

Abstract
The Staphylococcus aureus multidrug binding protein QacR binds to a broad spectrum of structurally dissimilar cationic, lipophilic drugs. Our previous structural analyses suggested that five QacR glutamic acid residues are critical for charge neutralization and specification of certain drugs. For example, E57 and E58 interact with berberine and with one of the positively charged moieties of the bivalent drug dequalinium. Here we report the structural and biochemical effects of substituting E57 and E58 with alanine and glutamine. Unexpectedly, individual substitutions of these residues did not significantly affect QacR drug binding affinity. Structures of QacR(E57Q) and QacR(E58Q) bound to dequalinium indicated that E57 and E58 are redundant for charge neutralization. The most significant finding was that berberine was reoriented in the QacR multidrug binding pocket so that its positive charge was neutralized by side chain oxygen atoms and aromatic residues. Together, these data emphasize the remarkable versatility of the QacR multidrug binding pocket, illustrating that the capacity of QacR to bind myriad cationic drugs is largely governed by the presence in the pocket of a redundancy of polar, charged, and aromatic residues that are capable of electrostatic neutralization.
Multidrug resistant bacteria represent a major global health threat that has in great part arisen through the action of multidrug efflux transporters. Capable of transporting a broad range of structurally and chemically diverse compounds from the cell, these proteins afford simultaneous resistance to multiple cytotoxic compounds and drugs. Despite this apparent promiscuity in substrate binding, each multidrug binding protein binds to a precise array of compounds. Uncovering the determinants that define this specificity in polyspecific substrate recognition may provide the key to combating multidrug resistance.
Important insights into multidrug recognition have been gained by examining the soluble regulatory proteins of bacterial multidrug efflux transporters, which are more amenable to high-resolution structural analyses and bind to many of the same compounds that are substrates for the pumps that they regulate. One such protein is QacR, the transcriptional repressor of the Staphylococcus aureus multidrug resistance efflux gene qacA. QacR is a member of the TetR family of repressors which share a helix−turn−helix DNA binding motif at their N-terminal regions but encode diverse C-terminal domains that are utilized in binding protein-specific inducing compounds (1–3). QacR binds to the IR1 DNA operator sequence, a large inverted repeat that overlaps the qacA promoter, as a pair of dimers (4,5) and is induced by numerous structurally dissimilar, monovalent and bivalent cationic, lipophilic compounds (6,7). Drugs bind to QacR in a one drug:QacR dimer stoichiometry (8), inducing a coil-to-helix transition of residues 89−93 within the drug-bound subunit of the dimer. Residues Y92 and Y93 are consequently expelled from the interior of the protein, leading to the formation of a large binding pocket (8). This conformational switch also leads to the relocation of the DNA binding domains, rendering the protein unable to bind the IR1 operator site, thus allowing upregulation of qacA transcription (8).
The QacR multidrug binding pocket created by this conformational switch is expansive and has been described as containing two distinct but partially overlapping binding sites, designated the rhodamine 6G (R6G)1 and ethidium (Et) sites after their respectively bound drugs. This pocket contains five glutamate, multiple aromatic and nonpolar, and several polar residues that mediate drug interactions (8). The majority of the drugs, including the monovalent plant alkaloid berberine (Be), bind in the R6G site. However, the binding of drugs is not restricted solely to the R6G or Et sites. For example, the monovalent dye malachite green (MG) contacts residues categorized to be within both sites, including the acidic residues E90 and E120, by binding in an intermediary region. The multifaceted nature of the QacR drug binding pocket also explains how it is able to bind bivalent drugs as exemplified by the structure of QacR bound to dequalinium (Dq). In this structure, each of the two positively charged aminomethylquinolinium moieties of Dq, which are connected by a 10-methylene carbon linker, inserts into one of the canonical sites, i.e., one into the R6G site and the other into the Et site (8). Notably, although each drug utilizes a different subset of QacR residues for binding, all, with the exception of pentamidine (Pt), which is complemented by residue E63, π−π, cation−π, and dipole−charge interactions, contact one or more of the four glutamate residues at positions 57, 58, 90, and 120 (8). The utilization of cation−π interactions has also been observed in the interaction of other multidrug binding proteins with drugs, most notably that of the RND-type multidrug transporter AcrB from Escherichia coli(9,10). Indeed, structures of AcrB bound to several drugs have highlighted the importance of cation−π and stacking interactions in the polyspecific binding of its ligands (10). While cation−π and stacking interactions appear to be important in multidrug binding, several studies suggest that negatively charged residues are likely to play a primary role in their recognition (11). This proposition has been supported by several biochemical studies involving the mutational analysis of acidic residues such as those described for the BmrR multidrug binding transcriptional regulator in Bacillus subtilis(11) and the multidrug resistance transport proteins MdfA and EmrE in E. coli(12,13), QacA in S. aureus(14,15), and human MRP1 (16). However, the relative contribution of each of the glutamate residues to the binding of different drugs in the QacR multidrug binding pocket is unclear.
To begin to address the role(s) of negatively charged residues in the multidrug binding pocket of QacR, we analyzed the effect of mutating two key acidic residues, E57 and E58, to alanine and glutamine on drug binding by QacR. Three drugs, Dq, Be, and MG, were assayed for their ability to bind these mutant proteins. These compounds were chosen because it was anticipated that their binding would be affected differently by these mutations. Specifically, since one of the two positively charged moieties of Dq interacts with residues E57 and E58, their substitution would be predicted to manifest some but not a drastic change in drug binding (8). On the other hand, residues E57 and E58 appear to provide the sole neutralization for the positive charge of Be, suggesting that mutating these residues is likely to affect its binding significantly (8). By contrast, MG makes no contacts with these residues, and thus, the binding of this aromatic monovalent cation to QacR would be predicted to be altered little, if any, by such mutations (8). In other words, MG served as a control, and it was expected that it would bind like the wild type (wt) to these mutant QacR proteins. In addition to the binding affinities of QacR(E57A/Q) and QacR(E58A/Q) for Be, Dq, and MG, the crystal structures of the QacR(E57Q)−Dq, QacR(E57Q)−MG, QacR(E58Q)−Be, QacR(E58Q)−Dq, and QacR(E58Q)−MG complexes were determined. The resulting data reveal unanticipated complexity in QacR multidrug binding and point to chemical redundancy as a key underlying principal of the ligand polyspecificity of this and likely other multidrug binding transcription regulators and efflux transporters.
