

Abstract
GroEL is an allosteric protein that facilitates protein folding in an ATP-dependent manner. Herein, the relationship between cooperative ATP binding by GroEL and the kinetics of GroE-assisted folding of two substrates with different GroES dependence, mouse dihydrofolate reductase (mDHFR) and mitochondrial malate dehydrogenase, is examined by using cooperativity mutants of GroEL. Strong intra-ring positive cooperativity in ATP binding by GroEL decreases the rate of GroEL-assisted mDHFR folding owing to a slow rate of the ATP-induced transition from the protein-acceptor state to the protein-release state. Inter-ring negative cooperativity in ATP binding by GroEL is found to affect the kinetic partitioning of mDHFR, but not of mitochondrial malate dehydrogenase, between folding in solution and folding in the cavity underneath GroES. Our results show that protein folding by this “two-stroke motor” is coupled to cooperative ATP binding.
The chaperonin GroEL and its cofactor GroES belong to a class of macromolecular assemblies collectively termed “protein machines” (1). In such machines, ordered conformational changes driven by nucleoside triphosphate binding and hydrolysis lead to highly organized spatial and temporal functions. GroEL facilitates protein folding in vivo and in vitro in an ATP-dependent manner (for reviews, see refs. 2–4). It is an oligomer of 14 identical subunits that form two heptameric rings, stacked back-to-back, with a cavity at each end (5). GroEL undergoes ATP-induced conformational changes (6) that are reflected in binding of ATP with positive cooperativity within rings (7–9) and negative cooperativity between rings (10). A nested model for cooperativity in ATP binding by GroEL has been proposed (11, 12) in which, in accordance with the Monod–Wyman–Changeux representation (13), each ring of GroEL is in equilibrium between a tense protein-acceptor (T) state, with low affinity for ATP and high affinity for nonfolded polypeptide substrates, and a relaxed protein-release (R) state, with high affinity for ATP and low affinity for nonfolded polypeptide substrates. In the absence of ligands, GroEL is predominantly in the TT state. In the presence of ATP, the equilibrium is shifted toward the TR state (L1 = [TR]/[TT]). Owing to inter-ring negative cooperativity, a further shift in the equilibrium from the TR state to the RR state (L2 = [RR]/[TR]) takes place only at higher ATP concentrations. The GroEL double-ring particle is thus in equilibrium between three allosteric states TT, TR, and RR, which have recently been visualized by using electron cryomicroscopy (14).
According to the current view of the GroEL reaction cycle, binding of ATP and GroES to a polypeptide substrate-bound GroEL ring creates a GroEL–GroES cis (“bullet”) complex and triggers release of the polypeptide into the space sequestered underneath GroES where it can fold (15, 16). Negative cooperativity between rings, with respect to ATP, helps to ensure that binding of an unfolded protein to the trans ring of an ADP bullet precedes binding of the encapsulating GroES molecule, thereby allowing a new reaction cycle to begin (17). Effects of the inter-ring negative cooperativity and the intra-ring positive cooperativity on the kinetics of polypeptide folding have, however, not been reported. Herein, we analyze the reactivation kinetics of two polypeptide substrates, mouse dihydrofolate reductase (mDHFR; ref. 18) and mitochondrial malate dehydrogenase (MDH; ref. 19), in the presence of either wild-type GroEL or various GroEL mutants with altered and well characterized allosteric properties. The GroEL mutants were chosen such that their allosteric constants span a wide range of values. The polypeptide substrates and refolding conditions were chosen such that reactivation by GroEL of one substrate (mDHFR) would require only ATP, whereas reactivation by GroEL of the other substrate (mitochondrial MDH) would require both ATP and GroES (18, 19).
Experimental Procedures
Materials.
Dihydrofolic acid, oxaloacetate, β-NADPH, and β-NADH were obtained from Sigma. Mitochondrial MDH from pig heart was also obtained from Sigma and used without further purification.
Molecular Biology and Biochemical Methods.
