

Abstract
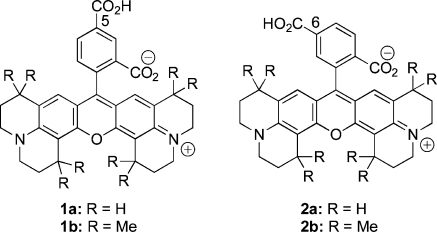
An efficient route is reported to 5- and 6-carboxy-X-rhodamines (compounds 1 and 2) that contain multiple n-propylene or γ,γ-dimethylpropylene groups bridging terminal nitrogen atoms and the central xanthene core. Gram quantities of these dyes are synthesized from inexpensive starting materials. The isolated products are activated by selective transformation of the carboxylic acid group into N-hydroxysuccinimidyl esters in situ and then conjugated with an amino group of a molecule of interest.
Rhodamine dyes are important because of their favorable photochemical and photophysical properties.(1) They have long wavelength absorption maxima, high molar absorptivities, and high quantum yields.(2) Compared to the fluorescein dyes, rhodamines are relatively resistant to photobleaching and their fluorescence spectra are pH independent over a range from 4 to 10.(3) For these reasons, they have broad application, not only in biotechnology for fluorescent labeling or single molecule detection but also in medicine for imaging living cells or live animals in preclinical research.6,7 In recent years, a wide variety of rhodamine dyes have been commercialized for conjugation with biomolecules.(2) Of these, 5- and 6-carboxy-X-rhodamines (1 or 2) are of special interest because of their highly intense absorption and emission. This is possibly due to the presence of the added structural rigidity introduced by multiple n-propylene bridges, which prevents fluorescence deactivation by nonradiative processes. The high cost of these dyes from commercial sources limits their use in biomedical research. Although different methods are available in the literature for the synthesis of diverse rhodamine derivatives,8−11 no report describes the synthesis of 5- and 6-carboxy-X-rhodamines. Therefore, as a part of our ongoing program to develop fluorescent ligands, we report the synthesis and structure of 5- and 6-carboxy-X-rhodamine dyes that contain repetitive n-propylene or γ,γ-dimethylpropylene bridging moieties. In addition, we describe an efficient route to activate and conjugate these dyes with the amino group of a molecule of interest to provide easy excess to fluorescent ligands that are useful for materials and biomedical and clinical research.
The alkylation of m-anisidine (3) using 1-chloro-3-methylbut-2-ene in the presence of K2CO3 gave 3-methoxy-N,N-bis(3-methylbut-2-enyl)aniline (4) in 69% yield. The treatment of compound 4 with concd HCl at 0 °C afforded 3-methoxy-N,N-bis(3-methylbut-2-enyl)aniline hydrochloride (5) in 99% yield. Intramolecular cyclization of compound 5 using neat MeSO3H at 95 °C gave 1,1,7,7-tetramethyl-8-methoxyjulolidine (6) in 65% yield, and O-desmethylation of compound 6 using BBr3 gave the corresponding 1,1,7,7-tetramethyl-8-hydroxyjulolidine (7b) in 80% yield. The 8-hydroxyjulolidine (7a) was synthesized using a reported protocol.(12) The synthesis of 5- or 6-carboxy-X-rhodamine dyes (1 or 2) requires a symmetrical condensation of 2 equiv of compound 7 with 1 equiv of 4-carboxyphthalic anhydride. The overall process requires two sequential Friedel−Crafts-type electrophilic aromatic substitution reactions for the formation of the xanthene skeleton. We first tried a traditional condensation protocol involving fusion of compound 7 (R = H or Me) with 4-carboxyphthalic anhydride at high temperature (180 °C). This reaction gave minimal yield, possibly due to considerable sublimation of the starting materials arround the vessel wall. Next, we used a catalytic amount of Lewis acid (ZnCl2), or a protic acid (H2SO4) that gave a poor yield of the desired products. However, the yield was dramatically increased by using a high-boiling weakly acidic solvent (n-PrCO2H, pKa 4.82) with a trace of 2 M H2SO4 under reflux (Scheme 1).
Scheme 1. Synthesis of Rhodamine-Based Fluorescent Dyes 1a,b and 2a,b.

The separation of the two isomers from the regioisomeric mixture [1a, 2a (R = H) or 1b, 2b (R = Me)] was achieved by flash chromatography. Extensive optimization was performed to develop conditions for the separation of significant quantities of 1 and 2. Silica gel 60 (230−450 mesh) was slurry packed into a 10 in. column (1 × 15 in.) and eluted with methanol/chloroform gradients at a flow rate of 5 mL/min. The progress of separation was monitored with a Spectroline ENF-280C UV detector (365 nm window). The best yield was obtained using 0.5 g of crude sample with relatively shallow gradients. In a typical experiment, we obtained 34% and 32% isolated yields for compounds 1a and 2a or 42% and 15% isolated yield for compounds 1b and 2b, respectively.
