

Abstract
Prostate specific antigen (PSA) molecules secreted by cancerous and normal prostate cells differ in their N-linked glycan composition, while the peptide backbone appears to be conserved. Antibodies selectively recognizing such differentially glycosylated PSA structures could form a basis for a new diagnostic assay for prostate cancer. Twenty-amino acid PSA fragments carrying di-, tri-, and tetrabranched complex-type glycans were prepared by total synthesis and conjugated to maleimide-modified keyhole limpet hemocyanin (KLH) carrier protein through backbone Cys residues. These glycopeptide/KLH conjugates were then used for antibody generation.
Introduction
The development of aberrant protein glycosylation patterns has long been recognized as one of the indicative changes that accompany malignant transformation of a cell.(1) In particular, the advanced branching of N-linked carbohydrates has been associated with the onset of metastasis of several types of cancer,(2) including breast,(3) colon,(4) and lung.(5) Our program, directed toward the fashioning and evaluation of anticancer vaccines, is based on exploiting differences in oligosaccharide epitope expression between normal and transformed cells.(6)
The work described herein seeks to take advantage of tumor-selective glycosylation patterns at a different level. Modified cellular glycosylation patterns may provide a potentially valuable, yet largely unexplored, opportunity for enhanced cancer diagnostics. A recent discovery of advanced branching in the glycoforms of prostate specific antigen (PSA) isolated from the LnCaP prostate cancer cell line, and not found in normal prostate cells, is potentially interesting from the perspective of diagnostics.(7) PSA, a 28-kDa glycoprotein secreted exclusively by the prostatic epithelium, consists of 237 amino acids and contains a single N-glycosylation site.(8) The high tissue selectivity of PSA renders it potentially valuable as a marker for emerging prostate cancer. The development of immunoassays to measure PSA levels has provided an important breakthrough in prostate cancer diagnostics.(9) Unfortunately, the very common problem of interpreting borderline PSA levels, necessary to differentiate between prostate cancer and benign prostatic hyperplasia, has consistently complicated the efforts of clinicians.(10) A number of methods have been suggested to overcome this problem, including determinations of free vs total PSA(11) (PSA index) and monitoring the increase of PSA levels for a particular patient(12) (PSA velocity); however, the utility of such potential measures remains unverified.(13)
All current state-of-the-art PSA-directed immunoassays employ antibodies that recognize only its peptide backbone. Such assays are therefore unable to differentiate between the various N-linked glycoforms of the protein. Thus, a potentially valuable criterion for discrimination between normal and transformed PSA remains invisible under the current methods.
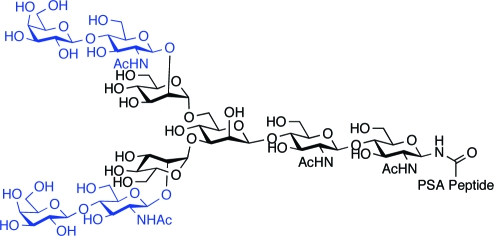
Analyses of PSA glycosylation patterns suggest that normal PSA variants contain only biantennary N-linked glycoforms,8,14 in which the mannose units of the pentasaccharide core are symmetrically glycosylated with N-acetyllactosamine units at the 2 and 2′ positions, as in glycopeptide 1 (Scheme 1). These lactosamines may be further modified through sialylation. By contrast, PSA samples isolated from prostate cancer cells appear to display tri- and even tetrabranched N-glycans,(15) such as in structures 2 and 3 (Scheme 1). In these glycopeptides, the core mannose segments are asymmetrically glycosylated with three or four N-acetyllactosamines at the 2, 2′, 4, and 6′ positions, as shown. While the exact structures of these transformed PSA glycans have not always been determined, these findings are certainly in line with the expectation of a higher degree of branching of N-linked glycoforms associated with neoplastic cells.(1)
Scheme 1. N-Linked Branching of Normal (1) and Transformed (2 and 3) Desialylated PSA Glycoforms.