Construction of the GroEL single mutants Arg-197 → Ala (10), Thr-522 → Ala (20), and Phe-44 → Trp (21) as well as the double mutant Arg-13 → Gly, Ala-126 → Val (22) has been described. Construction of the GroEL (Phe-44 → Trp, Arg-197 → Ala), GroEL (Phe-44 → Trp, Thr-522 → Ala), and GroEL (Phe-44 → Trp, Arg-501 → Ala) double mutants has also been described (23). Expression and purification of GroEL and GroES were achieved as before (24).
mDHFR was overexpressed in Escherichia coli TG2 cells and purified as described (25) but with some modifications. Cell pellets were lysed and centrifuged, and polyethyleneimine was then added to a final concentration of 0.125%. After centrifugation, the supernatant was subjected to an 85% (wt/vol) ammonium sulfate cutoff step. The pellet was resuspended in buffer A containing 40 mM KH2PO4, 2 mM EDTA, 1 mM DTT, 1 mM PMSF, and 100 mM KCl (pH 6.5) and applied to a 1 × 5-cm methotrexate-agarose column preequilibrated with the same buffer. Elution of mDHFR was carried out with buffer A containing 0.5 mM dihydrofolate. mDHFR-enriched fractions were pooled, concentrated, and desalted by using a PD-10 Sephadex column equilibrated with buffer B containing 50 mM Tris⋅HCl, 1 mM DTT, and 1 mM EDTA (pH 7.2). The sample was next applied to a Mono-Q HR 5/5 column preequilibrated with the same buffer. Unbound material was loaded on a Sephadex G-75 column equilibrated with buffer B. Pure mDHFR fractions were combined, concentrated, and stored at −80°C at a final concentration of 0.65 mM. Homogeneity of the mDHFR preparations was judged by SDS/PAGE and amino acid analysis to be > 99%. The concentration of mDHFR was determined by using ɛ280 = 25,180 cm−1⋅M−1.
Kinetic Methods.
Transient kinetic experiments were carried out by using an Applied Photophysics (Surrey, U.K.) SX.17MV stopped-flow apparatus. Refolding of mDHFR was initiated by rapid mixing of 1 volume of 6.25 μM protein, denatured in buffer C containing 50 mM Tris⋅HCl, 10 mM MgCl2, 10 mM KCl, 25 mM ATP, 1 mM DTT, and 4 M urea (pH 7.5) for 90 min at 25°C, with 24 volumes of the same buffer (without ATP and urea) containing wild-type or mutant GroEL, dihydrofolic acid, β-NADPH, and GroES (when appropriate). Mitochondrial MDH (6.25 μM) was denatured in buffer C containing 75 mM HCl for 90 min at 25°C and refolded by rapid mixing with 24 volumes of buffer C (without ATP and urea) containing wild-type or mutant GroEL, oxaloacetate, β-NADH, and GroES (when appropriate). The final concentrations of GroEL (oligomer), mDHFR, and MDH were 250 nM, and the final concentration of GroES (oligomer) was 250 or 500 nM. The final concentrations of dihydrofolic acid and β-NADPH were both 100 μM in the mDHFR assay, and those of β-NADH and oxaloacetate were both 300 μM in the MDH assay. The regain of activity of both enzymes was monitored at 25°C by following the change in absorbance at 340 nm by using a 1-cm path length and entrance as well as exit slit widths set to 7 nm. Kinetic traces were collected by using a linear time base with 1,000 sampling points and a time constant filter of 2 ms. Three to five independent traces were acquired for each experiment under exactly the same conditions and then averaged.
Data Analysis.
Values of the allosteric equilibrium constants L1 (which equals [TR]/[TT]) and L2 (which equals [RR]/[TR]) were determined as before (11). Rates of mDHFR folding (kf) were obtained by fitting data of the change in absorbance (A) at 340 nm as a function of time, in the presence or absence of the various GroEL mutants and GroES, to the following equation (26):
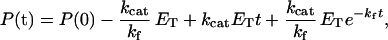 |
1 |
where P(t) is the concentration of the product at time t, kcat is the catalytic rate constant, and ET is the total enzyme concentration. This equation was derived by assuming an irreversible two-state folding transition for mDHFR and that dihydrofolate and β-NADPH interact only with fully folded mDHFR. In the case of mitochondrial MDH, the scheme was extended to include also a bimolecular dimerization step of the folded monomer (27). An analytical solution for such a scheme reduces to Eq. 1 if one assumes that the concentration of the folded monomer is at a steady state. Rates of mitochondrial MDH folding were obtained by fitting refolding traces of mitochondrial MDH, in the absence or presence of the various GroEL mutants, to Eq. 1. In the presence of both GroEL and GroES, refolding traces of mitochondrial MDH were fitted to the following equation derived by assuming that monomer folding and the subsequent dimerization step are kinetically uncoupled:
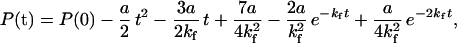 |
2 |
where a = kcatkdET2, ET is the total enzyme (dimer) concentration, and kd is the dimerization rate constant.