The isomeric purity of each of the isolated compounds was determined by HPLC analysis to be greater than 99%.
A complete structural analysis of compounds 1 and 2 was conducted using 1D and 2D NMR spectroscopy. There are three aromatic protons in each of these compounds that are characteristic for their differentiation by 1H NMR. For compound 1a, signals for two neighboring aryl protons at position-6 and position-7 (Harom-6 and Harom-7) were assigned as two separate doublets at 8.18 and 7.05 ppm, respectively, with a large vicinal coupling constant (J = 9.0 Hz). The assignment was confirmed by a 1H−1H COSY experiment, where Harom-6 showed a strong correlation with Harom-7 (Supporting Information). The proton located at position-4 (Harom-4) was assigned at 8.27 ppm and showed a weak correlation with Harom-6 in the COSY spectrum. This weak correlation indicates that Harom-4 is located at least four bonds from Harom-6 and supports the location of Harom-4 within two carboxyl groups of the aryl ring. In compound 2a, the resonances for the two neigboring aryl protons (Harom-4 and Harom-5) appeared as two doublets at 7.77 ppm and 8.04 ppm (J = 8.9 Hz), respectively. This assignment was confirmed by a 1H−1H COSY spectrum, in which Harom-4 showed a strong correlation with Harom-5. The sharp singlet located at 7.43 ppm was assigned as Harom-7, that has a weak correlation with Harom-5 in the COSY spectrum. This indicates that Harom-7 is located between the carboxyl-group and rhodamine scaffold. The chemical shifts for the two rhodaminyl/vinylic CH protons were assigned at 6.0 ppm. These two protons (H-8 and H-9) are equivalent. The CO2H proton on the aryl ring of compound 1a or 2a appeared at 13.22 ppm as a singlet. The chemical shift resonances and coupling patterns for the aliphatic protons of compounds 1a and 2a are almost identical. Details are described in the Supporting Information section.
Figure 1.

1H NMR spectra of compounds 1a and 2a dissolved in DMSO-d6 at 500 MHz.
We synthesized 4-methylbenzoylpiperazine hydrochloride (10) using a protocol described in the Scheme 2. Briefly, acylation of N-BOC piperazine by 4-methylbenzoyl chloride (8) in the presence of N,N-diisoproylethylamine gave 4-(4-methylphenylcarbonyl)piperazine-1-carboxylate (9) in 79% yield. The 4-(4-methylphenylcarbonyl)piperazine-1-carboxylate (9) was then deprotected to compound 10 using hydrogen chloride gas. This reaction proceeded in quantitative yield. The fluorescent compound 1a or 1b was activated using N,N,N,N-tetramethyl-O-(N-succinimidyl)uronium tetrafluoroborate (TSTU). The reaction was performed in the presence of ≥2 equiv of base, such as triethylamine, to give the corresponding N-hydroxysuccinimidyl ester which was generated in situ. The ester was then reacted with the amino group of compound 10. This coupling reaction proceeded efficiently with only a slight excess of TSTU. Air-free techniques were used for both activation and coupling reactions. The succinimidyl esterification was performed in an activator vessel under an inert atmosphere, and the activated dye was transferred via a cannula to the coupling vessel that contained compound 10 in dimethyl sulfoxide. The crude product was obtained by removal of solvent, and purification was conducted by a silica gel flash column chromatography using an isocratic elution of 35:7:1 CHCl3/MeOH/concd NH4OH. This procedure afforded 64% yield for compound 11. The structures were confirmed by 1H NMR and mass spectrometry. Compound 11 is a model for targeted small molecule−carboxy-X-rhodamine conjugates.
Scheme 2. Conjugate Chemistry.

