

Abstract
Chaperonins engulf other proteins and accelerate their folding by an unknown mechanism. Here, we combine all-atom molecular dynamics simulations with data from experimental assays of the activity of the bacterial chaperonin GroEL to demonstrate that a chaperonin’s ability to facilitate folding is correlated with the affinity of its interior surface for water. Our results suggest a novel view of the behavior of confined water for models of in vivo protein folding scenarios.
Protein folding in the cell often relies on the help of chaperonins, barrel-shaped enzymes that assist the folding of their substrates through an ATP-driven cycle of encapsulation and release.1,2 Water, which is crucial to the folding of all proteins, can become confined on the scale of a nanometer or less inside chaperonins and might therefore be expected to behave differently there than it does in bulk. Despite this, the prevailing view is that chaperonin function can be explained without explicitly considering the impact of the chaperonin on the water it encloses.(3) We have argued previously3,4 that a chaperonin cavity’s capacity for accumulating water near its surface should correlate with its success in catalyzing folding. Here, we test this prediction by analyzing simulations of mutants of the GroE complex whose activities were characterized in past experiments.
The crystal structure of GroEL+ES indicates that the complex undergoes a pronounced conformational change upon substrate encapsulation that significantly increases the number of polar residues projecting into the cavity’s interior.1,2 Past experiments have pointed to a role for these residues in chaperonin function; in two separate studies, the hydrophilicity of the cavity surface was shown to correlate qualitatively with the enzyme’s ability to assist in substrate folding.5,6 We hypothesized that polar residues on the inner surface of the chaperonin promote folding by causing water to accumulate in their vicinity, thereby producing a local environment in which an unfolded substrate pays a higher thermodynamic cost for failing to bury the hydrophobic parts of its surface that cannot hydrogen bond with the solvent.3,4
In order to test our hypothesis, we carried out all-atom, explicit solvent, molecular dynamics simulations of a series of GroEL mutants that were previously assayed for their ability to assist in the refolding of a slow-folding double mutant of maltose binding protein (DM-MBP).(6) Mutants of the single GroEL+ES closed complex were generated from the crystal structure 1PCQ in accordance with the mutations described in ref 6. Structures were solvated in a cube of water 20 nm per side with the KCl concentration following previous experiments, yielding a system with ∼242 000 solvent molecules. The size of the box was chosen to be the smallest one for which water density at the boundary was comparable to bulk density. Energies were minimized using steepest descent before molecular dynamics (MD) runs, and alpha carbons were harmonically restrained. NVT MD simulations were performed with the software package GROMACS version 3.3.1 with the Amber 2003 force-field and a TIP4P-EW water model. Temperature was fixed at 298 K using the Berendsen algorithm, and electrostatics were calculated with the particle mesh Ewald method. Trajectories of 850 ps were run for each mutant at 298 K with snapshots stored every 50 ps. The coordinates from the first 200 ps of the trajectory were discarded to allow the solvent to relax locally. Surface water oxygens were counted in a 5 nm vertical stack of 4 Å slices. For the alpha carbons in each slice, the rms distance d from the vertical axis (whose origin was the center of mass of the complex) was calculated, and waters were counted as near the surface if they were less than d from the vertical axis but greater than d − 1 nm. The layer width of 1 nm was chosen, since it is at roughly this range that we would expect solvent-mediated forces between the chaperonin and substrate surfaces to become important3,4 (see Supporting Information).
We calculated the average number of water molecules within 1 nm of the surfaces of different mutated GroEL complexes (Figure 1b) and compared this microscopic measure of surface hydrophilicity to the experimentally measured DM-MBP refolding rates inside each mutant (Figure 1a). As predicted, the DM-MBP refolding rate proved to be a strong predictor of cavity surface hydrophilicity (inset, corr. = 0.78). This correlation is notable because it constituted a substantial improvement over the performance of cavity surface charge (corr. = − 0.66; see Supporting Information), which accounts for much of the hydrophilicity but fails to capture effects arising from subtler differences in the spatial distribution of charge (a surface pocked with equal numbers of negative and positive point charges, for example, is obviously more hydrophilic than one displaying no charges at all). Our measure of hydrophilicity also performed far better than the area-weighted Kyte−Doolittle hydropathy(7) of the cavity surface (corr. = 0.06; see Supporting Information), which showed no correlation with folding rate whatsoever.
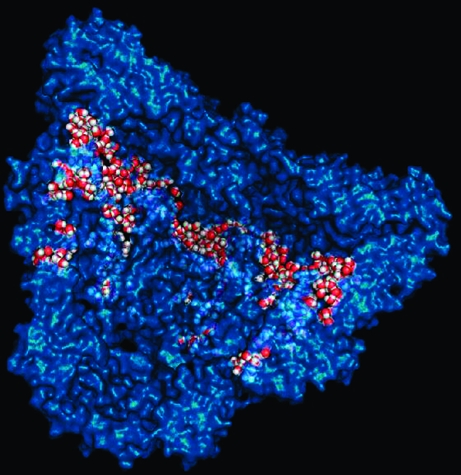
Figure 1.