The research described below(16) was driven by two goals. First, total synthesis of the PSA associable oligosaccharide patterns constitutes a formidable challenge to the state of the art of the field and, as such, would serve as a testing ground to evaluate operations at the perimeter of current feasibility. Moreover, the identification of antibodies sensitive to particular PSA glycoforms, accessed through total synthesis, could form the basis for the development of a new immunoassay capable of elucidating not just levels of PSA, but also the metastatic propensity of the disease. Our approach toward eliciting such distinguishing antibodies is summarized in Scheme 2. Animal immunization with glycosylated PSA fragments conjugated to a carrier protein (such as keyhole limpet hemocyanin (KLH)) and subsequent screening of serum could lead to the identification of the desired antibodies specific for single PSA glycoforms. It seemed that the total chemical synthesis of these types of complex and highly branched N-linked glycopeptides had never previously been accomplished. Yet, de novo synthesis would likely be the only way by which to gain access to completely homogeneous glycosylated PSA fragments in substantial quantities. To pursue this study, we elected to synthesize three glycopeptide fragments, each containing the uneicosapeptide portion of the PSA backbone flanking the glycosylation site (PSA(27−47)), and presenting either the di-, tri-, or tetrabranched glycans found in structures 1, 2, and 3, in unsialylated form. As sialylation adds a considerable degree of heterogeneity to glycoprotein, such samples are typically subjected to sialydase digestion prior to analysis.(17) Hence, our constructs did not contain sialic acids at their nonreducing termini, though in principle, they could be accommodated. As will be shown herein, we found that antibody binding to our synthetic glycopeptide constructs did exhibit modest selectivity, though binding was weaker than with peptide epitopes.
Scheme 2. Proposed Identification of Antibodies Sensitive to Neoplastic PSA Glycoforms.

Results and Discussion
The construction of complex homogeneous glycopeptides can generally be broadly divided into three synthetic tasks: (1) the preparation of the peptide sequence, (2) the synthesis of the glycan region, and (3) the merging of the two domains. While advances in automated peptide synthesis have greatly simplified the first task, the latter two still present daunting challenges to the synthetic organic chemist. In order to achieve efficient glycopeptide conjugation, we envisioned a route consisting of amination of the free reducing end of the carbohydrate, followed by coupling of the resulting β-amino group with a short Asp-containing peptide. Finally, native chemical ligation could be employed to further elongate the peptide chain.
We sought to design a highly convergent approach that could allow for access to all three carbohydrate domains from a single precursor. This would be of significant advantage, since sequential preparations of each separate domain would be likely to be highly “step intensive”. We hoped to achieve maximal efficiency through the simultaneous glycosylation of the pentasaccharide diol (5), triol (6), or tetraol (7) with either two, three, or four N-acetyllactosamine units (Scheme 3). In this way, the construction of both the symmetrically (2,2′) and asymmetrically (2,2′,4 and 2,4,2′,6′) branched glycans would ultimately be accomplished from a common building block. Each of the required pentasaccharides was to be derived from trisaccharide 4, which was deliberately designed to allow for either sequential or simultaneous exposure of the 3 and 6 hydroxyl groups of the terminal mannose.
Scheme 3. A Route Toward the Synthesis of Arbitrarily Branched PSA Glycopeptides from a Single Trisaccharide Precursor.

Two significant changes were built into our strategic plan for the total syntheses to be pursued under the PSA program. In our earlier glycopeptide syntheses, based on glycal assembly, the reducing end was carried to the end of the synthesis as a glycal linkage.(18) In the syntheses described here, the terminal glycal is converted at a much earlier stage (see compound 4) to the GlcNAc pattern, thereby avoiding the need to conduct such technically demanding steps at the end of the synthesis.