Results and Discussion
Effect of Intra-Ring Positive Cooperativity on the Rate of Refolding.
Values of the Hill coefficients for ATP binding, with respect to ATP, and of the rate constants for the TT → TR allosteric transition were recently measured for a series of GroEL mutants (23). In our study, the ability of these GroEL mutants to facilitate the folding of mDHFR was tested. Urea-denatured mDHFR was rapidly mixed with each of these GroEL mutants and ATP. Refolding was monitored by following the regain of enzyme activity. Values of refolding rates measured in this manner were found to be similar to the value determined by measuring binding of NADP+ to mDHFR released from GroEL by using fluorescence quenching (28). These rates correspond to a single-exponential process of folding of a nonnative late-folding intermediate of mDHFR (29).
An excellent linear correlation with a negative slope is observed between the rate constants of GroEL-mediated mDHFR refolding and values of the Hill coefficient, which are measures of the extent of intra-ring positive cooperativity in ATP binding (Fig. 1A). ATP binding and hydrolysis drive GroEL rings between T and R states (9, 12, 30). Strong intra-ring positive cooperativity in ATP binding indicates that the T state is thermodynamically favored relative to the R state. Such thermodynamic favoring may be due to a slow rate of the T → R transition, which, in turn, would lead to slow rates of protein release and folding. A linear correlation between the rate constants of GroEL-mediated mDHFR refolding and the rate constants of ATP-induced conformational changes in GroEL is indeed observed (Fig. 1B), in accordance with this proposed mechanism. This linear correlation implies that the conformational transition in GroEL is a rate-determining step in the refolding reaction of mDHFR. The rate constants of the ATP-induced conformational changes in GroEL were measured in the absence of unfolded substrates. Unfolded substrates bind and stabilize the T state of GroEL rings as reflected, for example, in the increase in the value of the Hill coefficient in the presence of nonfolded α-lactalbumin from about 2.5 to more than 3 (12). The presence of polypeptide substrate may, therefore, slow the rate of conformational change sufficiently to explain why it is found to control the rate of folding.
Figure 1.

Effect of intra-ring positive cooperativity in GroEL on the rate of GroEL-mediated folding of mDHFR. Values of the rates of refolding (kf) of mDHFR in the presence of different GroEL mutants were determined as described in Experimental Procedures. They are plotted against the values of the Hill coefficients (nH) (A) and rates (k1) of the TT → TR conformational transitions (B) of the respective GroEL mutants, which were determined previously (23). The values of the correlation coefficient (r) are 0.98 (A) and 0.96 (B). The errors are 1% in kf and about 5% in k1.
The folding of a lattice chain of single-bead residues in a confined environment was recently simulated (31). Folding times were calculated as a function of the hydrophobic–hydrophilic cycle time of the cavity walls for three different fractions of the overall time in which the cavity walls are hydrophobic. This fraction is directly related to the allosteric equilibrium constant for the T → R allosteric transition and, thus, to the extent of intra-ring positive cooperativity. In the case of cycling times that are comparable to the folding times of the lattice protein, an inverse correlation was found between the folding rate and the fraction of time in which the cavity walls are hydrophobic. This result from simulations is in good agreement with the experimental results shown in Fig. 1.
Effects of Inter-Ring Negative Cooperativity on the Rate of Refolding.
