferreira A. E.; Romao S.; Tomas A. M.; Freire A. P.; Cordeiro C. (2008) Catalysis and Structural Properties of Leishmania infantum Glyoxalase II: Trypanothione Specificity and Phylogeny. Biochemistry 47, 195–204. [DOI] [PubMed] [Google Scholar]
- Kim M. H.; Choi W. C.; Kang H. O.; Lee J. S.; Kang B. S.; Kim K. J.; Derewenda Z. S.; Oh T. K.; Lee C. H.; Lee J. K. (2005) The molecular structure and catalytic mechanism of a quorum-quenching N-acyl-l-homoserine lactone hydrolase. Proc. Natl. Acad. Sci. U.S.A. 102, 17606–17611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momb J.; Wang C.; Liu D.; Thomas P. W.; Petsko G. A.; Guo H.; Ringe D.; Fast W. (2008) Mechanism of the quorum-quenching lactonase (AiiA) from Bacillus thuringiensis. 2. Substrate Modeling and Active Site Mutations. Biochemistry 47, 7715–7725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P. W.; Stone E. M.; Costello A. L.; Tierney D. L.; Fast W. (2005) The quorum-quenching lactonase from Bacillus thuringiensis is a metalloprotein. Biochemistry 44, 7559–7569. [DOI] [PubMed] [Google Scholar]
- Momb J.; Thomas P. W.; Breece R. M.; Tierney D. L.; Fast W. (2006) The quorum-quenching metallo-γ-lactonase from Bacillus thuringiensis exhibits a leaving group thio effect. Biochemistry 45, 13385–13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Liu D.; Lepore B. W.; Petsko G. A.; Thomas P. W.; Stone E. M.; Fast W.; Ringe D. (2005) Three-dimensional structure of the quorum-quenching N-acyl homoserine lactone hydrolase from Bacillus thuringiensis. Proc. Natl. Acad. Sci. U.S.A. 102, 11882–11887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storoni L. C.; McCoy A. J.; Read R. J. (2004) Likelihood-enhanced fast rotation functions. Acta Crystallogr. D60, 432–438. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Numer 4 (1994) Acta Crystallogr. D50, 760–763. [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D53, 240–255. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr. D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M. (2008) A short history of SHELX. Acta Crystallogr. A64, 112–122. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. (2004) UCSF Chimera: A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Kleywegt G. J.; Jones T. A. (1996) xdlMAPMAN and xdlDATAMAN: Programs for reformatting, analysis and manipulation of biomacromolecular electron-density maps and reflection data sets. Acta Crystallogr. D52, 826–828. [DOI] [PubMed] [Google Scholar]
- Liu D.; Thomas P. W.; Momb J.; Hoang Q. Q.; Petsko G. A.; Ringe D.; Fast W. (2007) Structure and specificity of a quorum-quenching lactonase (AiiB) from Agrobacterium tumefaciens. Biochemistry 46, 11789–11799. [DOI] [PubMed] [Google Scholar]
- Harding M. M. (1999) The geometry of metal-ligand interactions relevant to proteins. Acta Crystallogr. D55, 1432–1443. [DOI] [PubMed] [Google Scholar]
- Wang L. H.; Weng L. X.; Dong Y. H.; Zhang L. H. (2004) Specificity and enzyme kinetics of the quorum-quenching N-acyl homoserine lactone lactonase (AHL-lactonase). J. Biol. Chem. 279, 13645–13651. [DOI] [PubMed] [Google Scholar]
- Scrofani S. D.; Chung J.; Huntley J. J.; Benkovic S. J.; Wright P. E.; Dyson H. J. (1999) NMR characterization of the metallo-β-lactamase from Bacteroides fragilis and its interaction with a tight-binding inhibitor: Role of an active-site loop. Biochemistry 38, 14507–14514. [DOI] [PubMed] [Google Scholar]
