

Abstract
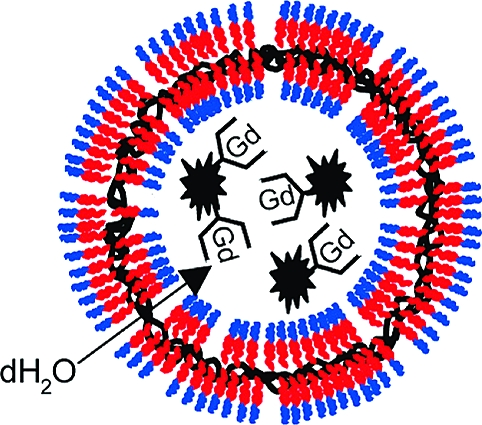
The ability of chelated Gd to serve as an effective magnetic resonance (MR) contrast agent largely depends on fast exchange rates between the Gd-bound water molecules and the surrounding bulk water. Because water diffuses slowly across lipid bilayers, liposomes with encapsulated chelated Gd have not been widely adopted as MR contrast agents. To overcome this limitation, we have synthesized chemically stabilized, porous polymersomes with encapsulated gadolinium (Gd) chelates. The polymerosmes, 125 nm in diameter, were produced from the aqueous assembly of diblock copolymers, PEO(1300)-b-PBD(2500) (PBdEO), and phospholipids, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC). The PBdEO was cross-linked using a chemical initiator and the POPC was extracted with surfactant, generating a highly porous outer membrane. The encapsulated Gd chelates were attached to dendrimers to prevent their leakage through the pores. It was estimated that, on average, nearly 44 000 Gd were encapsulated within each polymersome. As a result of the slower rotational correlation time of Gd-labeled dendrimers and the porous outer membrane, the paramagnetic porous polymersomes exhibited an R1 relaxivity of 7.2 mM−1 s1− per Gd and 315 637 mM−1 s−1 per vesicle. This corresponds to a relaxivity that is amplified by a factor of ∼105 compared with Gd−DTPA.
Introduction
Magnetic resonance (MR) imaging procedures have become a common practice in diagnostic clinical medicine owing to their ability to provide high-resolution three-dimensional images of soft tissue. Many of these diagnostic procedures utilize intravenous MR contrast agents, such as gadolinium (Gd), to improve tissue contrast and to provide important information about perfusion, vascular permeability, and extracellular volume.1–3 The recent development of targeted paramagnetic contrast agents promises to even further expand the utility of diagnostic MR imaging by providing a mechanism to probe the molecular profile of tissues. In order to compensate for the low signal enhancement generated by individual Gd ions, most targeted Gd compounds have relied on the development of nanoplatforms that can carry a high payload of chelated Gd and which exhibit high longitudinal relaxivities (R1). In general, the R1 of Gd-based contrast agents is dependent on two key features, the water-exchange rate between bulk water and water bound to the chelated Gd and the rotational correlation time of the Gd ions.(4)The rotational correlation lifetime is generally increased through conjugation of Gd chelates to macromolecular objects. A wide range of macromolecules and other nanoparticulate systems have already been tested as platforms for Gd labeling. Some examples include dendrimers,5,6 polymers,(7) liposomes,8–12 micelles,13,14 emulsions,(15) and silica16–18 nanoparticles. For these multimeric gadolinium complexes, it is not only the relaxivity per Gd that defines the effectiveness of the contrast agent but also the number of chelated Gd per nanoparticle. These two parameters can be represented as the relaxivity per nanoparticle.
