

Abstract
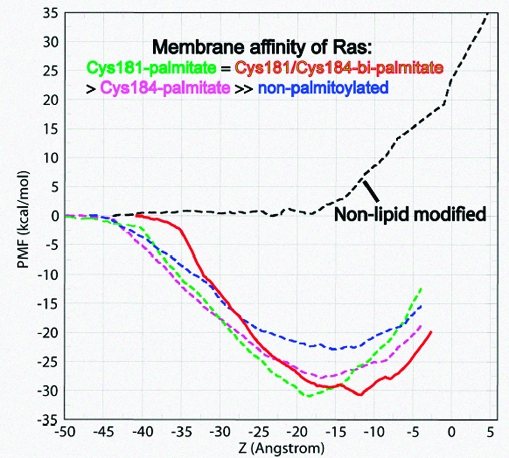
The functionally required membrane attachment of Ras is achieved through an invariant isoprenylation of a C-terminal Cys, supplemented by further lipid modification of adjacent Cys residues by one (N-ras) or two (H-ras) palmitoyls. However, whether the triply lipidated membrane anchor of H-ras has a higher membrane affinity than its doubly lipidated counterpart, or whether the affinity contribution of the two palmitates and the farnesyl is additive, was not known. To address this issue, we carried out potential of mean force (PMF or free energy profile) calculations on a hexadecylated but nonpalmitoylated anchor (Cys186-HD), hexadecylated and monopalmitoylated anchors (Cys181-monopalmitate and Cys184-monopalmitate), and a nonlipid-modified anchor. We found that the overall insertion free energy follows the trend Cys181/Cys184-bipalmitate (wild type) ≈ Cys181-monopalmitate > Cys184-monopalmitate ≫ nonpalmitoylated anchor. Consistent with suggestions from recent cell biological experiments, the computed PMFs, coupled with structural analysis, demonstrate that membrane affinity of the Ras anchor depends on both the hydrophobicity of the palmitate and the prenyl groups and the spacing between them. The data further suggest that while Cys181-palmitate and Cys186-farnesyl together provide sufficient hydrophobic force for tight membrane binding, the palmitoyl at Cys184 is likely designed to serve another, presumably functional, role.
Membrane targeting through posttranslational lipid modification is essential for the function of many signaling proteins, such as the GTPase Ras.(1) Lipid modification in Ras involves an irreversible isoprenylation of a C-terminal Cys supplemented by a polybasic domain (K-ras) or a reversible modification of adjacent Cys residues by one (N-ras) or two (H-ras) palmitoyls.(2) Kinetic studies have shown that the intervesicle transfer of doubly lipid-modified peptides is faster than their singly lipidated counterparts.(3) However, the notion that multiple lipidation invariably confers tighter membrane binding has not been systematically scrutinized. As a result, whether the triply lipidated membrane anchor of H-ras (residues Gly180-Cys181-Met182-Ser183-Cys184-Lys185-Cys186-OCH3) has a higher membrane affinity than its doubly lipidated counterpart, or whether the affinity contribution of Cys181-palmitate, Cys184-palmitate, and Cys186-farnesyl is additive, was not known. Furthermore, the regulatory function of the palmitoyl groups, such as in trafficking and membrane lateral segregation,4,5 raises the question whether the second palmitoyl is designed for enhancing affinity.
We addressed these issues by potential of mean force (PMF or free energy profile) calculations, performed by the Adaptive Biasing Force (ABF) method,(6) for the transfer of a series of H-ras anchor mutants from water to a DMPC bilayer. These include a hexadecylated (i.e., Cys186-HD, which models farnesyl (ref 7 and references therein)) but nonpalmitoylated anchor, hexadecylated and monopalmitoylated anchors (Cys181-monopalmitate and Cys184-monopalmitate), and a nonlipid-modified anchor. The results were compared with a previously reported(8) PMF for the hexadecylated and Cys181/Cys184-bipalmitoylated wild-type anchor (Figure 1A). The PMFs indicate that the overall insertion free energy follows the trend Cys181/Cys184-bipalmitate ≈ Cys181-monopalmitate > Cys184-monopalmitate ≫ nonpalmitoylated anchor. As a control, the profile of the nonlipidated anchor exhibits a sharp rise upon contact with the membrane surface (i.e., at ∼ −18 Å, or the average position of the lower leaflet DMPC P atoms). Such a PMF of the nonlipidated anchor indicates that lipid modification is absolutely required for membrane binding of the Ras anchor; the nonlipid modified side chains contribute to affinity only after insertion is achieved through the lipid-modified moieties. Note that all the PMFs do not include the full contribution of entropy to the unbound forms,9,10 but the relative free energies from the PMFs are still reliable.