Moreover, by the new scheme, the interior mannose (see ring C, compound 4) would be introduced by direct β-mannosylation. In our earlier approaches to complex mannose-based N-linked glycopeptides, the corresponding interior β-linked mannose moiety had been fashioned through epimerization of a β-glucoside precursor (by an oxidation−reduction sequence at C2).(18) By contrast, in the PSA synthesis, we would be employing direct β-mannosylation according to Crich,(19) using Kahne’s sulfoxide as the glycosyl donor.(20)
The decision was also made to equip the reducing end of 4 with a tert-butylsilyl (TBS)-protected alcohol, hoping to avoid the need for the glycal in the late stages of the synthesis. The stability of anomeric alcohols to the dissolving metal reduction conditions required for the global deprotection had been previously demonstrated in our laboratory.(21)
The synthesis of trisaccharide 4(22) commenced with the known disaccharide glycal 8, itself prepared through glycal assembly techniques developed in our laboratory. The double bond of 8 was subjected to iodosulfonamidation, and both resulting iodosulfonamide isomers were subsequently converted to the reducing disaccharide, 9 (Scheme 4). Interestingly, only the α-anomer of 9 was formed, possibly due to the stabilization of the axial hydroxide through intramolecular hydrogen bonding to the C2 sulfonamide. The α-configuration of the anomeric hydroxyl group could be preserved through silylation with excess TBSOTf. Deacetylation of the resultant disaccharide, under Zemplén conditions, provided the requisite acceptor 10 in 53−60% yield over four steps. In a defining step in the synthesis, direct β-mannosylation, under the Crich glycosylation protocol, was efficiently achieved through exposure to the presumed mannosyl triflate, generated in situ from the corresponding mannosyl sulfoxide, 11. Using a modest excess (1.5 equiv) of disaccharide 10, mannosyl coupling could be achieved with good levels of stereoselectivity (8:1, β:α), to provide trisaccharide 12 in 85−91% yields. The excess acceptor 10 could be recovered intact and reused. Deprotection of 12 provided the key trisaccharide building block, 4. Under this sequence, significant amounts of 4 were obtained, with the largest scale-up effort yielding 25 g of trisaccharide. Finally, the 4,6-benzylidene functionality of 4 could be reduced by the action of borane/tetrahydrofuran (THF) complex and dibutylboron triflate to afford 13. This compound, bearing the two required acceptor sites at C3 and C6 of the interior mannose, was destined to be a key building block as the synthesis unfolded.
Scheme 4. Synthesis of the Key Trisaccharide from Chitobiose Glycal.

Reagents and conditions: (a) I(coll)2ClO4, PhSO2NH2; (b) Et3N, H2O/THF; (c) TBSOTf, 2,6-lutidine, CH2Cl2; (d) NaOMe/MeOH; 53−60% for four steps; (e) i. Tf2O, DTBMP, CH2Cl2, −78 °C, ii. 11, 85%−91% (β/α = 8/1); (f) CAN, MeCN/H2O, 74% for two steps; (g) Bu2BOTf, BH3·THF, 72%.
The mannosylated chitobiose trisaccharide (13) was then fully deprotected to afford 14, which was used as a model for testing the viability of the Kochetkov amination/peptide coupling/native chemical ligation (NCL) sequence (Scheme 5). In the event, Kochetkov amination(23) of the trisaccharide proceeded smoothly, albeit over 6 days at room temperature, to give the β-glycosylamine (15). Because the starting material containing the free reducing end hydroxyl group and the product amine differ by only a single mass unit, the reaction was monitored by evaluating the product isotope ratios obtained via mass spectrometry of the reaction mixture. Removal of the solvent and the volatile salts was accomplished by repeated lyophilization. Care was taken to minimize the exposure of the mixture to aqueous conditions during lyophilization, since the amine is known to hydrolyze in solution to the anomeric alcohol.
Scheme 5. A Proof-of-Concept Aspartylation/Native Chemical Ligation Sequence with the Common Core Trisaccharide.

Reagents and conditions: (a) aq. NH4HCO3, 6 days; (b) i. HATU, NEt3, DMSO, 59% for two steps, ii. 20% piperidine/DMF, 68%; (c) 17, 18, PBS buffer pH 7.4, 78%.
Coupling of the trisaccharide glycosylamine (15) with a model pentapeptide (CADAS) proceeded uneventfully,(24) affording analytically pure (liquid chromatography/mass spectrometry (LCMS)) (fluorenylmethoxy)carbonyl (Fmoc)-protected N-linked trisaccharide in 59% yield (from the free glycan). Fmoc removal with 20% piperidine in dimethylformamide (DMF) provided the free trisaccharide pentapeptide (16) in 68% yield. Native chemical ligation(25) performed on an analytical scale with a 14-mer C-terminal thioester (17) showed substantial completion within 2−3 h, but technical challenges arose which, unaddressed, would have impeded the isolation of the desired product. LCMS indicated that the mercaptoethanesulfonate (MES) thioester derivative of the N-terminal coupling partner (used in excess) essentially coeluted with the desired material, as did its carboxylic acid derivative. Simply allowing the thioester to hydrolyze would be insufficient as a method for removing the 14-residue byproduct. Fortunately, trapping of the MES thioester intermediate with cysteamine proved to be an effective method for altering the compound’s polarity, thus allowing its effective separation from the desired product, even on larger scale reactions. By adding a 20-min cysteamine “quench” following reaction completion, it was possible to repeat the NCL on a large scale, and to isolate analytically pure N-linked trisaccharide nonadecapeptide (19) in 78% yield.