- Huntley J. J.; Fast W.; Benkovic S. J.; Wright P. E.; Dyson H. J. (2003) Role of a solvent-exposed tryptophan in the recognition and binding of antibiotic substrates for a metallo-β-lactamase. Protein Sci. 12, 1368–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntley J. J.; Scrofani S. D.; Osborne M. J.; Wright P. E.; Dyson H. J. (2000) Dynamics of the metallo-β-lactamase from Bacteroides fragilis in the presence and absence of a tight-binding inhibitor. Biochemistry 39, 13356–13364. [DOI] [PubMed] [Google Scholar]
- Garrity J. D.; Pauff J. M.; Crowder M. W. (2004) Probing the dynamics of a mobile loop above the active site of L1, a metallo-β-lactamase from Stenotrophomonas maltophilia, via site-directed mutagenesis and stopped-flow fluorescence spectroscopy. J. Biol. Chem. 279, 39663–39670. [DOI] [PubMed] [Google Scholar]
- Garcia-Saez I.; Mercuri P. S.; Papamicael C.; Kahn R.; Frere J. M.; Galleni M.; Rossolini G. M.; Dideberg O. (2003) Three-dimensional structure of FEZ-1, a monomeric subclass B3 metallo-β-lactamase from Fluoribacter gormanii, in native form and in complex with d-captopril. J. Mol. Biol. 325, 651–660. [DOI] [PubMed] [Google Scholar]
- Concha N. O.; Janson C. A.; Rowling P.; Pearson S.; Cheever C. A.; Clarke B. P.; Lewis C.; Galleni M.; Frere J. M.; Payne D. J.; Bateson J. H.; Abdel-Meguid S. S. (2000) Crystal structure of the IMP-1 metallo β-lactamase from Pseudomonas aeruginosa and its complex with a mercaptocarboxylate inhibitor: Binding determinants of a potent, broad-spectrum inhibitor. Biochemistry 39, 4288–4298. [DOI] [PubMed] [Google Scholar]
- Kurosaki H.; Yamaguchi Y.; Higashi T.; Soga K.; Matsueda S.; Yumoto H.; Misumi S.; Yamagata Y.; Arakawa Y.; Goto M. (2005) Irreversible inhibition of metallo-β-lactamase (IMP-1) by 3-(3-mercaptopropionylsulfanyl)propionic acid pentafluorophenyl ester. Angew. Chem., Int. Ed. 44, 3861–3864. [DOI] [PubMed] [Google Scholar]
- Kurosaki H.; Yamaguchi Y.; Yasuzawa H.; Jin W.; Yamagata Y.; Arakawa Y. (2006) Probing, inhibition, and crystallographic characterization of metallo-β-lactamase (IMP-1) with fluorescent agents containing dansyl and thiol groups. ChemMedChem 1, 969–972. [DOI] [PubMed] [Google Scholar]
- Nauton L.; Kahn R.; Garau G.; Hernandez J. F.; Dideberg O. (2008) Structural insights into the design of inhibitors for the L1 metallo-β-lactamase from Stenotrophomonas maltophilia. J. Mol. Biol. 375, 257–269. [DOI] [PubMed] [Google Scholar]
- Lu X.; Yuan Y.; Xue X. L.; Zhang G. P.; Zhou S. N. (2006) Identification of the critical role of Tyr-194 in the catalytic activity of a novel N-acyl-homoserine lactonase from marine Bacillus cereus strain Y2. Curr. Microbiol. 53, 346–350. [DOI] [PubMed] [Google Scholar]
- Davies A. M.; Rasia R. M.; Vila A. J.; Sutton B. J.; Fabiane S. M. (2005) Effect of pH on the active site of an Arg121Cys mutant of the metallo-β-lactamase from Bacillus cereus: Implications for the enzyme mechanism. Biochemistry 44, 4841–4849. [DOI] [PubMed] [Google Scholar]
- Llarrull L. I.; Fabiane S. M.; Kowalski J. M.; Bennett B.; Sutton B. J.; Vila A. J. (2007) Asp120 locates Zn2 for optimal metallo-β-lactamase activity. J. Biol. Chem. 282, 18276–18285. [DOI] [PubMed] [Google Scholar]
- Allen K. N.; Lavie A.; Glasfeld A.; Tanada T. N.; Gerrity D. P.; Carlson S. C.; Farber G. K.; Petsko G. A.; Ringe D. (1994) Role of the divalent metal ion in sugar binding, ring opening, and isomerization by d-xylose isomerase: Replacement of a catalytic metal by an amino acid. Biochemistry 33, 1488–1494. [DOI] [PubMed] [Google Scholar]
- Yim G.; Wang H. H.; Davies J. (2007) Antibiotics as signaling molecules. Philos. Trans. R. Soc. London, Ser. B 362, 1195–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.