Materials and Methods
Bacterial Strains and Growth Conditions
All cloning and overexpression procedures were performed in E. coli strain DH5α (17). This strain was transformed by standard procedures and cultured at 37 °C in Luria-Bertani broth or agar (17) containing, where appropriate, 100 μg/mL ampicillin.
DNA Isolation and Manipulations
The Quantum Prep plasmid miniprep kit (Bio-Rad) was employed to isolate plasmid DNA from E. coli. Restriction enzymes, T4 DNA ligase, calf intestinal alkaline phosphatase, and Taq DNA polymerase (all from New England Biolabs), were each used according to the manufacturer’s instructions. Oligonucleotides were purchased from GeneWorks. PCR products were purified with the Wizard PCR Prep kit (Promega), and DNA fragments were isolated from agarose gels using the GFX PCR and gel band purification kit (Amersham Biosciences).
Construction of QacR Mutants
The single QacR mutants E57A, E57Q, E58A, and E58Q and the double QacR mutant variant E57Q/E58Q were obtained by generating a PCR-derived fragment encompassing the residue to be mutated and replacing the wt sequence. This was achieved by subcloning into the pTTQ18-based qacR clone pSK5676 (7), which has a six-histidine tag incorporated into the C-terminus of the expressed QacR protein. Automated DNA sequencing, performed at the Australian Genome Research Facility (Brisbane, Australia), was employed to verify the presence of the desired mutations and the absence of spurious mutations that may have been introduced during PCR.
Protein Purification
Mutant QacR proteins were overexpressed from cells carrying plasmid derivatives of pSK5676, purified by Ni2+-NTA metal chelate affinity chromatography (Invitrogen ProBond resin), and dialyzed against buffer A [1 M NaCl, 20 mM Tris-HCl, and 5% (v/v) glycerol (pH 7.5)] as previously described (7). The purified proteins were more than 99% pure as estimated from Coomassie-stained SDS−PAGE gels, and their concentrations were determined using the Coomassie plus protein assay reagent (Pierce).
Tryptophan Fluorescence Measurements
Fluorescence intensity measurements of QacR proteins were performed and Kd values calculated as described previously (7) with the exception that ligand stocks (2.5 mM) were dissolved in Milli-Q water and dilutions made as required in buffer B [100 mM NaCl, 15 mM Tris-HCl, and 2.5% (v/v) glycerol (pH 7.5)]. Each of the reported Kd values represents an average of three independent experiments (Figure S1). Intrinsic fluorescence could not be used to assess QacR binding to Dq because Dq displays significant fluorescence at 340 nm, and therefore, Dq binding was assessed by isothermal titration calorimetry.
Isothermal Titration Calorimetry
Isothermal titration calorimetry (ITC) was used to determine the affinity of QacR proteins for Dq and carried out as described previously (18) (see ref (18) and Figure S2). Dq was dissolved in buffer B, and ligand and protein concentrations were determined spectrophotometrically. Protein and ligand samples were degassed before being loaded into the cell and syringe of the VP-ITC from MicroCal Inc. (Northampton, MA) at concentrations of 8 and 280 μM, respectively. The stirring speed was set to 300 rpm and the power to 10 μcal/s, and the experimental data were collected at 23 °C. Data analyses were performed with Origin 5.0 (MicroCal Inc.).
Crystallization, Data Collection, and Structure Determination of Mutant QacR−Drug Complexes
Purified QacR mutant proteins were subjected to reductive alkylation of lysines, which allows reproducible crystallization of QacR−drug complexes (8,19). The reductive alkylation was terminated by the addition of 200 mM glycine. The resulting alkylated proteins were concentrated to 10−15 mg/mL, during which the protein was buffer exchanged into a solution of 50 mM Tris (pH 7.5), 50 mM imidazole, 300 mM NaCl, and 5% glycerol. Reductive alkylation of QacR has been demonstrated to have no detrimental effect on its ability to bind drugs (8). QacR complexes were crystallized at room temperature using the hanging drop vapor diffusion method. QacR(E57Q)−MG, QacR(E58Q)−MG, and QacR(E58Q)−Be complexes were produced via addition of drug to the protein solution to final concentrations ranging from 100 to 200 μM and overnight incubation. Because Dq displays low solubility, QacR(E57Q)−Dq and QacR(E58Q)−Dq complexes were formed via addition of a small amount of solid Dq to the protein solution and incubation for several hours at 4 °C. The mixture was then centrifuged, and the supernatant, which contained the QacR−Dq complex, was used for crystallization. Each QacR−drug complex was mixed 1:1 with the crystallization solution of 2.9 M ammonium sulfate and 50−150 mM sodium acetate (pH 4.6) and sealed over a 1 mL reservoir of the same solution. Data-quality crystals were obtained for the QacR(E57Q)−Dq, QacR(E57Q)−MG, QacR(E58Q)−Dq, QacR(E58Q)−MG, and QacR(E58Q)−Be complexes. Despite numerous attempts, the QacR(E57Q)−Be and QacR(E57Q/E58Q)−drug complexes did not crystallize or data-quality crystals could not be obtained. X-ray intensity data were collected at 100 K at ALS beamline 4.2.2 and the data processed with MOSFLM and scaled with SCALA (20,21). All crystals took the native P42212 space group. However, because of slight changes in the cell edges of the mutant structures compared to those of the wild type, the wt coordinates were optimally positioned into the new unit cells using the molecular replacement program MOLREP as implemented in CCP4. The structures were refined in CNS (22). O was utilized to visualize experimental electron density maps and for model building (23). Each structure was validated with PROCHECK (24). Table 1 lists selected data collection and refinement statistics for each of the complexes. Figure 1 was made with PyMol (25), while Figures 4–6 were made with Chimera (26). The atomic coordinates and structure factors for each structure have been deposited in the Protein Data Bank as entries 3BT9 for the QacR(E57Q)−Dq complex, 3BTC for the QacR(E57Q)−MG complex, 3BTI for the QacR(E58Q)−Be complex, 3BTJ for the QacR(E58Q)−Dq complex, and 3BTK for the QacR(E58Q)−MG complex.