Figure 1.

Insertion free energy and membrane interaction of Ras. (A) Potential of mean force (PMF) for the insertion of wild-type and mutant H-ras anchors into a DMPC bilayer. The computed PMF is plotted against the average distance of backbone C and O atoms’ center-of-geometry from the bilayer center (r). (B) Evolution of anchor−DMPC nonpolar contacts. Contacts are defined as the number of selected Ras heavy atoms within 4 Å of DMPC tail carbon atoms. Contacts by Ras lipid tail (heavy lines), Met182 side chain (medium thick lines), and Lys side chain (thin lines) are shown.
Recent biochemical experiments found that, qualitatively, the membrane affinity of Cys181-monopalmitate is equivalent to that of Cys181/Cys184-bipalmitate (i.e., wild-type), whereas Cys184-monopalmitate is weaker.(4) Although no quantitative comparison is possible, the fact that the calculated insertion free energy of the wild-type anchor and that of Cys181-monopalmitate are almost identical, while Cys184-monopalmitate is weaker by ∼4 kcal/mol (Figure 1A), indicates that our results are consistent with this observation. This result thus confirms that membrane affinity of the Ras anchor depends on both the hydrophobicity of the palmitate and prenyl groups and the spacing between them. Below we discuss the physical and structural underpinnings of this surprising result.
The free energy difference between the monopalmitoylated and nonpalmitoylated variants can be readily explained by the difference in the total number of vdW interactions. vdW interactions are estimated from the number of anchor−DMPC nonpolar contacts (Figure 1B; see also ref 11). For example, contacts of the Ras acyl carbons with those of DMPC are 1.5 to 2 times more numerous in Cys181- and Cys184-monopalmitates than in Cys186-HD, respectively. However, variation in nonpolar contacts alone does not explain why the membrane affinity of Cys181-monopalmitate is higher than that of Cys184-monopalmitate (Figure 1). Differences in the structure of the peptide (Figure 2) affect the bilayer localization of the backbone, as well as the polar (Ser183 and Lys185) and nonpolar (Met182 and the unmodified Cys) side chains. For example, the orientation of Met182 and Lys185 in Cys184-monopalmitate (Figure 2, middle) is energetically costly because the neighboring lipids at positions 184 and 186 drag the charged amino group of Lys185 into the core while the apolar Met182 is still in water or at the headgroup region. This is manifested in the delayed initial contacts of the Met182 side chain with DMPC tails (Figure 1B). In contrast, the shape of Cys181-monopalmiate allows favorable interactions between most of the anchor nonlipid atoms and DMPC (Figure 2, top). The distribution of backbone amide nitrogen atoms around the phosphate groups, as shown by the radial distribution functions (Figure 2), illustrates this point.
Figure 2.
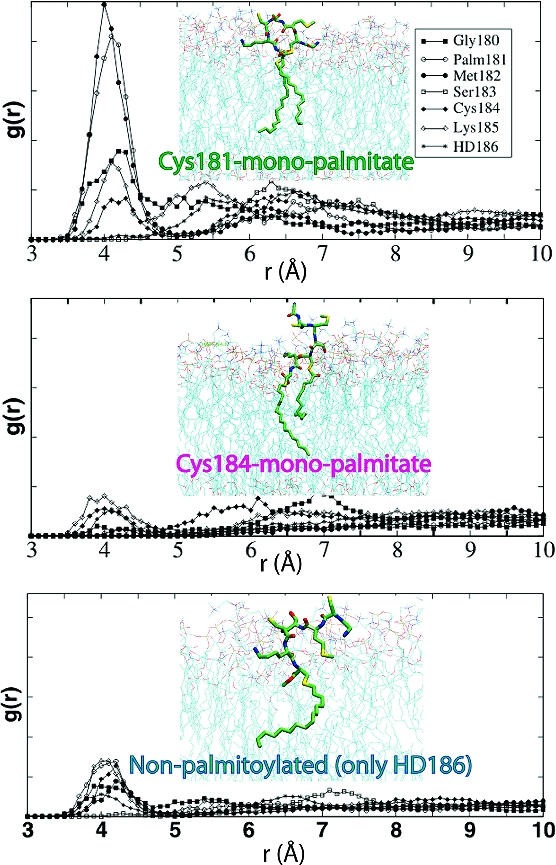
Radial distribution functions (g(r)) for backbone nitrogen atoms and DMPC phosphorus atoms. Snapshots representing the distribution of anchor atoms at the approximate location of the free energy minimum are provided as insets. Color code: carbon, green; nitrogen, blue; oxygen, red; sulfur, yellow.