Having demonstrated the feasibility of the amination/ligation sequence with the model trisaccharide system, we sought to prepare the normal, dibranched PSA glycopeptide, 1. It would first be necessary to prepare the pentasaccharide diol, 5, through two-fold glycosylation of trisaccharide 13. This transformation would require two high-yielding glycosylation events. A number of protocols were assessed for their ability to provide maximal yield and minimal orthoester formation. Ultimately, bis-glycosylation of 13 under Sinay radical cation activation conditions(26) with excess α-mannosyl donor, 20, proved to be most efficient, furnishing the pentasaccharide adduct in 74% yield, with little or no concomitant orthoester formation. Saponification of the two benzoyl groups afforded the pentasaccharide diol, 5. Subsequent two-fold glycosylation with disaccharide donor 21 under MeOTf activation proceeded smoothly. Following conversion of the phthalimide functionalities to acetates and liberation of the anomeric hydroxyl group, compound 22, representing the fully protected nonasaccharide domain of the target system (1), was in hand. At this stage, we were able to take advantage of an earlier remarkable finding to the effect that the integrity of the “free” reducing end hemiacetals in such systems can be maintained even under Birch debenzylation conditions. Global deprotection, followed by amine-specific diacetylation, yielded the free glycan 23 as a mixture of anomers. The latter was convertable to the free β-glycosylamine, 24, under Kochetkov amination conditions. Coupling of 24 with excess equivalents of hexapeptide 25, followed by removal of the Fmoc and 4,4-dimethyl-2,6-dioxocyclohex-1-ylidene (ivDde) protecting groups, provided glycoconjugate 26. Finally, compound 26 was subjected to NCL with pentadecapeptide 27, as shown, to provide construct 1, representing the fully deprotected normal PSA(27−47) glycopeptide fragment (Scheme 6).
Scheme 6. Synthesis of Normal PSA Glycopeptide 1.

Reagents and conditions: (a) 20, (BrC6H4)3NSbCl6, MeCN, 74%; (b) NaOMe/MeOH, 89% for two steps; (c) MeOTf, DTBP, CH2Cl2, 60%; (d) i. ethylenediamine, n-BuOH/toluene, 90 °C, ii. Ac2O/py, iii. NaOMe/MeOH, 72%; (e) TBAF/AcOH, THF, 76%; (f) i. Na/NH3, −78 °C, ii. Ac2O, iii. NaOMe/MeOH, 65%; (g) NH4HCO3/H2O; (h) 25, HATU, Hünig’s base, DMSO, 61% from 23; (i) (NH2)2, piperidine, DMF, 62%; (j) 27, MES-Na, pH 7.4, 17%.
Having successfully synthesized the nontransformed, dibranched glycopeptide fragment of PSA, we next set out to synthesize the significantly more challenging tri- and tetrabranched transformed glycopeptides, 2 and 3. The first task would be the preparation of α-mannosyl donors 30 and 31, which would be required for the assembly of the pentasaccharides 6 and 7. Toward this end, the α-mannosyl unit, 28, was selectively benzylated at the 3 position, as shown. The resultant intermediate, 29, could be readily converted to donor 30, possessing benzoylation at the 2 and 4 positions, or to donor 31, in which the 2 and 6 positions were selectively benzoylated (Scheme 7).
Scheme 7. Synthesis of Selectively Benzoylated Mannosyl Donors.

Reagents and conditions: (a) i. Bu2SnO, toluene, ii. BnBr (89%); (b) i. NaBH3CN, HCl, THF, 77%, ii. BzCl, DMAP, 98%; (c) i. Bu2BOTf, BH3 THF, 97%, ii. BzCl, DMAP 84%.