(a) Gray bars report the experimentally measured folding rates of DM-MBP when encapsulated inside different mutants of SR-EL, a single ring variant of GroEL,(6) and blue bars show the best linear fit of the number of surface waters to the measured refolding rate from the inset scatter plot (corr. = 0.78). Error bars for the surface waters are calculated for 95% confidence based on the standard error of the sample. The mutants are colored and ordered left to right according to their interior cavity surface charge, with the most negative being leftmost. The nomenclature for the mutants is taken from the experimental reference. (b) The blue, partly transparent surface is a cut-away view of wild type GroEL+ES. The waters counted as near the surface for this snapshot are colored red and white. The image was generated using MacPymol.
Strikingly, our atomistic calculation of cavity hydrophilicity is able to distinguish between the activities of nearly equivalent mutants. Of the four pairs of mutants of the same type (D to N, −DE to KKK, etc.) that differed only in the locations of the mutated residues, three of them gave the correct rank ordering for the impact on folding rate. The predicted order is also correct for the three mutants with equal surface charge (KKK1, KKK2, and 3N3Q), accurately placing a six-residue charged-to-neutral mutant in between two different triple-lysine substitutions that one might have initially guessed would closely resemble each other given the extreme similarity of their sequences.
It is possible that the differences in measured hydrophilicity between seemingly equivalent mutants arose from the variable thickness of the water layer at different heights in the cavity (see Supporting Information). When we examined the crystal structure in detail at the mutation sites, however, we found that these differences might also be explained as a result of the spatial arrangement of charges at each site. Both experimentally measured refolding rates and hydrophilicity calculated from simulation proved to be more sensitive to mutations of the GroEL cavity at sites near aspartate 359 than at sites near aspartate 253. As Figure 2a demonstrates, the residues at locus 253 point in roughly the same direction on a relatively less curved surface and largely border other charged residues. In contrast, Figure 2b shows that, at locus 359, the three mutated residues project in different directions on a more curved surface and have more neighboring residues that are electrically neutral. An equal number of charged residues should be expected to have a larger total impact on hydrophilicity when spread farther apart in space. Charges crammed closer together are forced to exert their influence on the same small volume of solvent, and solvent that is already at elevated density and depressed entropy from the influence of one charge should be less able to respond to the influence of another charge (Figure 2, left side). This provides an explanation for the difference between locus 253 and locus 359; the latter is more sensitive to mutation because the sites mutated exert better-separated influences on different regions in the aqueous solvent, and each individual residue therefore has a larger impact on hydrophilicity.
Figure 2.

Surface maps of the crystal structure of GroEL+ES are colored by residue. The three residues altered in the mutants D253K, QNQ, D253N, and KKK1 (a) and in D359K, D359N, NNQ, and KKK2 (b) are colored in orange, pink, and yellow. Charged residues are colored in red for negative and blue for positive. To the left, a schematic illustrates the greater overlap of hydration spheres for charged residues arranged closer together in space.
The data presented here advance a new framework for understanding the mechanism of chaperonin function. In this view, accelerated folding occurs, at least in part, because the solvent environment inside the chaperonin is altered by the cavity walls in order to provide a stronger drive for the decrease in substrate surface hydrophobicity that accompanies folding. This mechanism of chaperonin action is consistent with past experiments demonstrating that hydrophobic-to-polar surface mutants of GroES have enhanced activity.(5) It also helps to explain the ability of chaperonins to promote folding in a wide variety of proteins (most of which, presumably, are stabilized by the enhanced hydrophobic effect inside the chaperonin), as well as the enrichment of GroEL substrates in TIM barrel folds(8) (whose folding mechanisms are thought to be particularly dependent on the formation of “hydrophobic clusters” of residues forming long-range contacts(9)). It is our hope that this first success for confined solvent effects in shedding light on one aspect of intracellular protein folding may serve as a guide for future attempts to understand the impact of nonbulk-like water on a wide variety of in vivo folding scenarios ranging from the crowded cytosol to the ribosome.
Acknowledgments
This publication and the project described was supported by the National Institutes of Health through the NIH Roadmap for Medical Research (PN2 EY016525). The authors also relied on NSF Award CNS-0619926 for computer resources. J.E. thanks Drs. Judith Frydman and Daniel Kaganovich for helpful comments and the Fannie and John Hertz Foundation for financial support.
Supporting Information Available
Experimental details. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hartl F. U.; Hayer-Hartl M. Science 2002, 295, 1852–1858. [DOI] [PubMed] [Google Scholar]
- Fenton W. A.; Horwich A. L. Q. Rev. Biophys. 2002, 36, 229–256. [DOI] [PubMed] [Google Scholar]
- England J.; Lucent D.; Pande V. Curr. Opin. Struct. Biol. 2008, 18, 163–169. [DOI] [PubMed] [Google Scholar]
- England J.; Pande V. Biophys. J.2008, published online July 3, doi: 10.1529/biophysj.108.131037..
- Wang J. D.; Herman C.; Tipton K. A.; Gross C. A.; Weissman J. S. Cell. 2002, 111, 1027–1039. [DOI] [PubMed] [Google Scholar]
- Tang Y.; Chang H.; Roeben A.; Wischnewski D.; Wischnewski N.; Kerner M. J.; Hartl F. U.; Hayer-Hartl M. Cell 2006, 125, 903–914. [DOI] [PubMed] [Google Scholar]
- Kyte J.; Doolittle R. F. J. Mol. Biol. 1982, 157, 105–132. [DOI] [PubMed] [Google Scholar]
- Kerner M.; Naylor D.; Ishihama Y.; Maier T.; Chang H.-C.; Stines A.; Georgopoulos C.; Frishman D.; Hayer-Hartl M.; Mann M.; Hartl F. U. Cell 2005, 122, 209–220. [DOI] [PubMed] [Google Scholar]
- Selvaraj S.; Gromiha M. M. Biophys. J. 2003, 84, 1919–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.