Table 1. Selected Data Collection and Refinement Statistics.
| QacR(E57Q)−Dq | QacR(E57Q)−MG | QacR(E58Q)−Be | QacR(E58Q)−Dq | QacR(E58Q)−MG | |
|---|---|---|---|---|---|
| space group | P42212 | P42212 | P42212 | P42212 | P42212 |
| unit cell constants (Å) | a = b = 171.24 | a = b = 171.44 | a = b = 171.5 | a = b = 171.9 | a = b = 171.8 |
| c = 94.2 | c = 94.74 | c = 94.4 | c = 94.5 | c = 94.8 | |
| resolution (Å) | 81.6−2.75 | 76.8−2.90 | 76.7−2.85 | 81.7−2.98 | 76.8−2.90 |
| overall Rsym (%)a | 4.5/38.4 | 4.7/33.6 | 8.1/47.5 | 6.8/47.8 | 5.5/31.7 |
| overall I/σ(I) | 12.0/2.0 | 12.3/2.1 | 5.7/1.8 | 9.3/1.8 | 8.9/2.4 |
| total no. of reflections | 128283/7823 | 196681/14856 | 116739/16219 | 102268/12907 | 286661/12390 |
| no. of unique reflections | 36964/2617 | 31866/2200 | 33379/4779 | 29386/4125 | 31835/4561 |
| completeness (%) | 96.5/72.9 | 99.3/99.3 | 99.8/99.8 | 99.6/99.7 | 99.4/99.4 |
| Refinement Statistics | |||||
| Rwork/Rfree (%)b | 23.9/29.0 | 24.0/28.8 | 21.7/27.5 | 21.2/26.4 | 22.2/26.4 |
| root-mean-square deviation | |||||
| bond angles (deg) | 1.25 | 1.16 | 1.03 | 1.26 | 1.16 |
| bond lengths (Å) | 0.009 | 0.009 | 0.008 | 0.009 | 0.009 |
| B-values (Å2) | 1.6 | 1.5 | 1.6 | 1.7 | 2.5 |
| Ramachandran analysis | |||||
| most favored (%, no.) | 87.1/610 | 88.4/616 | 87.1/610 | 86.4/605 | 87.1/610 |
| additional allowed (%, no.) | 11.6/81 | 10.0/70 | 11.7/82 | 12.0/84 | 11.6/81 |
| generously allowed (%, no.) | 0.6/4 | 0.9/6 | 0.4/3 | 0.9/6 | 0.6/4 |
| disallowed (%, no.) | 0.7/5 | 0.7/5 | 0.7/5 | 0.7/5 | 0.7/5 |
Rsym = ∑∑|Ihkl − Ihkl(j)|/∑NIhkl, where Ihkl(j) is the observed intensity and Ihkl is the final average value of intensity.
Rwork and Rfree = ∑||Fobs| − |Fcalc||/∑Fobs, where Fobs is the observed structure factor amplitude and Fcalc the calculated structure factor amplitude for the working and test sets, respectively. Data for highest-resolution shells given after the slash.
Figure 1.

Structure of the QacR(E58Q)−Be complex. A ribbon diagram of QacR(E58Q) bound to berberine with the drug-bound subunit colored yellow and the drug-free subunit gray. Secondary structural elements and the N- and C-termini are labeled. Residues E57 and E58 are shown as CPK. Berberine is shown as orange sticks.
Figure 4.

Views of the multidrug binding pockets of superimposed wt QacR (light blue) and QacR(E57Q) (orange) in complex with (a) dequalinium (Dq) and (b) malachite green (MG). For clarity, only key residues are shown. Nitrogen and oxygen atoms are colored dark blue and red, respectively. Helices containing residues interacting with the drug are labeled.
Figure 6.

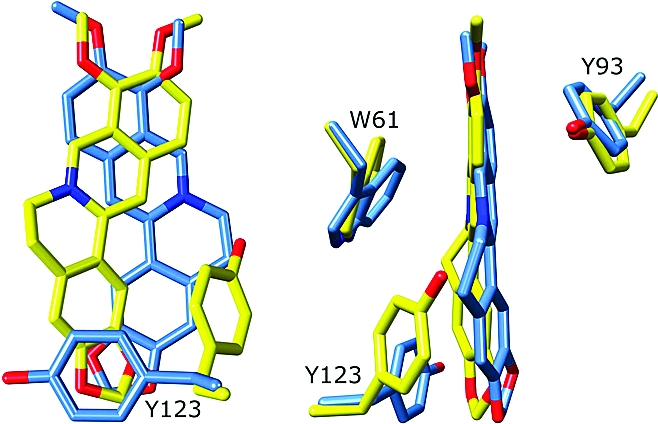
Magnification of the multidrug binding pockets of superimposed wt QacR (blue) and QacR(E58Q) (yellow) in complex with berberine. Shown are two views related by an ∼90° rotation. Note the striking reorientation of the drug in the E58Q mutant protein and the altered location of the Y123 side chain in the mutant structure.