The reason for the almost identical free energy, or affinity, of wild-type and Cys181-monopalmitate anchors is not apparent from the structures. For example, the average number of acyl−acyl contacts in the wild-type is ∼25, which is larger than the corresponding value in the Cys181-monopalmitate anchor (Figure 1B). It is likely that the extra interaction energy of the triply lipidated anchor is effectively canceled by the comparatively higher cost of untangling the interwoven solution structure of the Ras lipids.(9)
Another interesting observation from Figure 1A is that, relative to the wild-type, the PMF in the other anchors begins to decline when the backbone is >5 Å from the bilayer. This is because the lipid tails in the bi- and monolipidated anchors remain largely extended (not shown). This is in contrast to the more compact structure that three lipids would make by winding around each other.(9) An extended lipid tail is able to reach and insert while the backbone is still far from the bilayer surface. This structural difference is also responsible for the different slopes of the PMFs. Furthermore, the free energy minimum of Cys181-monopalmitate is shifted to the left and coincides with the mean location of the lower leaflet phosphorus atoms, whereas those of Cys184-monoplamitate and Cys186-HD are shallow. The left shift in the free energy minimum of the former arises from the sharply bent backbone conformation (Figure 2, inset); the average distance between Cα atoms of residues 180 and 186 is 9.8 ± 1.1, 13.3 ± 2.6, and 13.5 ± 1.6 Å in Cys181-monopalmitate, Cys184-monopalmitate, and the wild-type anchor, respectively. It is more expensive to insert a structure of larger surface area past the interfacial headgroup. The preferred location of the backbone is also reflected in its interaction with the bilayer, where the backbone amides form hydrogen bonds preferentially with the glycerol carbonyl oxygen atoms in the wild-type,8,11 but with phosphate oxygen atoms in the Cys181-monopalmitate anchor (Figure 2, top). The amide−glycerol and amide−phosphate interactions in the wild-type and Cys181-monopalmitate, respectively, also narrow the free energy minimum. These interactions are much weaker in Cys184-monopalmitate and Cys186-HD (Figure 2, middle and bottom).
The individual contribution of each of the palmitoyls to affinity, in the context of a dually lipidated heptapeptide, can be obtained by subtracting the free energy of Cys186-HD(12) from that of Cys181- or Cys184-monopalmiate. We find that palmitoylation at Cys181 increases membrane affinity by ∼9 kcal/mol. The corresponding increase in affinity due to palmitoylation at Cys184 is 5 kcal/mol. The former compares well with the ∼9.0 kcal/mol of heptane−water partition free energy of a 16 carbon free fatty acid (FFA)(13) or the −8.8 to −9.8 kcal/mol for the association of palmitoylated lipopeptides with 90:10 phosphatidylcholine/phosphatidylglycerol vesicles.(3) The nonoptimal affinity contribution of Cys184-palmitate implies that the role of palmitoyls to affinity is dependent on their spacing from the farnesyl moiety. This conclusion is reinforced by the fact that the affinity contribution of the palmitates at Cys181 and Cys184 is not additive; the free energy difference between the tri- and monolipidated anchors (∼9 kcal/mol) is less than the contribution of the two palmitates together (9 + 5 = 14 kcal/mol). Furthermore, the intriguing similarity between the free energies of wild-type and Cys181-monopalmitate suggests that the purpose of Cys184 palmitoylation might not be to enhance membrane affinity.