With these differentially protected monosaccharide units in hand, we had the key building blocks required for the completion of the syntheses of our target glycopeptides, 2 and 3. The objective would now be the assembly of pentasaccharide units 6 and 7, presenting appropriately positioned free hydroxyl groups, which would ultimately serve as the sites of tri- or tetrabranching. In the event, treatment of trisaccharide 4 with the α-mannosyl donor 30, followed by regiospecific reductive cleavage of the 4,6-benzylidene moiety, afforded tetrasaccharide intermediate 32, possessing two sites of benzoylation and a free C-6 hydroxyl group, as shown.
Tetrasaccharide 32 served as a common intermediate in the syntheses of pentasaccharides 6 and 7. The synthesis of 6, a key intermediate en route to the tribranched glycopeptide (2), was accomplished through glycosylation of 32 with thiomannoside 20, again by implementation of the Sinay radical cation activation protocol. Saponification of the benzoate protecting groups afforded the requisite pentasaccharide triol acceptor, 6. In a similar fashion, 32 was readily advanced to the pentasaccharide tetraol, 7, an intermediate in the synthesis of the tetrabranched glycopeptide (3). Mannosylation of 32 with the 2,6-di-O-benzoyl donor, 31, provided pentasaccharide 34, which readily underwent saponification to afford tetraol 7. The program, which had been devised to reach pentasaccharides with well-identified sites for concurrent glycosylation, was implementable in a relatively straightforward way (Scheme 8).
Scheme 8. Synthesis of Pentasaccharides with Selectively Exposed Branching Points.

Reagents and conditions: (a) 30, (BrC6H4)3NSbCl6, MeCN, 71%; (b) BH3·THF, Bu2BOTf, THF, 85%; (c) 20, (BrC6H4)3NSbCl6, MeCN; (d) NaOMe/MeOH, 72% for two steps; (e) 31, (BrC6H4)3NSbCl6, MeCN, 74%; (f) NaOMe/MeOH, 92%.
The synthesis of the tribranched glycopeptide, 2, from pentasaccharide 6 is summarized in Scheme 9. Three-fold β-lactosylation was achieved in 41% yield through exposure of 6 to excess β-lactosamine donor 21. The steps required for the transformation of 35 to 2 are analogous to those described for the dibranched glycopeptide 1 (Scheme 6). The phthalimide protecting groups were replaced with N-acetyl functionalities, and the anomeric hydroxyl group was liberated, as shown. Global benzyl deprotection proceeded uneventfully to yield intermediate 37, which smoothly underwent Kochetkov amination to afford the free β-glycosylamine, 38. Coupling of 38 with hexapeptide 25 and subsequent NCL proceeded without incident to yield the tribranched glycopeptide unit 2.
Scheme 9. Synthesis of Tribranched Transformed PSA Glycopeptide 2.

Reagents and conditions: (a) MeOTf, DTBP, CH2Cl2, 41%; (b) i. ethylenediamine, n-BuOH/toluene, 90 °C, ii. Ac2O/py, iii. NaOMe/MeOH, 77%; (c) TBAF/AcOH, THF, 95%; (d) i. Na/NH3, −78 °C, ii. Ac2O, iii. NaOMe/MeOH, 94%; (e) NH4HCO3/H2O; (f) 25, HATU, Hünig’s base, DMSO, 25% from 37; (g) (NH2)2, piperidine, DMF, 66%; (h) 27, MES-Na, pH 7.4, 38%.
The assembly of the tetrabranched glycopeptide (3) was accomplished through a route analogous to those described above for the syntheses of the di- and tribranched systems. Thus, as summarized in Scheme 10, the pentasaccharide tetraol, 7, underwent four-fold glycosylation with 21 to yield 40. As would be expected, this high complexity-building transformation proceeded in modest yield (19%). Nonetheless, the subsequent Kochetkov amination/aspartylation/NCL sequence proceeded smoothly to provide the tridecasaccharide−uneicosapeptide glycoconjugate 3 in homogeneous form as judged by MS and NMR analysis.
Scheme 10.

Reagents and conditions: (a) MeOTf, DTBP, CH2Cl2, 19%; (b) i. ethylenediamine, n-BuOH/toluene, 90 °C, ii. Ac2O/py, iii. NaOMe/MeOH, 79%; (c) TBAF/AcOH, THF, 97%; (d) i. Na/NH3, −78 °C, ii. Ac2O, iii. NaOMe/MeOH, 81%; (e) NH4HCO3/H2O; (f) 25, HATU, Hünig’s base, DMSO, 25% from 42; (g) (NH2)2, piperidine, DMF, 52% for three steps; (h) 27, MES-Na, pH 7.4, 65%.