The rigid rhodamine dyes 1a,b and 2a,b exhibited strong fluorescence properties in aqueous buffer. Compounds 1a,2a [1a: λexit = 580 nm (ε = 3.6 × 104 M−1 cm−1), λemit = 604 nm (ϕ = 0.94); 2a: λexit = 581 nm (ε = 3.6 × 104 M−1 cm−1), λemit = 605 nm (ϕ = 0.96)] and 1b,2b [1b: λexit = 582 nm (ε = 4.1 × 104 M−1 cm−1), λemit = 605 nm (ϕ = 0.91); 2b: λexit = 583 nm (ε = 4.1 × 104 M−1 cm−1), λemit = 606 nm (ϕ = 0.92)] demonstrated similar photophysical properties. Also, their absorption and emission maxima are essentially identical with those of their conjugates 11 [λexit = 581 nm (ε = 3.6 × 104 M−1 cm−1), λemit = 604 nm (ϕ = 0.92)]. Compared with a rhodamine dye [rhodamine B: λexit = 554 nm (9.7 × 104 M−1 s−1), λemit = 573 nm (ϕ = 0.36)](13) that lacks such rigidity, compounds 1, 2, or 11 exhibited red-shifted (30−40 nm) absorption and emission maxima and higher fluorescence quantum yields. Octamethyl substitution (1a,2a → 1b,2b) modestly increases the molar absorptivity and quantum yields.
In summary, we report the first synthesis of 5- and 6-carboxy-X-rhodamines having multiple n-propylene or γ,γ-dimethylpropylene bridging moieties that provide rigidity for preventing nonradiative fluorescence deactivation processes. Synthesis of gram quantities of these fluorescent dyes is conducted from inexpensive starting materials. We describe the structural analysis and methodology for activation followed by conjugation of the dye with a molecule of interest. These dyes and their conjugates exhibit red-shifted absorption and emission maxima and high quantum yields for fluorescence. The availability of a facile and inexpensive route to these fluorophores should enable their adoption for a broad range of applications in material sciences, biotechnology, and biomedical imaging.
Acknowledgments
We are grateful to Brenda C. Crews and Dr. Carol Rouzer of the Department of Biochemistry for critical readings of this manuscript and to Gert-Jan Kremers of the Department of Molecular Physiology and Biophysics for quantum yield measurements. We gratefully acknowledge the National Institutes of Health (U54CA105296) for financial support.
Supporting Information Available
Complete description of the experimental details and product characterization. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a He J.; Ritalahti K. M.; Yang K.-L.; Koenigsberg S. S. Loffler Nature 2003, 424, 62. [DOI] [PubMed] [Google Scholar]; b George A.; Mottola-Hartshorn C.; Warner A.; Fields B.; Chen L. B. Proc. Natl. Acad. Sci. U.S.A. 1990, 87, 6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hougland R. P.Handbook of Fluorescent Probes and Research Products, 9th ed.; Molecular Probes Inc.: Eugene, OR, 2002. [Google Scholar]
- a Lakowicz J. R.Principles of Fluorescnce Spectroscopy, 2nd ed.; Plenum Publishing: New York, 1999. [Google Scholar]; b Valeur B.Molecular Fluorescence: Principles and Applications; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Kreis T. E.; Geiger B.; Schlessinger J. Cell 1982, 29, 835. [DOI] [PubMed] [Google Scholar]
- a Kwon J. Y.; Jang Y. J.; Lee Y. J.; Kim K. M.; Seo M. S.; Nam W.; Yoon J. J. Am. Chem. Soc. 2005, 127, 10107. [DOI] [PubMed] [Google Scholar]; b Yang Y.-K.; Yook K.-J.; Tae J. J. Am. Chem. Soc. 2005, 127, 16760. [DOI] [PubMed] [Google Scholar]
- a Johnson L. V.; Walsh M. L.; Chen L. B. Proc. Natl. Acad. Sci. U.S.A. 1980, 77, 990. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Belmont L. D.; Hyman A. A.; Sawin K. E.; Mitchison T. J. Cell 1990, 62, 579. [DOI] [PubMed] [Google Scholar]
- Marnett L. J.; Uddin M. J.; Crews B. C. US Patent Application WO/2007149546, 2007.
- Scala-Valero C.; Doizi D.; Guillaumet G. Tetrahedron Lett. 1999, 40, 4803. [Google Scholar]
- Kojima H.; Hirotani M.; Urano Y.; Kikuchi K.; Higuchi T.; Nagano T. Tetrahedron Lett. 2000, 41, 69. [Google Scholar]
- Jiao G.-S.; Castro J. C.; Thoresen L. H.; Burgess K. Org. Lett. 2003, 5, 3675. [DOI] [PubMed] [Google Scholar]
- Liu J.; Giwu Z.; Leung W.-Y.; Lu Y.; Patch B.; Haugland R. P. Tetrahedron Lett. 2003, 44, 4355. [Google Scholar]
- Gompel J. V.; Schuster G. B. J. Org. Chem. 1987, 52, 1465. [Google Scholar]
- Nguyen T.; Francis M. B. Org. Lett. 2003, 5, 3245. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.