Among the many nanoparticulate systems, those that are self-assembled from amphiphilic molecules (i.e., liposomes, micelles, and emulsions) are particularly attractive due to their unique structure wherein the hydrophobic domain serves as a natural carrier environment for hydrophobic drugs and the exterior surface provides a platform for attaching targeting ligands. Amphiphilic nanoparticles are easily transformed into paramagnetic contrast agents by either encapsulating chelated Gd within the hollow core (in the case of liposomes) or through the immobilization of chelated Gd onto the outer membrane surface. Although, liposomes with encapsulated Gd-chelates have previously been used for contrast-enhanced MR imaging, the slow flux of water across the membrane bilayer does impair the water exchange rate with encapsulated Gd and thus leads to a significant reduction in relaxivity.(8) While this can be partially overcome by increasing the surface-to-volume ratio (i.e., decreasing the size of the vesicle), even liposomes 100 nm in diameter have been shown to exhibit a relaxivity (per Gd) that is 62% lower than free chelated Gd.8,12 Reagents that are commonly used to increase the stability of liposomes, such as cholesterol. even further reduce the relaxivity of encapsulated Gd.(9) Because of these concerns, vesicles with encapsulated Gd chelates have been all but abandoned as MR contrast agents and liposomes, micelles, and perfluorocarbon emulsions with Gd immobilized on the surface have become the preferred embodiment for MR imaging applications.10,13–15 In this study, we revive the possibility of using Gd-encapsulated vesicles with the development of a new porous polymersome nanoplatform, in which Gd relaxivity is not compromised following encapsulation but, in fact, improved. By taking advantage of the large intraparticle volume, we also show that much larger payloads of Gd per particle can be achieved compared with vesicles of similar size containing surface-immobilized Gd. Porous polymersomes are composed of three key features: (1) a stabilized polymer shell, (2) a porous membrane structure that allows for improved water flux across the bilayer, and (3) a dendrimer-attached Gd complex that is loaded inside the porous vesicles for MRI detection.
Experimental Section
Materials
PEO(1300)-b-PBD(2500) (PBdEO) was purchased from Polymer Source (Dorval, Quebec, Canada). 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (Rhod-PE) were obtained from Avanti Polar Lipids (Alabaster, AL). PAMAM dendrimers (ethylenediamine core, generation 2) were from Aldrich Chemical Co. as 20% w/v solutions in methanol. 5-(and-6)-Carboxyfluorescein (CF) and tetramethylrhodamine-5-(and-6)-isothiocyanate (TRITC) were obtained from Molecular Probes. Diethylenetriaminepentaacetic acid dianhydride (DTPA dianhydride), gadolinium(III) chloride, potassium persulfate, sodium metabisulfite, and iron(II) sulfate heptahydrate were obtained from Sigma-Aldrich. All other chemical were used as received. All of the buffer solutions were prepared with DI water.
Synthesis of PAMAM−DTPA−Gd
A 126 mg portion of PAMAM was dissolved with 5 mL of sodium bicarbonate buffer (0.1 M, pH 9.5) and reacted with 400 mg of DTPA dianhydride. The reaction solutions were maintained at pH 9.5 with NaOH over the reaction time of 10 h. The PAMAM−DTPA was purified by centrifugal filter devices (Amicon Ultra-4, 5000 MWCO, Millipore Corp.). The purified PAMAM−DTPA conjugates were mixed with 100 mg of GdCl3 in 0.1 M citrate buffer (pH 5.6) for overnight at 42 °C. The unreacted Gd3+ was removed by centrifugal filter devices (Amicon Ultra-4, 5000 MWCO, Millipore Corp.) while simultaneously changing the buffer to 0.1 M PBS buffer. To ensure complete removal of unreacted Gd3+, the Gd content in the eluent was checked after each centrifugation by performing a Xylenol Orange assay until no Gd3+ was detectable.(19) The purified PAMAM−DTPA−Gd conjugates were used for vesicle encapsulation.
Preparation of Giant Vesicles
Giant vesicles were prepared as previously described.20,21 Briefly, appropriate amounts of PBdEO, POPC, and Rhod-PE at a molar ratio of 85:14:1 were dissolved in chloroform. The solvent was then removed using a direct stream of nitrogen prior to vacuum desiccation for a minimum of 4 h. The giant vesicles were formed upon the addition of 285 mOsm sucrose solution to dried film and incubated in the 65 °C water bath for more than 24 h.