In order to elicit a sustained immune response against the tribranched transformed glycopeptide 2, we chose KLH as the protein carrier. KLH has proven to be effective in numerous vaccine applications because of its many foreign epitopes, large molecular mass, and poor solubility. Using the unique thiol on the glycopeptide as a convenient handle for conjugation to a maleimide-functionalized KLH, we prepared an extensively functionalized KLH system. In order to prevent precipitation due to resilient thiol (from cysteine) cross-linking, KLH was first reacted with maleimidobutyric acid in order to block the free thiols (Scheme 11). This preliminary step allowed for a higher degree of maleimide functionalization, and also provided stability to the construct. The lysines of KLH were derivatized using a large excess of succinimidyl 4-[N-maleimidomethyl]cyclohexane-1-carboxylate (SMCC). Up to 1000 maleimide moieties were grafted on KLH using this protocol, as assessed by 35S-cysteine labeling. Coupling of glycopeptide 2 to this activated KLH yielded a conjugate bearing 300 residues, as assessed by carbohydrate high-performance liquid chromatography analysis after total hydrolysis (data not shown). The conjugating efficiency was 20%, which is relatively standard in the field.
Scheme 11. Conjugation of Glycopeptides to KLH.

Reagents and Conditions: a) maleimidobutyric acid, PBS EDTA pH 7.0, rt; b) LC-SMCC, PBS EDTA pH 7.0, rt; c) glycopeptide, PBS EDTA pH 6.8, rt.
BALB/c mice were immunized with glycopeptide 2 conjugated to KLH, and high-titer antisera were elicited for the antigen. About 1500 hybridoma supernatants were screened for the presence of antibodies recognizing glycopeptide 2 in a standard enzyme-linked immunosorbent assay (ELISA). About 10% of the hybridomas were found to be positive and were tested for recognition of the nontransformed glycopeptide 1. Our goal was to select antibodies binding preferentially to the tribranched, transformed glycopeptide 2 compared to the normal, dibranched glycopeptide 1, thereby allowing us to discard antibodies that were likely to be directed against the peptidic moiety of the antigen. When tested in parallel in at least two independent experiments, a selectivity ranging from 2- to 4-fold in favor of the tribranched glycopeptide 2 compared to the dibranched glycopeptide 1 was confirmed for 17 clones, which were selected for further experiments (Figure 1).
Figure 1. Binding of selected hybridoma supernatants to the tribranched glycopeptide 2 compared to the dibranched glycopeptide 1. The data presented are from one representative experiment.

Selected hybridoma supernatants were evaluated for binding to LnCaP tumor PSA in a sandwich ELISA. These tests were done on hybridoma supernatant fluids and not purified monoclonal antibodies, and antibodies generated against carbohydrates are often of lower affinity. Therefore, absolute titers of individual hybridomas cannot be compared. In addition, we had purposely immunized the mice against the PSA glycopeptide 2, which is devoid of any sialyl group, in order to prevent the generation of antibodies directed against this ubiquitous moiety. LnCap PSA carbohydrates, however, are likely to be sialylated, which could affect both of the recognized epitopes through masking or conformational changes. Antibodies recognizing nonsialylated PSA carbohydrates might therefore not bind to their sialylated counterparts. To test this hypothesis, we measured the binding of our hybridoma supernatants to desialylated LnCap PSA. A positive control monoclonal antibody included in these tests, H117, which is reactive with the peptide moiety of PSA,(27) showed strong binding to both sialylated and desialylated peptides, as expected. Small increases in the binding to LnCap PSA after desialylation were observed for four of the six tested supernatants, sometimes resulting in up to a 2-fold increase in binding (data not shown). This result suggests that some of the generated antibodies recognized a desialylated carbohydrate moiety of native LnCap PSA better than the sialylated forms; however, only small changes were observed. The small difference in binding observed after desialylation of LnCap PSA is consistent with a report in which LnCap PSA has little sialic acid.(28) Desialylation of LnCap PSA would therefore be expected to affect only moderately the binding of the antibodies that we have generated against the desialylated glycopeptide 2. Interestingly, when we measured the binding of the 17 selected hybridoma supernatants to the serum of a patient with prostate cancer, we observed an increase in binding after desialylation for 13 of 17 supernatants tested ranging from 2- to 3-fold (data not shown). While this result could not be repeated due to the limited amounts of hybridoma supernatant fluids available, we have identified a trend suggesting that the antibodies raised against the nonsialylated glycopeptide 2 can recognize the desialylated glycan moiety of PSA from a patient serum.