An excellent linear relationship with a negative slope is observed between the logarithm of the rate of folding of mDHFR, in the absence of GroES, and the extent of inter-ring negative cooperativity [log(L1/L2)], with respect to ATP (Fig. 2). Owing to inter-ring negative cooperativity, ATP binding to a polypeptide substrate-bound ring will be inhibited by the binding of ATP to the other ring. ATP-dependent substrate release and folding are, therefore, expected to be slower in the case of strong inter-ring negative cooperativity, as indeed found in this study (Fig. 2). Addition of GroES is found to have two effects on the folding of mDHFR. These effects are not observed if GroES is added in the absence of ATP (data not shown). First, folding of mDHFR is accelerated, probably because binding of GroES to one ring reduces the affinity of the opposite ring for polypeptide substrates (17, 24), thereby facilitating substrate release. Second, a linear relationship between the logarithm of the folding rate of mDHFR and the extent of inter-ring negative cooperativity is observed but with a positive slope (Fig. 2). Owing to inter-ring negative cooperativity, binding of ATP and GroES to a polypeptide substrate-bound ring will be inhibited by binding of ATP to the other ring. Folding of mDHFR may, thus, be faster in the case of strong inter-ring negative cooperativity because of the lower probability for trapping the polypeptide substrate in a cis ternary complex underneath GroES.
Figure 2.

Rates of GroEL-mediated folding of mDHFR as a function of the extent of negative cooperativity. Values of the rates of refolding (kf) of mDHFR, in the presence of different GroEL mutants and in the absence (○) or presence of 250 nM (●) or 500 nM (■) GroES oligomer, were determined as described in Experimental Procedures. Values of the allosteric equilibrium constants L1 (which equals [TR]/[TT]) and L2 (which equals [RR]/[TR]) were determined by fitting data of initial rates of ATPase activity as a function of ATP concentration as described (11). Values of logkf are plotted against the values of log(L1/L2), which are measures of the extent of negative cooperativity (11). The value of the correlation coefficient (r) is 0.99 for each of the three linear fits. The errors are up to 1% in kf and about 10% in log(L1/L2).
The above explanation for the effect of inter-ring negative cooperativity on the rate of folding of mDHFR relies on the fact that folding of this substrate, under the conditions examined in this study, does not require GroES. Two predictions follow from this explanation. One prediction is that the effect of inter-ring negative cooperativity on formation of cis ternary complexes, and thus on the rate of folding, may be overcome by increasing the concentration of GroES. It may be seen in Fig. 2 that a two-fold increase in the concentration of GroES does indeed reduce the effect of inter-ring negative cooperativity on the rate of folding. A further increase in the concentration of GroES may, however, accelerate folding by preventing cis complex formation. A second prediction is that the rate of folding of a GroES-dependent substrate will decrease (or change very little) with increasing inter-ring negative cooperativity. This prediction is based on the assumption that the overall rate of encapsulation decreases with increasing inter-ring negative cooperativity. It may be seen in Fig. 3 that the rate of folding of mitochondrial MDH, a GroES-dependent substrate (19), depends only very weakly on the extent of inter-ring negative cooperativity, both in the presence and in the absence of GroES. This finding suggests that, in the case of the GroEL mutants studied, the rate of encapsulation is fast relative to the folding rate of mitochondrial MDH.
Figure 3.

Rates of GroEL-mediated folding of mitochondrial MDH as a function of the extent of negative cooperativity. Values of the rates of refolding (kf) of MDH, in the presence of different GroEL mutants and in the absence (○) or presence of 250 nM (●) or 500 nM (■) GroES oligomer, were determined as described in Experimental Procedures. Values of logkf are plotted against the values of log(L1/L2), which were calculated by fitting data of initial rates of ATPase activity as a function of ATP concentration as described (11). The errors are up to 10% in kf and about 10% in log(L1/L2).
The linear fits for the data of the logarithm of the rate of mDHFR folding as a function of the extent of negative cooperativity, in the presence and in the absence of GroES, intersect at a value of log(L1/L2) ≈ 0 (Fig. 2). In other words, the presence of GroES (at 1:1 or 2:1 molar ratio with respect to GroEL) does not affect the rate of folding of mDHFR when negative cooperativity is absent. This conclusion is further confirmed by the finding that the rates of mDHFR folding mediated by the GroEL mutant Arg-13 → Gly, Ala-126 → Val, in which inter-ring negative cooperativity with respect to ATP is absent (22), are similar in the presence and in the absence of GroES and coincide with the point of intersection. In the case of a GroES-dependent substrate such as mitochondrial MDH, the rate of folding is affected by GroES even if negative cooperativity is absent.
Conclusions.