Results
Structural analyses of QacR in complex with nine cationic drugs (8,27,28) suggested that the formal negative charge carried by glutamate residues that line the QacR multidrug binding pocket is pivotal in their neutralization and, thus, to binding affinity of these positively charged drugs. Here we examined the effect of substituting glutamate residues E57 and E58, which lie proximal to the positively charged nitrogen atoms of the bound drugs Be and Dq but are distal to the positive charge of MG (8), with the uncharged residues alanine and glutamine (Figure 1). The double glutamine substitution, QacR(E57Q/E58Q), was also constructed and examined. Drug binding affinities of mutant proteins were determined, and structures of QacR(E58Q) bound to Be, Dq, and MG and QacR(E57Q) bound to Dq and MG were determined.
Drug Binding Affinities
Utilizing the intrinsic fluorescence quenching of tryptophan residues, the binding affinities of wt and mutant QacR proteins were determined for MG and Be. Because Dq exhibited significant fluorescence at 340 nm, ITC was used to obtain the binding affinities for this compound. As predicted, there was no significant difference between the binding affinities of mutant QacR proteins for MG compared to wt QacR, with changes not exceeding 2.4-fold (Table 2). However, unexpectedly, this was also true for the majority of the QacR mutants with Be and Dq; i.e., nearly all displayed binding affinities for these drugs within 2-fold of that of wt QacR (Table 2). The notable exceptions were QacR(E58Q) and the double mutant QacR(E57Q/E58Q), which remarkably displayed a 4- and 9-fold increases in affinity for Be, respectively. By contrast, the QacR(E57Q/E58Q) double mutant exhibited a 4-fold decrease in binding affinity for Dq (Table 2).
Table 2. Effect of Substitutions of QacR Residues E57 and E58 on Drug Binding Affinity (Kd) in Vitro.
| compound (μM) |
|||
|---|---|---|---|
| QacR | MGa | Bea | Dqa |
| wt | 1.23 ± 0.14 | 2.86 ± 0.62 | 1.30 ± 0.30 |
| E57A | 2.98 ± 0.23 | 1.88 ± 0.23 | NDb |
| E57Q | 1.77 ± 0.17 | 1.39 ± 0.15 | 1.95 ± 0.18 |
| E58A | 0.81 ± 0.19 | 1.35 ± 0.10 | NDb |
| E58Q | 1.09 ± 0.07 | 0.72 ± 0.08 | 2.40 ± 0.20 |
| E57Q/E58Q | 1.99 ± 0.08 | 0.32 ± 0.15 | 5.20 ± 3.80 |
Values represent an average of three separate experiments.
Not determined.
Structural Determination of QacR(E57Q)− and (E58Q)−Drug Complexes
To understand the molecular basis for the unexpected drug binding affinities exhibited by the QacR(E57Q) and QacR(E58Q) mutants, we conducted X-ray crystallographic experiments with these QacR mutants in complexes with Be, Dq, and MG. Crystal structures were obtained for QacR(E57Q) bound to Dq or MG and for QacR(E58Q) in complex with Be, Dq, or MG. Structures of QacR(E57Q) bound to MG and Dq were refined to final Rwork and Rfree values of 24.0 and 28.8% to 2.90 Å resolution and 23.9 and 29.0% to 2.75 Å resolution, respectively. Structures of QacR(E58Q) bound to Be, MG, and Dq had Rwork and Rfree values of 21.7 and 27.5%, 22.2 and 26.4%, and 21.2 and 26.4% and were resolved to 2.85, 2.90, and 2.98 Å resolution, respectively. Electron density for each drug was clear (Figures 2a,b and 3a−c). Selected data collection statistics, refinement statistics, and the Ramachandran plot values of each complex are given in Table 1. Consistent with the crystallographic isomorphism of the mutant−drug complexes and corresponding wt QacR−drug complexes, root-mean-square deviations (rmsd) of <0.70 Å were obtained after superimposition of all corresponding Cα atoms, demonstrating that no global structural changes were produced by the E57Q or E58Q substitution (Figures 1, 4, and 5).
Figure 2.

Electron density maps (2Fo − Fc) of QacR(E57Q) in complex with (a) malachite green (MG) and (b) dequalinium (Dq). The molecules are shown as sticks with carbon, nitrogen, and oxygen atoms colored yellow, blue, and red, respectively. The maps were calculated after refinement of the protein structure and prior to addition of the drugs. The contour level is 0.8σ.
Figure 3.

Electron density maps (2Fo − Fc) of QacR(E58Q) in complex with (a) malachite green (MG), (b) dequalinium (Dq), and (c) berberine (Be). The molecules are shown as sticks with carbon, nitrogen, and oxygen atoms colored yellow, blue, and red, respectively. The maps were calculated after refinement of the protein structure and prior to addition of the drugs. The contour level is 0.7σ.
Figure 5.