We note also that the determination of the actual glycobiology and structural chemistry of these glycans is still a work in progress.(29) As firmer answers to these highly complex structural issues are available, the synthetic chemistry addressed to simulating nuances of the various PSA related antigens can be adapted. In this way, one can hope to improve the correspondence of the chemical construct with the cell surface, thereby achieving more selective and more discerning induced antibody−natural antigen affinities.
Conclusion
In summary, we have shown the feasibility of preparation of highly complex N-linked glycopeptides through entirely chemical means. In this case, di-, tri-, and tetrabranched glycans have been synthesized from a single trisaccharide precursor with most diversity introduction performed at the end of the oligosaccharide assembly. Amination of such glycans followed by coupling/NCL sequence allows attachment of long peptide motifs. We have also demonstrated that significant glycopeptide loading on carrier proteins can be achieved through maleimide modification/Michael addition. The possibility of selective recognition of the 11-mer carbohydrate vs the 9-mer as well as a significant increase in binding upon sialidase digestion of PSA suggests the potential of this methodology for providing a path toward a new, more selective prostate cancer diagnostic assay. While a trend suggesting binding of the newly generated antibodies to both LnCap PSA and serum from a prostate cancer patient after desialylation has been detected, further optimization of both antigen structure and antibody preparation is required before a practical use of the assay can be projected.
Acknowledgments
Support for this work was provided by the NIH (CA23766 and CA55349 to D.A.S. and CA103823 to S.J.D.). We thank Dr. H. Lilja for the gift of the H117 antibody for PSA assays, Dr. G. Ragupathi for assistance on KLH conjugate analysis, and the Monoclonal Antibody Core Facility for help with antibody development. J.S.M. is the P.I. of DAMD 17-03-1-0016.
Supporting Information Available
Experimental procedures and compound characterization data. This information is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Dennis J. W.; Granovsky M.; Warren C. E. Biochim. Biophys. Acta 1999, 1473, 21. [DOI] [PubMed] [Google Scholar]
- Dennis J. W.; Laferte S.; Waghorne C.; Breitman M. L.; Kerbel R. S. Science 1987, 236, 582. [DOI] [PubMed] [Google Scholar]
- Seberger P. J.; Chaney W. G. Glycobiology 1999, 9, 235. [DOI] [PubMed] [Google Scholar]
- Fernandes B.; Sagman U.; Auger M.; Demetrio M.; Dennis J. W. Cancer Res. 1991, 51, 718. [PubMed] [Google Scholar]
- Lu Y.; Pelling J. C.; Chaney W. G. Clin. Exp. Metastasis 1994, 12, 47. [DOI] [PubMed] [Google Scholar]
- a Danishefsky S. J.; Allen J. R. Angew. Chem., Int. Ed. 2000, 39, 836. [DOI] [PubMed] [Google Scholar]; b Ouerfelli O.; Warren J. D.; Wilson R. M.; Danishefsky S. J. Exp. Rev. Vaccines 2005, 4, 677. [DOI] [PubMed] [Google Scholar]
- a Armbruster D. A. Clin. Chem. 1993, 39, 181. [PubMed] [Google Scholar]; b Abrahamsson P. A.; Lilja H.; Oesterling J. E. Urol. Clin. N. Am. 1997, 24, 353. [DOI] [PubMed] [Google Scholar]; c Egawa S. Biomed. Pharmacother. 2001, 55, 130. [DOI] [PubMed] [Google Scholar]