Preparation of Nanometer-Sized Vesicles
Nanometer-sized vesicles were prepared using the film hydration technique. An aqueous solution (0.1 M PBS, pH 7.0) was added to dried PBdEO film with 15% POPC or without POPC and incubated in a 65 °C water bath for 0.5 h and then sonicated for another 1 h at the same temperature. Samples were subjected to 10 freeze−thaw−vortex cycles in liquid nitrogen and warm H2O (65 °C), followed by extrusion 21 times through two stacked 100 nm Nuclepore polycarbonate filters using a stainless steel extruder (Avanti Polar Lipids).
For dye encapsulation, 1 mL of 100 mM CF or 0.5 mL of PAMAM−TRITC (40 mg/mL PAMAM) in 0.1 M sodium phosphate (pH 7.0) was added to the dried PBdEO/POPC (mole ratio 85/15, 10 mg of PBdEO) films and freeze−thaw and extrusion were performed as described. Nonentrapped CF and PAMAM-TRITC were removed via size exclusion chromatography using Sepharose CL-4B (Sigma-Aldrich) and rehydration buffer as the eluent.
For PAMAM−DTPA−Gd encapsulation, 1 mL of PAMAM−DTPA−Gd was added to the dried PBdEO/POPC (mole ratio 85/15, 100 mg of PBdEO). Nonentrapped PAMAM−DTPA−Gd was removed through repeated washing on centrifugal filter devices (Amicon Ultra-4, 100K MWCO, Millipore Corp.). To ensure complete removal of PAMAM−DTPA−Gd, the T1 relaxation time of the eluent was checked after each centrifugation until no Gd was detectable, i.e. until the T1 relaxation time was equivalent to that of sodium phosphate (pH 7.0) buffer (∼1000 ms). T1 relaxation times were determined using a Bruker mq60 MR relaxometer operating at 1.41 T (60 MHz).
Polymersome Polymerization
Polybutadiene in the cores of the vesicle membranes was cross-linked by free radical polymerization in solution.22–25 Specifically, 100 mg of potassium persulfate was added into polymersome solution and then stirred for 1 h. The polymerization was initiated by injecting appropriate amounts of redox couple, Na2S2O5/FeSO4·7H2O, in the sequence of sodium metabisulfite and ferrous sulfate. All cross-linking polymerizations were performed at fixed weight ratios of 1:1:0.5:0.02 between the PBdEO, potassium persulfate, sodium metabisulfite, and ferrous sulfate.
POPC Extraction
To remove POPC from polymerized vesicles, Triton X-100 was added into polymerized vesicles and incubated for 10 min with shaking. The final concentration of Triton X-100 is 10 mM. Triton X-100 was subsequently removed through repeated washing on centrifugal filter devices (Amicon Ultra-4, 100K MWCO, Millipore Corp.)
Quantification of Gd Encapsulation
To determine the number of Gd per porous polymersome, it was assumed that none of the 100 mg of polymer was lost during the synthesis process. The number of polymersomes in the purified sample was then calculated by determining the amount of polymer in each vesicle. For this calculation, the average diameter of each polymersome was taken to be 125 nm. Further, the average area occupied by single polymer molecules in a bilayer has previously been determined to be ∼1 nm2.(22) The average area occupied by single POPC molecules in a bilayer has previously been determined to be ∼0.65nm2,(26) and the thickness of the polymersome bilayer has previously been determined to be ∼10 nm.(27) The amount of Gd in the polymersome sample was measured by ICP-AES.
Instrumentation
Dynamic light scattering (DLS) measurements were performed on a Zetasizer Nano from Malvern Instruments. The scattering angle was held constant at 90°. Microscopy measurements were performed on an Olympus IX 81 motorized inverted fluorescence microscope equipped with an Andor DU897 EMCCD, an X-Cite 120 excitation source (EXFO), and Sutter excitation and emission filter wheels. The image of Rhod-PE was acquired using the filter sets HQ560/55, HQ645/75, and Q595LP (Chroma). The exposure time was 100 ms. Fluorescence spectra measurements were done on a SPEX FluoroMax-3 spectrofluorometer (Horiba Jobin Yvon). T1 relaxation times were determined using a Bruker mq60 MR relaxometer operating at 1.41 T (60 MHz). ICP-AES was performed by VHG Laboratories (Manchester, NH).