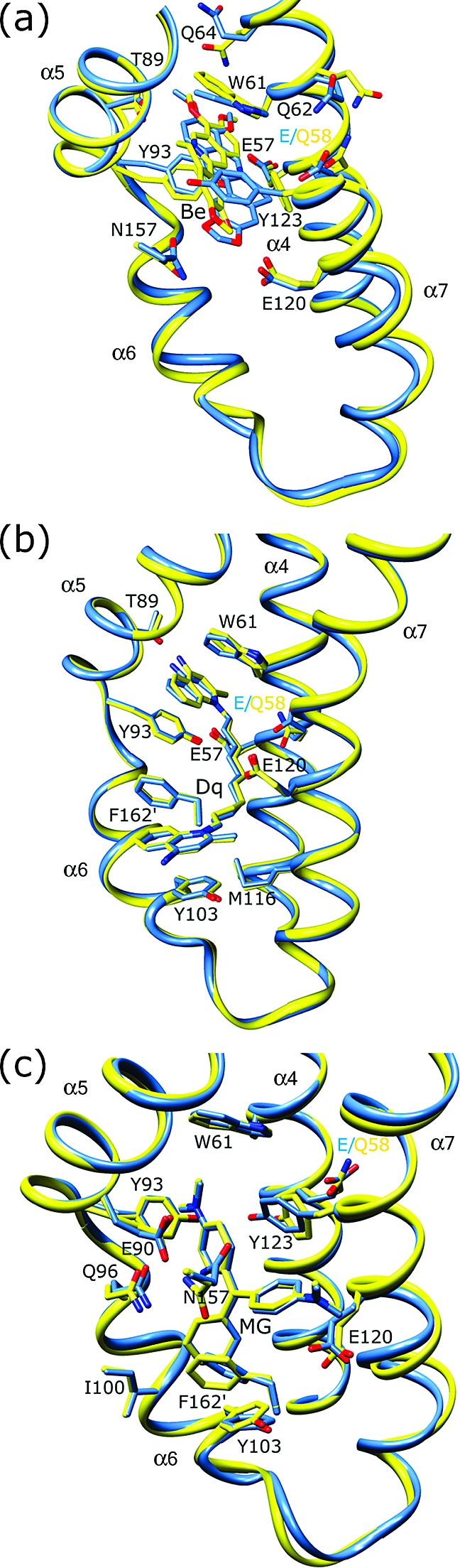
Views of the multidrug binding pockets of superimposed wt QacR (light blue) and QacR(E58Q) (yellow) in complex with (a) berberine (Be), (b) dequalinium (Dq), and (c) malachite green (MG). For clarity, only key residues are shown. Nitrogen and oxygen atoms are colored dark blue and red, respectively. Helices containing residues interacting with the drug are labeled.
Because E57 and E58 make no contacts with MG and their mutations have little effect on drug binding, we predicted that the structures of these mutants in complex with MG would remain wt. Indeed, the binding site occupied by MG in both the QacR(E57Q)−MG and QacR(E58Q)−MG structures was found to be identical to that in the wt QacR−MG structure with the same protein−drug interactions being made (Figures 4b and 5c and Table 3). These included contacts from QacR residues E90 and E120 to the two dimethylamino groups of MG, at which the delocalized positive charge largely rests. In addition, critical aromatic contacts provided by QacR residues W61, Y93, Y103, Y123, and F162′ (where the prime indicates the other subunit of the dimer) were conserved between the mutant and wt structures. In the QacR(E57Q)−Dq and QacR(E58Q)−Dq structures, Dq was also bound essentially in the same manner as in wt QacR, with the majority of the protein−drug contacts maintained (Figures 4a and 5b and Table 3). Specifically, Dq spanned the R6G and Et subpockets such that one positively charged aminomethylquinolinium group remained neutralized by E120 and the other largely by the unsubstituted carboxylate of either E57 or E58. Thus, in the case of Dq, there appears to be a functional redundancy of the charged groups of residues E57 and E58 such that in the Dq structure only one of these carboxylate side chains was necessary to interact with, and thereby neutralize, the positively charged aminomethylquinolinium group that was inserted into the R6G pocket. In addition, W61 and Y93 also provide cation−π interactions to aid in charge neutralization of this aminomethylquinolinium group. This was consistent with binding data showing that when both E57 and E58 were changed to glutamine there was a reduction, albeit modest (4-fold), in the level of QacR binding to Dq, while single substitutions of each residue had no discernible effect on the QacR−Dq binding affinity or binding mode (Table 2).
Table 3. Distances between Residues E57, E58, E90, and E120 and Berberine (Be), Malachite Green (MG), and Dequalinium (Dq) in the wt and QacR(E57Q) and QacR(E58Q) Mutant−Drug Complexesa.
| distance (Å) |
|||||
|---|---|---|---|---|---|
| charge center | QacR | E/Q57 | E/Q58 | E90 | E120 |
| Be (N1) | wt | 5.6 | 4.9 | ||
| E58Q | 8.7 | 8.6 | |||
| MG (N2) | wt | 3.4 | |||
| E57Q | 3.8 | ||||
| E58Q | 3.5 | ||||
| MG (N3) | wt | 3.6 | |||
| E57Q | 4.3 | ||||
| E58Q | 3.7 | ||||
| Dq (N1) | wt | 4.8 | |||
| E57Q | 4.4 | ||||
| E58Q | 4.9 | ||||
| Dq (N2) | wt | 4.8 | 4.9 | ||
| E57Q | 4.3 | 5.9 | |||
| E58Q | 4.7 | 5.7 | |||
The distances (in angstroms) are from the atom in the specified residue that is closest to the listed atom in the drug.