The rate constant of GroEL-mediated mDHFR refolding is found in this study to be lower when the value of the Hill coefficient, which measures the extent of intra-ring positive cooperativity in ATP binding, is higher. The reason for this result is probably a slow rate of the T → R transition in the case of strong intra-ring positive cooperativity, which, in turn, leads to a slow rate of protein release and folding. This finding may explain why the value of the Hill coefficient for ATP binding by wild-type GroEL, with respect to ATP, is only about 2.5 (7, 11), which is relatively low for a heptamer. Similar values have been found for hemoglobin (32) and yeast glyceraldehyde-3-phosphate dehydrogenase (33), although they are tetramers. It is likely that the value of the Hill coefficient for ATP binding by wild-type GroEL reflects evolutionary optimization of both the rate of folding, which has pushed its value down, and the equilibrium distribution between polypeptide substrate acceptor (T) and release (R) states, which has pushed its value up. The relative extent of intra-ring positive cooperativity in ATP binding, with respect to ATP, in each of the two rings of GroEL determines the extent of negative cooperativity between rings. Inter-ring negative cooperativity in ATP binding by GroEL is found to affect the kinetic partitioning of mDHFR between folding in solution and folding in the cavity underneath GroES. In the absence of inter-ring negative cooperativity, GroES does not affect the folding rate of mDHFR. In contrast, the GroES-dependent folding rate of mitochondrial MDH is found to be almost independent of the extent of inter-ring negative cooperativity. Optimization of the allosteric properties of GroEL must, therefore, also reflect the cell's need to handle a wide range of substrates with different folding rates, propensities to aggregate, and affinities for GroEL's T state. One intriguing question that remains open is whether the allosteric properties of GroEL are also related to the cooperativity in protein folding.
Acknowledgments
This work was supported by the Israel Science Foundation administered by the Israel Academy of Sciences and Humanities and by the Minerva Foundation.
Abbreviations
- mDHFR
mouse dihydrofolate reductase
- T state
protein-acceptor state
- R state
protein-release state
- MDH
malate dehydrogenase
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.040449997.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.040449997
References
- 1.Alberts B. Cell. 1998;92:291–294. doi: 10.1016/s0092-8674(00)80922-8. [DOI] [PubMed] [Google Scholar]
- 2.Sigler P B, Xu Z, Rye H S, Burston S G, Fenton W A, Horwich A L. Annu Rev Biochem. 1998;67:581–608. doi: 10.1146/annurev.biochem.67.1.581. [DOI] [PubMed] [Google Scholar]
- 3.Ranson N A, White H E, Saibil H R. Biochem J. 1998;333:233–242. doi: 10.1042/bj3330233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Horovitz A. Curr Opin Struct Biol. 1998;8:93–100. doi: 10.1016/s0959-440x(98)80015-8. [DOI] [PubMed] [Google Scholar]
- 5.Braig K, Otwinowski Z, Hegde R, Boisvert D C, Joachimiak A, Horwich A L, Sigler P B. Nature (London) 1994;371:578–586. doi: 10.1038/371578a0. [DOI] [PubMed] [Google Scholar]
- 6.Roseman A M, Chen S, White H, Braig K, Saibil H R. Cell. 1996;87:241–251. doi: 10.1016/s0092-8674(00)81342-2. [DOI] [PubMed] [Google Scholar]
- 7.Gray T E, Fersht A R. FEBS Lett. 1991;292:254–258. doi: 10.1016/0014-5793(91)80878-7. [DOI] [PubMed] [Google Scholar]