Results and Discussion
An overview of the approach used to synthesize chemically stabilized, porous polymersomes is depicted in Figure 1. Preparation of the porous polymersomes required generation-2 dendrimers to first be labeled with Gd−DTPA. The Gd-labeled dendrimers were then encapsulated into vesicles assembled from a mixture of polymerizable amphiphillic diblock copolymer [PEO(1300)-b-PBD(2500), PBdEO] and nonpolymerizabe native phospholipid (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, POPC). To confirm that nonpolymerizable phospholipids were integrated into the polymer bilayer during vesicle formation, a low molar percentage of fluorescent phospholipid was used during fabrication. A classical swelling technique was also adopted to form giant vesicles that could be observed by microscopy.20,21 As shown in Figure 2, incorporation of the fluorescent lipid into the polymersome bilayer was evident at the resolution of the optical microscope. This result demonstrates the feasibility of forming polymer−lipid hybrid vesicles.
Figure 1.

Schematic diagram illustrating the approach used to synthesize paramagnetic porous polymersomes. (a) Vesicles consisting of the diblock copolymer PBdEO and the phospholipid POPC at a molar ratio of 85:15 were prepared. Second-generation dendrimer conjugated Gd chelates were encapsulated within the aqueous interior during vesicle formation. (b) Cross-linking of PBdEO within the vesicle bilayer was induced by free radical polymerization. (c) Pores were formed in the polymersome bilayer by extracting the POPC with the surfactant Triton X-100.
Figure 2.

Microscopy images of giant vesicles composed of diblock copolymers and lipids. The vesicles were fluorescently labeled by incorporating the phospholipid rhodamine-PE into the membrane bilayer: (a) phase contrast image and (b) fluorescence image. Fluorescence is seen in individual giant vesicles. The scale bar represents 25 μm.
Following the formation of vesicles, the average hydrodynamic diameter was reduced to ∼125 nm (±2.5 nm standard deviation) by subjecting the sample to multiple freeze−thaw cycles and extrusion through a 100 nm polybicarbonate filter. The hydrodynamic diameter was measured by dynamic light scattering. Though vesicles were extruded through 100 nm pores, the resulting vesicles are larger than the corresponding pore diameter. This difference may result from the interplay between the mechanical constraints of the extrusion and the energetic constraints of the vesicles. It should be noted that the polymer vesicle does possess a relatively thick hydrophobic domain (∼8 nm).(28)
Once the extrusion process was complete, the diblock copolymers were cross-linked via free radical polymerization, and the stability of the polymerized vesicles was examined by surfactant solubilization.22–25 Specifically, polymerized and unpolymerized vesicles were treated with Triton X-100 at a high molar ratio of Triton X-100-to-polymer. Treatment of unpolymerized vesicles with Triton X-100 led to complete vesicle dissolution (data not shown), as determined by dynamical light scattering (DLS). The vesicles dissolve and form mixed micelles when the surfactant concentration exceeds its critical concentration. When the chemically cross-linked polymersomes were treated with a high concentration of Triton X-100, the mean vesicle diameter was essentially unchanged compared with vesicles that had not been treated with Triton X-100 (Figure 3). The second peak centered at 10 nm can be attributed to Triton X-100 micelles in the same solution. A similar peak at 10 nm was observed in samples containing just Triton X-100. The ability to impart structural stability into vesicle bilayer architectures is likely to prove highly beneficial in biological applications that require nanoplatforms with long in vivo half-lives.(29) The diversity of applications in which many phospholipid-based vesicles can be used is often limited due to their low stability in circulation.(30) These findings suggest that polymerized polymersomes may provide a stable, alternative platform to liposomes.
Figure 3.

Intensity-weighted hydrodynamic diameter measurements of cross-linked polymersomes before and after adding the surfactant Triton X-100 (final concentration = 8.3 mM). The measuring angle of DLS is 90°. When the chemically cross-linked polymersomes were treated with Triton X-100, the mean vesicle diameter (∼125 nm) was unchanged compared with vesicles that had not been treated with Triton X-100. The second peak that arises at 10 nm can be attributed to Triton X-100 micelles in the same solution.