By sharp contrast to the essentially wt modes of binding of MG and Dq to QacR(E57Q) and QacR(E58Q), the mode of binding of Be to QacR(E58Q) was quite different from that observed in the wt QacR−Be complex (Figures 5a and 6 and Table 2). In the QacR(E58Q) structure, the Be molecule occupied roughly the same position in the pocket relative to its location in wt QacR; however, the drug was rotated approximately 180° along its long axis (Figure 6a,b). In this new position, the delocalized positive charge, which is centered on N1 of Be, moved 3 and 4 Å from residues 57 and 58, respectively, and was now located 3.8 Å from the hydroxyl side chain of residue T89. Interestingly, this new location placed the N1 atom of Be 2.5 Å closer to the carboxylate side chain of E90 as compared to its wt QacR position (new distance of ∼7 Å), conferring a new long-range electrostatic interaction that may contribute to the neutralization of the positive charge of the drug (Table 3). However, charge neutralization of Be in this new orientation was mediated primarily by aromatic and polar residues (Figures 5a and 6b). These include more favorable stacking interactions from the side chain of Y123. Indeed, the side chain of residue Y123 was rotated in the mutant structure from its wt conformation to make these interactions (Figures 5a and 6a,b). In addition to providing enhanced stacking interactions, the rotation of the Y123 side chain was coupled to the flipped conformation of Be in the QacR(E58Q) protein as the drug and Y123 side chain would clash if either maintained its wt binding mode. In this new binding position, Be was also able to make more favorable interactions with the side chain of residue W61. Finally, in its new position, the Be molecule makes van der Waals interactions with the side chain of S68. These new and enhanced interactions between Be and QacR(E58Q) are consistent with the modestly improved binding affinity of this mutant for Be compared to wt QacR (Table 2).
Discussion
The presence of charged residues in the ligand binding pockets of multidrug binding proteins has emerged as an important drug binding determinant. Indeed, substitution of a key glutamate residue (E253) in the ligand binding pocket of the C-terminal binding domain of the multidrug transcription regulator, BmrR, resulted in the abolition of binding to the majority of representative cationic co-activators (11). In the S. aureus multidrug binding repressor protein QacR, which binds to a diverse yet precise array of cationic compounds, it was predicted that glutamate residues lining the multidrug binding pocket would play a key role in drug binding affinity. Evidence supporting this prediction was previously provided by crystallographic and binding analyses of QacR binding simultaneously to the two drugs Et and proflavine. These data indicated that disruption of the interaction of Et with E120 significantly reduced its binding affinity (28).
The QacR E57 and E58 residues were predicted from earlier crystallographic studies to be important for drug binding. These residues interact with one of the positively charged aminomethylquinolinium moieties of the bivalent drug Dq and with the positively charged nitrogen atom of Be (8). In this study, we examined the effect of mutating these residues to both alanine and the isosteric residue glutamine, the side chains of which do not carry a formal negative charge, on drug binding by QacR. Contrary to expectations, our results demonstrated that the formal negative charge of the E57 and E58 side chains alone does not contribute significantly to drug binding affinity (Table 2). Noticeably, this near-isoenergetic drug binding displayed by the QacR E57 and E58 mutants appears to be primarily attributable to the functional and chemical redundancy of residues that are capable of electrostatic neutralization in the QacR multidrug binding pocket.
This mechanism was clearly evident in the structures of QacR(E57Q) and QacR(E58Q) bound to Dq and QacR(E58Q) bound to Be. In both the QacR(E57Q)−Dq and QacR(E58Q)−Dq complex structures, movement of Dq in the multidrug binding pocket was negligible (Figures 4a and 5b), with the immediate functional redundancy provided by E57 and E58 in the R6G subpocket apparently being sufficient to permit the essentially isoenergetic binding mode of Dq. Consistent with this, mutation of both residues to glutamine resulted in a measurable decrease in binding affinity (Table 2). The fact that the E57Q/E58Q double mutant displayed an only 4-fold reduction in Dq binding affinity is attributed in great part to the retention of the E120−aminomethylquinolinium interaction and the large number of contacts between QacR and Dq in this preferred binding mode. Indeed, the large size and extended nature of the Dq molecule appear to constrain it to adopt only one conformation within the pocket.
A more complex form of redundancy in the QacR multidrug binding mechanism is observed in the QacR(E58Q)−Be structure. Strikingly, in this complex, Be reorients in the multidrug binding pocket such that the positive charge centered on N1 of Be is no longer immediately complemented by either E57 or E58, or indeed by any other acidic residue (Figures 5a and 6a,b). Moreover, this new position allows Be to make more favorable stacking interactions with the side chains of aromatic residues W61, Y93, and Y123, thereby providing π−π and cation−π complementation of the drug. In addition, the negative dipole of the side chain oxygen atom of residue T89 makes a direct contact with the N1 atom of Be, providing further charge stabilization. These combined changes likely explain the 4-fold increase in binding affinity of Be for the QacR(E58Q) mutant compared to wt. A similar mechanism of drug reorientation with alternative charge complementation and enhanced contacts may explain the 9-fold increase in binding affinity for Be by the double mutant QacR(E57Q/E58Q) (Table 2), the complex of which has failed to yield crystals.
Collectively, these data suggest that the involvement of acidic residues E57 and E58 in the QacR multidrug binding pocket is not absolutely required for the neutralization or binding affinity of either cationic drug Be or Dq. This is in direct contrast to observations made regarding other multidrug binding proteins such as EmrE, MdfA, and BmrR, for which acidic residues have been shown to directly mediate a significant energetic contribution to ligand binding (11–15). Indeed, a recent study of the TetR-family multiligand binding transcription repressor, TtgR, indicated that binding of phloretin to the high-affinity site of TtgR was dependent upon its interaction with a specific arginine residue (29). Glycine substitution of this residue destroyed binding to this site, whereas notably, TtgR has a second “general” binding site to which phloretin and other drugs bind with lower affinity, which is not dependent upon charged residues (29). Nevertheless, the unusual mode of Be neutralization by the QacR(E58Q) mutant is somewhat similar to what has been observed in the QacR−Pt complex (27). Like Dq, Pt is a bivalent drug, and in the QacR−Pt structure, one of the positively charged benzamidine moieties of Pt was neutralized by the carboxylate side chain of residue E63 while the other was electrostatically complemented not by glutamate or aspartate residues, but by stacking and dipole−charge interactions from residues S86, Y127, A153, and N157 (27). Thus, the QacR(E57Q)−Be and QacR−Pt structures indicate that aromatic residues can play a central role in drug neutralization because they can complement both the lipophilic and charged nature of the drugs. Similar contacts are thought to be important in the interactions of ligands and multidrug efflux transport proteins Pgp and LmrA (30), neither of which contains acidic residues within its transmembrane domain, but they both still selectively interact with and export cationic compounds (31,32). This contention is supported by the finding that site-directed substitution of aromatic residues in the transmembrane domain of Pgp had a negative impact on transport activity and drug resistance (33,34). Additionally, structures of drug-bound AcrB have revealed an aromatic rich binding pocket containing many candidates for hydrophobic or π−π interactions (10).