That both palmitoyls serve more purposes than passively sticking into membranes has been demonstrated previously:4,5 Cys181-palmitate is required and sufficient for H-ras trafficking to the plasma membrane while Cys184-palmitate, once targeted to the plasma membrane, supports correct GTP-regulated lateral segregation.(4) Although the current calculations lack the catalytic domain and therefore cannot address nucleotide-dependent effects, they support our recent proposal that lateral segregation is modulated by shifting the location of the free energy minimum (or the depth of membrane insertion).7,8,11
In summary, the computed PMFs and structural analysis of Ras anchor mutants demonstrate that Cys181-palmitate and Cys186-farnesyl alone provide sufficient hydrophobic force for tight membrane binding of H-ras. The palmitoyl at Cys184 does not provide extra affinity. We propose that Cys184-palmitate regulates lateral segregation by altering the location of the free energy minimum, which, as discussed before,7,8,11 is modulated by the linker and the activation state of the catalytic domain. The data presented here thus suggest a new twist in the role of protein lipidation, namely, lipid-modified moieties serving not only as “sticky fingers”(14) or “greasy handles”(15) but also as strategically placed “navigators” in the sea of membrane. In response to biochemical cues from distant regions, they may help “direct” the molecule to the right destination.
Acknowledgments
We acknowledge NCSA, SDSC, CTBP, and NBCR for computational resources and the NSF, NIH, and HHMI for financial support.
Funding Statement
National Institutes of Health, United States
References
- Resh M. D. Nat. Chem. Biol. 2006, 2, 584–90. [DOI] [PubMed] [Google Scholar]
- Hancock J. F.; Magee A. I.; Childs J. E.; Marshall C. J. Cell 1989, 57, 1167–77. [DOI] [PubMed] [Google Scholar]
- Shahinian S.; Silvius J. R. Biochemistry 1995, 34, 3813–22. [DOI] [PubMed] [Google Scholar]
- Roy S.; Plowman S.; Rotblat B.; Prior I. A.; Muncke C.; Grainger S.; Parton R. G.; Henis Y. I.; Kloog Y.; Hancock J. F. Mol. Cell. Biol. 2005, 25, 6722–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Rocks O.; Peyker A.; Bastiaens P. I. Curr. Opin. Cell Biol. 2006, 18, 351–7. [DOI] [PubMed] [Google Scholar]; b Rocks O.; Peyker A.; Kahms M.; Verveer P. J.; Koerner C.; Lumbierres M.; Kuhlmann J.; Waldmann H.; Wittinghofer A.; Bastiaens P. I. Science 2005, 307, 1746–52. [DOI] [PubMed] [Google Scholar]
- a Henin J.; Chipot C. J. Chem. Phys. 2004, 121, 2904. [DOI] [PubMed] [Google Scholar]; b Rodriguez-Gomez D.; Darve E.; Pohorille A. J. Chem. Phys. 2004, 120, 3563. [DOI] [PubMed] [Google Scholar]
- Gorfe A. A.; Hanzal-Bayer M.; Abankwa D.; Hancock J. F.; McCammon J. A. J. Med. Chem. 2007, 50, 674–84. [DOI] [PubMed] [Google Scholar]
- Gorfe A. A.; Babakhani A.; McCammon J. A. Angew. Chem., Int. Ed. 2007, 46, 8234–8237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorfe A. A.; Baron R.; McCammon J. A.. Biophys. J., in press. [DOI] [PMC free article] [PubMed]
- Swanson J. M.; Henchman R. H.; McCammon J. A. Biophys. J. 2004, 86, 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorfe A. A.; Babakhani A.; McCammon J. A. J. Am. Chem. Soc. 2007, 129, 12280–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- As noted in ref 8, insertion free energy of farnesyl (∼ −7 kcal/mol3) is smaller than what can be inferred for the hexadecyl group from Figure 1.
- Kampf J. P.; Cupp D.; Kleinfeld A. M. J. Biol. Chem. 2006, 281, 21566–74. [DOI] [PubMed] [Google Scholar]
- Magee T.; Hanley M. Nature 1988, 335, 114–5. [DOI] [PubMed] [Google Scholar]
- Magee A. I.; Seabra M. C. Biochem. J. 2003, 376, e3–4. [DOI] [PMC free article] [PubMed] [Google Scholar]