Following polymerization of the diblock copolymers, the nonpolymerizable phospholipids were removed by extraction with Triton X-100, leaving pores within the membrane bilayer. A visible loss of the rhodamine-labeled phospholipid was observed, while dynamic light scattering confirmed that the polymersomes retained their structure (Figure 3). To confirm that pores were, in fact, created, chemically stabilized porous polymersomes similar to those described above were synthesized with carboxyfluorescein encapsulated within the core. Following the extraction of the phospholipids with Triton X-100, the polymersomes were centrifuged on a Microcon centrifugal filtering device (Millipore) with a molecular weight cutoff of 100 kDa. The liquid that flowed through the filter was then tested for fluorescence. It was hypothesized that if pores were present in the polymersome, the encapsulated carboxyfluorescein would diffuse across the membrane bilayer and pass through the filter during centrifugation. Alternatively, if no pores were present, no fluorescence would be detected in the flow-through because the carboxyfluorescein would remain entrapped within the polymersomes, which are too large to pass through the filter. As seen in Figure 4A, when polymerized polymersomes were treated with Triton X-100, 4 times more carboxyfluorescein was detected in the flow-through compared with vesicles that had not been treated with Triton X-100. Samples that were not treated with Triton X-100 likely exhibit a weak fluorescent signal due to a small amount of free carboxyfluorescein that was not removed during polymersome purification. When analogous studies were performed on polymersomes that had not been doped with phospholipids (i.e., no pores), only 1.2 times more carboxyfluorescein fluorescences was detected in the flow-through following treatment with Triton X-100 (Figure 4B). This small increase is due to the effect of Triton X-100 on carboxyfluorescein fluorescence. These results clearly demonstrate that highly porous vesicles can be created though the surfactant-mediated extraction of lipids from polymerized hybrid vesicles.
Figure 4.

Evaluation of pore formation within cross-linked polymersomes following surfactant solubilization. The water-soluble, low-molecular-weight dye carboxyfluorescein was encapsulated into chemically cross-linked polymersomes with and without 15% phospholipid. The polymerized vesicles were treated with an equivalent volume of buffer or Triton X-100 (final concentration = 10 mM) and then centrifuged on a Microcon centrifugal filtering device with a 100 kDa MWCO membrane (Millipore). The liquid that flowed through the filter was tested for fluorescence. (a) Cross-linked polymersomes doped with 15% phospholipid. (b) Cross-linked polymersomes without phospholipids.
Although diffusion of water across the polymersome bilayer is desirable, the leakage of Gd-labeled dendrimer from the vesicle would be detrimental to the utility of the probe. To confirm that second-generation dendrimers were too large to pass through the porous membrane of the polymerized vesicles, TRITC-labeled dendrimers were encapsulated within the core of the chemically stabilized porous polymersomes. Following centrifugation on the Microcon filtering device, no fluorescence was detected in the flow-through, suggesting that the dendrimer is retained within the porous vesicle (Figure 5). A similar assay was also conducted with polymersomes containing Gd-labeled dendrimers, where T1 measurements of the flow-through were conducted to confirm the absence of Gd. As expected, the T1 relaxation time of the flow-through was similar to that of pure phosphate buffer, ∼1000 ms. Assuming, that between 50 and 100% of the amines on each PAMAM dendrimer were labeled with DTPA−Gd, the estimated molecular weight of PAMAM−DTPA−Gd is between 7500 and 12 000 Da.
Figure 5.

Retention of generation-2 dendrimer within cross-linked porous polymersomes. TRITC-labeled dendrimer was encapsulated into chemically cross-linked polymersomes containing 15% phospholipid. The polymerized vesicles were treated with an equivalent volume of buffer or Triton X-100 (final concentration = 10 mM) and then centrifuged on a Microcon centrifugal filtering device with a 100 kDa MWCO membrane (Millipore). The liquid that flowed through the filter was tested for fluorescence. The emission spectrum of vesicles treated with Triton X-100 was nearly identical to that of the vesicles treated by buffer, indicating that the dendrimer is retained within the porous vesicles.