One interpretation of these data is that the acidic residues in the QacR multidrug binding pocket contribute primarily to polyspecific binding capability and drug orientation. This is supported by the finding that upon abolition of the formal negative charge at position 58 in the structure of the QacR(E58Q)−Be complex, Be moved away from a favorable electrostatic interaction with residue E57 to a new position in the pocket that is essentially equidistant, yet distal, 7.8 and 8.7 Å from E90 and E57, respectively. Furthermore, mutational analyses of acidic residues involved in the charge complementation of structurally homologous proteins recognized by the T-cell antigen CD2 suggested that this receptor utilizes energetically neutral electrostatic interactions to create specificity in low-energy protein−protein recognition (35). Indeed, this method of recognition has been proposed to be highly appropriate for biological interactions that require lower affinities, uncoupling increases in specificity from increases in affinity. This premise may apply to the glutamate residues of QacR and, perhaps also, to the multidrug recognition mechanisms of other proteins, which settle for relatively low-affinity binding to their substrates to permit recognition of a myriad of structurally dissimilar ligands. In general support of this concept, although acidic residues appear to provide a strong energetic contribution to cationic ligand binding by MdfA and EmrE, it has been suggested that the transport rate mediated by these multidrug transporters may be inversely related to substrate binding affinity, and hence, adverse consequences, i.e., inhibition of drug efflux, could arise from affinities that are too high (13,36).
In conclusion, this study demonstrates that seemingly unfavorable mutations of acidic residues in the QacR multidrug binding pocket can be compensated either by an immediate redundancy of functionally equivalent residues or by shifts in drug orientation to permit alternative sets of contacts, which result in near-isoenergetic and in some cases even more favorable binding modes and further attest to the remarkable plasticity of the QacR multidrug binding pocket.
Supporting Information Available
Results showing the determination of binding affinity of wt QacR and QacR(E58Q) for malachite green (MG) by quenching of intrinsic tryptophan fluorescence (Figure S1) and the ITC binding isotherm for QacR(E58Q) binding to dequalinium (Dq) (Figure S2). This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Footnotes
Abbreviations: Be, berberine; Et, ethidium; Dq, dequalinium; MG, malachite green; Pt, pentamidine; R6G, rhodamine 6G; rmsd, root-mean-square deviation; wt, wild type; ITC, isothermal calorimetry.
Supplementary Material
References
- Ramos J. L.; Martinez-Bueno M.; Molina-Henares A. J.; Teran W.; Watanabe K.; Zhang X.; Gallegos M. T.; Brennan R.; Tobes R. (2005) The TetR family of transcriptional repressors. Microbiol. Mol. Biol. Rev. 69, 326–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grkovic S.; Brown M. H.; Skurray R. A. (2002) Regulation of bacterial drug export systems. Microbiol. Mol. Biol. Rev. 66, 671–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouch D. A.; Cram D. S.; DiBerardino D.; Littlejohn T. G.; Skurray R. A. (1990) Efflux-mediated antiseptic resistance gene qacA from Staphylococcus aureus: Common ancestry with tetracycline- and sugar-transport proteins. Mol. Microbiol. 4, 2051–2062. [DOI] [PubMed] [Google Scholar]
- Grkovic S.; Brown M. H.; Schumacher M. A.; Brennan R. G.; Skurray R. A. (2001) The staphylococcal QacR multidrug regulator binds a correctly spaced operator as a pair of dimers. J. Bacteriol. 183, 7102–7109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher M. A.; Miller M. C.; Grkovic S.; Brown M. H.; Skurray R. A.; Brennan R. G. (2002) Structural basis for cooperative DNA binding by two dimers of the multidrug binding protein QacR. EMBO J. 21, 1210–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grkovic S.; Brown M. H.; Roberts N. J.; Paulsen I. T.; Skurray R. A. (1998) QacR is a repressor protein that regulates expression of the Staphylococcus aureus multidrug efflux pump QacA. J. Biol. Chem. 273, 18665–18673. [DOI] [PubMed] [Google Scholar]
- Grkovic S.; Hardie K. M.; Brown M. H.; Skurray R. A. (2003) Interactions of the QacR multidrug-binding protein with structurally diverse ligands: Implications for the evolution of the binding pocket. Biochemistry 42, 15226–15236. [DOI] [PubMed] [Google Scholar]
- Schumacher M. A.; Miller M. C.; Grkovic S.; Brown M. H.; Skurray R. A.; Brennan R. G. (2001) Structural mechanisms of QacR induction and multidrug recognition. Science 294, 2158–2163. [DOI] [PubMed] [Google Scholar]
- Yu E. W.; McDermott G.; Zgurskaya H. I.; Nikaido H.; Koshland D. E. Jr. (2003) Structural basis of multiple drug-binding capacity of the AcrB multidrug efflux pump. Science 300, 976–980. [DOI] [PubMed] [Google Scholar]
- Murakami S.; Nakashima R.; Yamashita E.; Matsumoto T.; Yamaguchi A. (2006) Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature 443, 173–179. [DOI] [PubMed] [Google Scholar]
- Vázquez-Laslop N.; Markham P. N.; Neyfakh A. A. (1999) Mechanism of ligand recognition by BmrR, the multidrug-responding transcriptional regulator: Mutational analysis of the ligand-binding site. Biochemistry 38, 16925–16931. [DOI] [PubMed] [Google Scholar]