- Okada T.; Sato Y.; Kobayashi N.; Sumida K.; Satomura S.; Matsuura S.; Takasaki M.; Endo T. Biochim. Biophys. Acta 2001, 1525, 149. [DOI] [PubMed] [Google Scholar]
- Ward A. M.; Catto J. W. F.; Hamdy F. C. Ann. Clin. Biochem. 2001, 38, 633. [DOI] [PubMed] [Google Scholar]
- a Semjonow A.; Hertle L. Urol.-Ausg. A 1995, 34, 290. [PubMed] [Google Scholar]; b Semjonow A.; Brandt B.; Oberpenning F.; Roth S.; Hertle L. Prostate 1996, 3. [PubMed] [Google Scholar]
- Demura T.; Shinohara N.; Tanaka M.; Enami N.; Chiba H.; Togashi M.; Ohashi N.; Nonomura K.; Koyanagi T. Cancer 1996, 77, 1137. [DOI] [PubMed] [Google Scholar]
- Thiel R.; Pearson J. D.; Epstein J. I.; Walsh P. C.; Carter H. B. Urology 1997, 49, 716. [DOI] [PubMed] [Google Scholar]
- Catalona W. J.; Southwick P. C.; Slawin K. M.; Partin A. W.; Brawer M. K.; Flanigan R. C.; Patel A.; Richie J. P.; Walsh P. C.; Scardino P. T.; Lange P. H.; Gasior G. H.; Loveland K. G.; Bray K. R. Urology 2000, 56, 255. [DOI] [PubMed] [Google Scholar]
- a Belanger A.; Vanhalbeek H.; Graves H. C. B.; Grandbois K.; Stamey T. A.; Huang L. H.; Poppe I.; Labrie F. Prostate 1995, 27, 187. [DOI] [PubMed] [Google Scholar]
- Prakash S.; Robbins P. W. Glycobiology 2000, 10, 173. [DOI] [PubMed] [Google Scholar]
- Dudkin V. Y.; Miller J. S.; Danishefsky S. J. J. Am. Chem. Soc. 2004, 126, 736. [DOI] [PubMed] [Google Scholar]
- Hilz H.; Noldus J.; Hammerer P.; Buck F.; Luck M.; Huland H. Eur. Urol. 1999, 36, 286. [DOI] [PubMed] [Google Scholar]
- Wang Z.-G.; Warren J. D.; Dudkin V. Y.; Zhang X.; Iserloh U.; Visser M.; Eckhardt M.; Seeberger P. H.; Danishefsky S. J. Tetrahedron 2006, 62, 4954. [Google Scholar]
- a Crich D.; Sun S. X. J. Am. Chem. Soc. 1998, 120, 435. [Google Scholar]; b Crich D.; Sun S. X. Tetrahedron 1998, 54, 8321. [Google Scholar]
- Kahne D.; Walker S.; Cheng Y.; Van Engen D. J. Am. Chem. Soc. 1989, 111, 6881. [Google Scholar]
- Iserloh U.; Dudkin V. Y.; Wang Z. G.; Danishefsky S. J. Tetrahedron Lett. 2002, 43, 7027. [Google Scholar]
- Dudkin V. Y.; Miller J. S.; Danishefsky S. J. Tetrahedron Lett. 2003, 44, 1791. [Google Scholar]
- Likhosherstov L. M.; Novikova O. S.; Derevitskaja V. A.; Kochetkov N. K. Carbohydr. Res. 1986, 146, C1. [DOI] [PubMed] [Google Scholar]
- Cohen-Anisfeld S. T.; Lansbury P. T. J. Am. Chem. Soc. 1993, 115, 10531. [Google Scholar]
- a Dawson P. E.; Muir T. W.; Clark-Lewis I.; Kent S. B. H. Science 1994, 266, 776. [DOI] [PubMed] [Google Scholar]; b Tolbert T. J.; Wong C. H. J. Am. Chem. Soc. 2000, 122, 5421. [Google Scholar]
- Zhang Y. M.; Mallet J. M.; Sinay P. Carbohyd. Res. 1992, 236, 73. [DOI] [PubMed] [Google Scholar]
- Leinonen J.; Wu P.; Stenman U.-H. Clin. Chem. 2002, 48, 2208. [PubMed] [Google Scholar]
- Peracaula R.; Tabarés G.; Royle L.; Harvey D. J.; Dwek R. A.; Rudd P. M.; de Llorens R. Glycobiology 2003, 13, 457. [DOI] [PubMed] [Google Scholar]
- Tabarés G.; Radcliffe C. M.; Barrabés S.; Ramírez M.; Núria A.; Hoesel W.; Dwek R. A.; Rudd P. M.; Peracaula R.; de Llorens R. Glycobiology 2006, 13, 132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.