To assess the loading efficiency and paramagnetic properties of the chemically stabalized porous polymersomes, the amount of Gd in the polymersome sample was determined by ICP-AES. The relaxivity of the Gd within the polymersomes was then calculated as the slope of the curves 1/T1 vs Gd concentration, as shown in Figure 6. T1 relaxation times were determined using a Bruker mq60 MR relaxometer operating at 1.41 T (60 MHz) and at 40 °C. It was found that the R1 relaxivity per Gd was 7.2 mM−1 s−1. Polymersomes with encapsulated Gd-labeled dendrimer but no pores exhibited an R1 relaxivity of 3.1 mM−1 s−1 per Gd. Assuming no polymer was lost during the synthesis process and an average polymersome diameter of 125 nm, it was estimated that there were approximately 43 838 Gd per porous polymersome. The relaxivity per porous polymersome was then calculated, giving an R1 equal to 315 637 mM−1s−1. For comparison, chelated Gd alone was determined to possess an R1 relaxivity of 3.9 mM−1 s−1. Therefore, the polymersomes generate a relaxivity that is amplified by a factor of ∼105 for T1-weighted imaging.
Figure 6.

Relaxivity determination for Gd−dendrimer conjugates encapsulated within cross-linked porous polymersomes. Comparisons are made to Gd−dendrimer conjugates encapsulated within cross-linked polymersomes that do not contain pores and to Gd−DTPA. Measurements were acquired at 1.41 T (60 MHz) at 40 °C.
The paramagnetic properties of the polymersomes reported here compare very favorably with Gd-based agents that have previously been reported in the literature. For example, Gd-labeled shell-cross-linked nanoparticles (40 nm diameter) exhibit an R1 of 39 mM−1 s−1 per Gd (0.47 T) but possess only 510 Gd per particle, which results in an R1 of 2 × 104 mM−1 s−1 per nanoparticle.(14) This relaxivity is significantly lower than the paramagnetic porous polymersomes presented here.
Paramagnetic silica nanoparticles (∼100 nm) have been found to exhibit an R1 of 9.0 mM−1 s−1 per Gd (4.7 T) and contain 16 000 Gd per nanoparticle, which results in an R1 of 1.4 × 105 mM−1 s−1 per nanoparticle.(18) While these properties are similar to those of the paramagnetic polymersomes, polymersomes possess a hydrophobic domain that presents a natural carrier environment for hydrophobic drugs. This may allow paramagnetic porous polymersomes to be more readily adapted for combined targeted drug delivery and MR imaging.
Perfluorocarbon nanoparticles have a reported R1 of 25.3 mM−1 s−1 per Gd (1.5 T) and 94 200 Gd per particle, which results in an R1 of 2.38 × 106 mM−1 s−1 per nanoparticle. Although this relaxivity is higher than that of the 125 nm polymersomes reported here, it should be noted that the perfluorocarbon particles are much larger with a diameter of 273 nm.(15) Scaling with volume, a porous polymersome of this size would possess 456 681 Gd and exhibit an R1 of 3.3 × 106 mM−1 s−1 per polymersome. Also, while perfluorocarbons only exhibit a half-life of ∼1 h, polymersomes ∼100 nm in diameter have a reported plasma half-life of >15 h.27,31 The smaller size and extended half-life of polymersomes could provide important advantages in molecular imaging applications, including improved tissue penetration and increased target site accumulation.(32)
Conclusion
In conclusion, the preparation of chemically stabilized porous polymersomes for magnetic resonance imaging applications has been described. The porous vesicle membrane leads to a significant improvement in the water-exchange rate of the encapsulated Gd due to the faster flux of water across the bilayer. The relaxivity of the nanoparticle is also improved due to the slower rotational correlation lifetime of the encapsulated Gd−dendrimers. In addition, the polymersomes possess an unobstructed outer surface that may be used for attaching targeting ligands and a hydrophobic domain that presents a natural carrier environment for hydrophobic drugs. Therefore, the porous polymersomes described here provide a powerful new platform for combined targeted drug delivery and MR imaging.