- 8.Bochkareva E S, Lissin N M, Flynn G C, Rothman J E, Girshovich A S. J Biol Chem. 1992;267:6796–6800. [PubMed] [Google Scholar]
- 9.Jackson G S, Staniforth R A, Halsall D J, Atkinson T, Holbrook J J, Clarke A R, Burston S G. Biochemistry. 1993;32:2554–2563. doi: 10.1021/bi00061a013. [DOI] [PubMed] [Google Scholar]
- 10.Yifrach O, Horovitz A. J Mol Biol. 1994;243:397–401. doi: 10.1006/jmbi.1994.1667. [DOI] [PubMed] [Google Scholar]
- 11.Yifrach O, Horovitz A. Biochemistry. 1995;34:5303–5308. doi: 10.1021/bi00016a001. [DOI] [PubMed] [Google Scholar]
- 12.Yifrach O, Horovitz A. J Mol Biol. 1996;255:356–361. doi: 10.1006/jmbi.1996.0028. [DOI] [PubMed] [Google Scholar]
- 13.Monod J, Wyman J, Changeux J P. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 14.White H E, Chen S, Roseman A M, Yifrach O, Horovitz A, Saibil H R. Nat Struct Biol. 1997;4:690–694. doi: 10.1038/nsb0997-690. [DOI] [PubMed] [Google Scholar]
- 15.Weissman J S, Hohl C M, Kovalenko O, Kashi Y, Chen S, Braig K, Saibil H R, Fenton W A, Horwich A L. Cell. 1995;83:577–587. doi: 10.1016/0092-8674(95)90098-5. [DOI] [PubMed] [Google Scholar]
- 16.Mayhew M, da Silva A C R, Martin J, Erdjument-Bromage H, Tempst P, Hartl F-U. Nature (London) 1996;379:420–426. doi: 10.1038/379420a0. [DOI] [PubMed] [Google Scholar]
- 17.Rye H S, Roseman A M, Chen S, Furtak K, Fenton W A, Saibil H R, Horwich A L. Cell. 1999;97:325–338. doi: 10.1016/s0092-8674(00)80742-4. [DOI] [PubMed] [Google Scholar]
- 18.Viitanen P V, Donaldson G K, Lorimer G H, Lubben T H, Gatenby A A. Biochemistry. 1991;30:9716–9723. doi: 10.1021/bi00104a021. [DOI] [PubMed] [Google Scholar]
- 19.Staniforth R A, Cortes A, Burston S G, Atkinson T, Holbrook J J, Clarke A R. FEBS Lett. 1994;344:129–135. doi: 10.1016/0014-5793(94)00348-3. [DOI] [PubMed] [Google Scholar]
- 20.Aharoni A, Horovitz A. Proc Natl Acad Sci USA. 1997;94:1698–1702. doi: 10.1073/pnas.94.5.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yifrach O, Horovitz A. Biochemistry. 1998;37:7083–7088. doi: 10.1021/bi980370o. [DOI] [PubMed] [Google Scholar]
- 22.Aharoni A, Horovitz A. J Mol Biol. 1996;258:732–735. doi: 10.1006/jmbi.1996.0282. [DOI] [PubMed] [Google Scholar]
- 23.Yifrach O, Horovitz A. J Am Chem Soc. 1998;120:13262–13263. [Google Scholar]
- 24.Inbar E, Horovitz A. Biochemistry. 1997;36:12276–12281. doi: 10.1021/bi9714870. [DOI] [PubMed] [Google Scholar]
- 25.Prendergast N J, Delcamp T J, Smith P L, Freisheim J H. Biochemistry. 1988;27:3664–3671. doi: 10.1021/bi00410a022. [DOI] [PubMed] [Google Scholar]
- 26.Gray T E, Fersht A R. J Mol Biol. 1993;232:1197–1207. doi: 10.1006/jmbi.1993.1471. [DOI] [PubMed] [Google Scholar]
- 27.Jaenicke R, Rudolph R, Heider R. Biochemistry. 1979;18:1217–1223. doi: 10.1021/bi00574a016. [DOI] [PubMed] [Google Scholar]
- 28.Clark A C, Frieden C. J Mol Biol. 1997;268:512–525. doi: 10.1006/jmbi.1997.0969. [DOI] [PubMed] [Google Scholar]
- 29.Clark A C, Frieden C. J Mol Biol. 1999;285:1777–1788. doi: 10.1006/jmbi.1998.2403. [DOI] [PubMed] [Google Scholar]
- 30.Todd M J, Viitanen P V, Lorimer G H. Science. 1994;265:659–666. doi: 10.1126/science.7913555. [DOI] [PubMed] [Google Scholar]
- 31.Betancourt M R, Thirumalai D. J Mol Biol. 1999;287:627–644. doi: 10.1006/jmbi.1999.2591. [DOI] [PubMed] [Google Scholar]
- 32.Edelstein S J. Nature (London) 1971;230:224–227. doi: 10.1038/230224a0. [DOI] [PubMed] [Google Scholar]
- 33.Kirschner K, Gallego E, Schuster I, Goodall D. J Mol Biol. 1971;58:29–50. doi: 10.1016/0022-2836(71)90230-0. [DOI] [PubMed] [Google Scholar]