- Edgar R.; Bibi E. (1999) A single membrane-embedded negative charge is critical for recognizing positively charged drugs by the Escherichia coli multidrug resistance protein MdfA. EMBO J. 18, 822–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muth T. R.; Schuldiner S. (2000) A membrane-embedded glutamate is required for ligand binding to the multidrug transporter EmrE. EMBO J. 19, 234–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen I. T.; Brown M. H.; Littlejohn T. G.; Mitchell B. A.; Skurray R. A. (1996) Multidrug resistance proteins QacA and QacB from Staphylococcus aureus: Membrane topology and identification of residues involved in substrate specificity. Proc. Natl. Acad. Sci. U.S.A. 93, 3630–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell B. A.; Paulsen I. T.; Brown M. H.; Skurray R. A. (1999) Bioenergetics of the staphylococcal multidrug export protein QacA: Identification of distinct binding sites for monovalent and divalent cations. J. Biol. Chem. 274, 3541–3548. [DOI] [PubMed] [Google Scholar]
- Zhang D. W.; Cole S. P.; Deeley R. G. (2001) Identification of a nonconserved amino acid residue in multidrug resistance protein 1 important for determining substrate specificity: Evidence for functional interaction between transmembrane helices 14 and 17. J. Biol. Chem. 276, 34966–34974. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E. F., and Maniatis T. (1989) Molecular Cloning: A Laboratory Manual, 2nd ed., Cold Spring Harbor Laboratory Press, Plainview, NY. [Google Scholar]
- Brooks B. E.; Piro K. M.; Brennan R. G. (2007) Multidrug-binding transcription factor QacR binds the bivalent aromatic diamidines DB75 and DB359 in multiple positions. J. Am. Chem. Soc. 129, 8389–8395. [DOI] [PubMed] [Google Scholar]
- Rayment I. (1997) Reductive alkylation of lysine residues to alter crystallization properties of proteins. Methods Enzymol. 276, 171–179. [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994) The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D50, 760–763. [DOI] [PubMed] [Google Scholar]
- Leslie A. G. W. (1992) Recent changes to the MOSFLM package for processing film and image plate data. Newsletter of Protein Crystallography 26, . [Google Scholar]
- Brunger A. T.; Adams P. D.; Clore G. M.; DeLano W. L.; Gros P.; Grosse-Kunstleve R. W.; Jiang J. S.; Kuszewski J.; Nilges M.; Pannu N. S.; Read R. J.; Rice L. M.; Simonson T.; Warren G. L. (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D54, 905–921. [DOI] [PubMed] [Google Scholar]
- Jones T. A.; Zou J. Y.; Cowan S. W.; Kjeldgaard M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 47, Part 2110–119. [DOI] [PubMed] [Google Scholar]
- Laskowski R. A.; MacArthur M. W.; Moss D. S.; Thornton J. M. (1993) PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291. [Google Scholar]
- Delano W. L. (2006) The PyMOL Molecular Graphics System, DeLano Scientific, Palo Alto, CA.
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. (2004) UCSF Chimera: A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Murray D. S.; Schumacher M. A.; Brennan R. G. (2004) Crystal structures of QacR-diamidine complexes reveal additional multidrug-binding modes and a novel mechanism of drug charge neutralization. J. Biol. Chem. 279, 14365–14371. [DOI] [PubMed] [Google Scholar]
- Schumacher M. A.; Miller M. C.; Brennan R. G. (2004) Structural mechanism of the simultaneous binding of two drugs to a multidrug-binding protein. EMBO J. 23, 2923–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alguel Y.; Meng C.; Teran W.; Krell T.; Ramos J. L.; Gallegos M. T.; Zhang X. (2007) Crystal structures of multidrug binding protein TtgR in complex with antibiotics and plant antimicrobials. J. Mol. Biol. 369, 829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Veen H. W.; Higgins C. F.; Konings W. N. (2001) Molecular basis of multidrug transport by ATP-binding cassette transporters: A proposed two-cylinder engine model. J. Mol. Microbiol. Biotechnol. 3, 185–192. [PubMed] [Google Scholar]
- Poelarends G. J.; Konings W. N. (2002) The transmembrane domains of the ABC multidrug transporter LmrA form a cytoplasmic exposed, aqueous chamber within the membrane. J. Biol. Chem. 277, 42891–42898. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Clarke D. M. (1999) The transmembrane domains of the human multidrug resistance P-glycoprotein are sufficient to mediate drug binding and trafficking to the cell surface. J. Biol. Chem. 274, 24759–24765. [DOI] [PubMed] [Google Scholar]
- Ueda K.; Taguchi Y.; Morishima M. (1997) How does P-glycoprotein recognize its substrates?. Semin. Cancer Biol. 8, 151–159. [DOI] [PubMed] [Google Scholar]
- Kwan T.; Loughrey H.; Brault M.; Gruenheid S.; Urbatsch I. L.; Senior A. E.; Gros P. (2000) Functional analysis of a tryptophan-less P-glycoprotein: A tool for tryptophan insertion and fluorescence spectroscopy. Mol. Pharmacol. 58, 37–47. [DOI] [PubMed] [Google Scholar]
- Davis S. J.; Davies E. A.; Tucknott M. G.; Jones E. Y.; van der Merwe P. A. (1998) The role of charged residues mediating low affinity protein-protein recognition at the cell surface by CD2. Proc. Natl. Acad. Sci. U.S.A. 95, 5490–5494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewinson O.; Bibi E. (2001) Evidence for simultaneous binding of dissimilar substrates by the Escherichia coli multidrug transporter MdfA. Biochemistry 40, 12612–12618. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.