Acknowledgments
This work was supported in part by the National Institute of Health (NCI) (R21 CA-125088), the American Cancer Society (RSG-07-005-01), and the Transdisciplinary Program in Translational Medicine and Therapeutics. We would like to thank Daniel Hammer, Natalie Christian, and Greg Robbins for their helpful discussions.
Funding Statement
National Institutes of Health, United States
References
- Caravan P.; Ellison J. J.; McMurry T. J.; Lauffer R. B. Chem. Rev. 1999, 99 (9), 2293–352. [DOI] [PubMed] [Google Scholar]
- Lee D. C.; Klocke F. J. Curr. Cardiol. Rep. 2006, 8 (1), 59–64. [DOI] [PubMed] [Google Scholar]
- Uematsu H.; Maeda M. Eur. Radiol. 2006, 16 (1), 180–6. [DOI] [PubMed] [Google Scholar]
- Caravan P. Chem. Soc. Rev. 2006, 35 (6), 512–23. [DOI] [PubMed] [Google Scholar]
- Wiener E. C.; Brechbiel M. W.; Brothers H.; Magin R. L.; Gansow O. A.; Tomalia D. A.; Lauterbur P. C. Magn. Reson. Med. 1994, 31 (1), 1–8. [DOI] [PubMed] [Google Scholar]
- Bryant L. H.; Brechbiel M. W.; Wu C.; Bulte J. W.; Herynek V.; Frank J. A. J. Magn. Reson. Imaging 1999, 9 (2), 348–52. [DOI] [PubMed] [Google Scholar]
- Duarte M. G.; Gil M. H.; Peters J. A.; Colet J. M.; Elst L. V.; Muller R. N.; Geraldes C. F. Bioconjugate Chem. 2001, 12 (2), 170–7. [DOI] [PubMed] [Google Scholar]
- Tilcock C.; Unger E.; Cullis P.; MacDougall P. Radiology 1989, 171 (1), 77–80. [DOI] [PubMed] [Google Scholar]
- Tilcock C.; MacDougall P.; Unger E.; Cardenas D.; Fajardo L. Biochim. Biophys. Acta 1990, 1022 (2), 181–6. [DOI] [PubMed] [Google Scholar]
- Parac-Vogt T. N.; Kimpe K.; Laurent S.; Pierart C.; Elst L. V.; Muller R. N.; Binnemans K. Eur. Biophys. J. 2006, 35 (2), 136–44. [DOI] [PubMed] [Google Scholar]
- Unger E.; Shen D. K.; Wu G. L.; Fritz T.. Magn. Reson. Med. 1991, 22 (2), 304−8(discussion 313). [DOI] [PubMed] [Google Scholar]
- Fossheim S. L.; Fahlvik A. K.; Klaveness J.; Muller R. N. Magn. Reson. Imaging 1999, 17 (1), 83–9. [DOI] [PubMed] [Google Scholar]
- Amirbekian V.; Lipinski M. J.; Briley-Saebo K. C.; Amirbekian S.; Aguinaldo J. G.; Weinreb D. B.; Vucic E.; Frias J. C.; Hyafil F.; Mani V.; Fisher E. A.; Fayad Z. A. Proc. Natl. Acad. Sci. U.S.A. 2007, 104 (3), 961–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner J. L.; Pan D. P. J.; Plummer R.; Chen Z. Y.; Whittaker A. K.; Wooley K. L. Adv. Funct. Mater. 2005, 15 (8), 1248–1254. [Google Scholar]
- Morawski A. M.; Winter P. M.; Crowder K. C.; Caruthers S. D.; Fuhrhop R. W.; Scott M. J.; Robertson J. D.; Abendschein D. R.; Lanza G. M.; Wickline S. A. Magn. Reson. Med. 2004, 51 (3), 480–6. [DOI] [PubMed] [Google Scholar]
- Lin Y. S.; Hung Y.; Su J. K.; Lee R.; Chang C.; Lin M. L.; Mou C. Y. J. Phys. Chem. B 2004, 108 (40), 15608–15611. [Google Scholar]
- Rieter W. J.; Kim J. S.; Taylor K. M.; An H.; Lin W.; Tarrant T. Angew. Chem., Int. Ed. 2007, 46 (20), 3680–2. [DOI] [PubMed] [Google Scholar]
- Santra S.; Bagwe R. P.; Dutta D.; Stanley J. T.; Walter G. A.; Tan W.; Moudgil B. M.; Mericle R. A. Adv. Mater. 2005, 17 (18), 2165–2169. [Google Scholar]
- Kamaly N.; Kalber T.; Ahmad A.; Oliver M. H.; So P. W.; Herlihy A. H.; Bell J. D.; Jorgensen M. R.; Miller A. D. Bioconjugate Chem 2008, 19 (1), 118–29. [DOI] [PubMed] [Google Scholar]
- Ghoroghchian P. P.; Frail P. R.; Susumu K.; Blessington D.; Brannan A. K.; Bates F. S.; Chance B.; Hammer D. A.; Therien M. J. Proc. Natl. Acad. Sci. U.S.A. 2005, 102 (8), 2922–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoroghchian P. P.; Frail P. R.; Susumu K.; Park T. H.; Wu S. P.; Uyeda H. T.; Hammer D. A.; Therien M. J. J. Am. Chem. Soc. 2005, 127 (44), 15388–90. [DOI] [PubMed] [Google Scholar]
- Discher B. M.; Bermudez H.; Hammer D. A.; Discher D. E.; Won Y. Y.; Bates F. S. J. Phys. Chem. B 2002, 106 (11), 2848–2854. [Google Scholar]
- Reiner J. E.; Wells J. M.; Kishore R. B.; Pfefferkorn C.; Helmerson K. Proc. Natl. Acad. Sci. U.S.A. 2006, 103 (5), 1173–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won Y. Y.; Davis H. T.; Bates F. S. Science 1999, 283 (5404), 960–3. [DOI] [PubMed] [Google Scholar]
- Won Y. Y.; Paso K.; Davis H. T.; Bates F. S. J. Phys. Chem. B 2001, 105 (35), 8302–8311. [Google Scholar]
- Lantzsch G.; Binder H.; Heerklotz H. J. Fluoresc. 1994, 4 (4), 339–343. [DOI] [PubMed] [Google Scholar]
- Photos P. J.; Bacakova L.; Discher B.; Bates F. S.; Discher D. E. J. Controlled Release 2003, 90 (3), 323–34. [DOI] [PubMed] [Google Scholar]
- Discher B. M.; Won Y. Y.; Ege D. S.; Lee J. C.; Bates F. S.; Discher D. E.; Hammer D. A. Science 1999, 284 (5417), 1143–6. [DOI] [PubMed] [Google Scholar]
- LeDuc P. R.; Wong M. S.; Ferreira P. M.; Groff R. E.; Haslinger K.; Koonce M. P.; Lee W. Y.; Love J. C.; McCammon J. A.; Monteiro-Riviere N. A.; Rotello V. M.; Rubloff G. W.; Westervelt R.; Yoda M. Nat. Nanotechnol. 2007, 2 (1), 3–7. [DOI] [PubMed] [Google Scholar]
- Kirby C.; Clarke J.; Gregoriadis G. Biochem. J. 1980, 186 (2), 591–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flacke S.; Fischer S.; Scott M. J.; Fuhrhop R. J.; Allen J. S.; McLean M.; Winter P.; Sicard G. A.; Gaffney P. J.; Wickline S. A.; Lanza G. M. Circulation 2001, 104 (11), 1280–5. [DOI] [PubMed] [Google Scholar]
- Moghimi S. M.; Hunter A. C.; Murray J. C. Pharmacol. Rev. 2001, 53 (2), 283–318. [PubMed] [Google Scholar]