

Abstract
Novel quinolinonyl diketo acids were designed to obtain integrase (IN) inhibitors selectively active against the strand transfer (ST) step of the HIV integration process. Those new compounds are characterized by a single aryl diketo acid (DKA) chain in comparison to 4, a bifunctional diketo acid reported by our group as an anti-IN agent highly potent against both the 3′-processing and ST steps. Compound 6d was the most potent derivative in IN enzyme assays, while 6i showed the highest potency against HIV-1 in acutely infected cells. The selective inhibition of ST suggested the newly designed monofunctional DKAs bind the IN−DNA acceptor site without affecting the DNA donor site.
Introduction
Three different classes of chemotherapeutic agents are generally combined to block the replication of human immunodeficiency virus type 1 (HIV-1a) responsible for AIDS and to prevent the occurrence of resistance: reverse transcriptase inhibitors (RTI), protease inhibitors (PRI), and fusion inhibitors. This widespread triple combination therapy is referred to as HAART (highly active antiretroviral therapy).(1) HAART effectively inhibits HIV replication to such an extent that the virus becomes undetectable in the blood. However, it fails to eradicate viruses that are integrated in the host genome or that persist in cellular and anatomical “reservoirs”. In addition, prolonged drug exposure led to HIV drug resistance, thus reducing patients’ therapeutically available options.(2) The above considerations and the toxicity of a number of antiretroviral agents have fueled the discovery of drugs against additional targets. Among them, HIV integrase (IN), which has no cellular counterpart, has been intensely studied over the past 15 years.3–5 IN has recently been fully validated as a therapeutic target with the first FDA approved IN inhibitor raltegravir.(6)
IN catalyzes the insertion of the viral cDNA (generated by reverse transcription of the viral RNA) into the host cell genome. Integration occurs via a sequence of reactions, which start with the IN-mediated cleavage of terminal dinucleotide from the 3′-end of the viral cDNA (termed “3′-processing”, 3′-P) shortly after reverse transicription in the cytoplasm. Following transfer of the resulting processed viral cDNA into the nucleus, IN catalyzes the insertion of both ends into target cellular host DNA. That second reaction is referred as “strand transfer” (ST).(4) In the past 15 years, a range of natural and synthetic compounds have been identified as inhibitors of recombinant IN enzyme in biochemical assays. Interestingly, polyhydroxylated aromatics and diketo compounds were among the first inhibitors identified.3,7–9 However, those early polyhydroxylated derivatives were later demonstrated to inhibit viral entry or to be too toxic to be pursued as therapeutic IN inhibitors.(10) More recently, the Merck and Shionogi companies discovered aryl diketo acid (DKA) derivatives as selective anti-HIV agents that block the viral replication cycle via IN inhibition in vivo. Those compounds are typified by 1-(5-chloroindol-3-yl)-3-hydroxy-3-(2H-tetrazol-5-yl)propenone (5CITEP, 1) synthesized by Shionogi & Co. Ltd.(11) and by the pyrrole derivative L-731,988 (2) developed by Merck Research Laboratories(12) (Figure 1). They are characterized by their ability to preferentially inhibit ST versus 3′-P. Chemically, DKA is characterized by a diketo acid moiety (α and γ ketone and a carboxylic acid), which is believed to be essential for the inhibitory activity, although the carboxylic group can be effectively replaced by a bioisoster azole ring (triazole, tetrazole) (i.e., 1)(11) and the 1,3-diketo acid moiety can be mimicked by a 8-hydroxy-[1,6]naphthyridine ring(13) (i.e., compound 3, Figure 1).
Figure 1.

Structures of HIV-1 IN inhibitors belonging to the mono- and bifunctional DKA class and related 8-hydroxy[1,6]naphthyridine bioisoster.
Recent studies on quinolinonyl diketo acid derivatives led us to discover the bifunctional compound 4 as a potent IN inhibitor for both 3′-P and ST.(14) Moreover, 4 inhibits HIV-1 replication in acutely infected cells.(14) Docking studies on the binding mode of 4 to the IN catalytic site also suggested a peculiar interaction of the drug involving both the acceptor and the donor DNA binding sites of the enzyme. This hypothesis was confirmed by our recent cross-linking experiment studies that pointed out a specific interaction of 4 with K156 and K159 amino acid residues of the IN catalytic core domain.(15) This binding mode could account for the high potency of 4 against both 3′-P and ST.
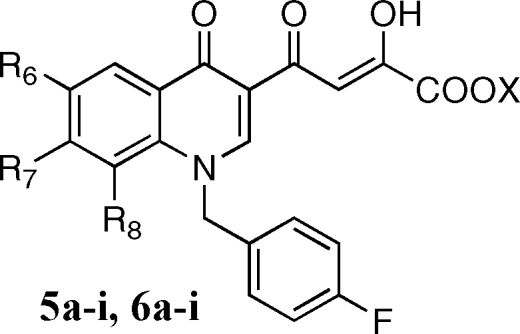
The aim of the present project was the design of new quinolinone derivatives endowed with a selective activity against ST. This project could provide new information regarding the interactions of quinolinonyl diketo acids with the IN active site and consequently increase our knowledge about the catalytic mechanisms of IN, which in the absence of structural information on IN−DNA and drug molecular structures are still far from being totally elucidated. In the present manuscript, we describe novel quinolinonyl diketo acid derivatives 5a−i, 6a−i, 7a,b, and 8a,b designed by replacement of the 6-diketo acid chain of 4, responsible for binding to the donor DNA binding site, with smaller substituents in the 6-, 7-, or 8-position of the quinolinone ring (Figure 2). These substituents should exhibit reduced binding to the donor viral cDNA site due to their small size/length or to their limited ability to form hydrogen bonds. Thus, these structural modifications should provide novel quinolinonyl diketo acid derivatives with increased ST selectivity.
Figure 2.

Structures of newly designed monofunctional quinolinonyl diketo acid derivatives as HIV-1 IN inhibitors.
Results and Discussion
Chemistry
Synthesis of derivatives 5a−i, 6a−i, 7a,b, and 8a,b is outlined in Schemes 1–3. 3-Acetyl-4(1H)-quinolinones 9a−h were prepared by reaction of the proper aniline with ethyl orthoformate and ethyl acetoacetate, which were thermally condensed in the presence of an inert heating medium (Dowtherm A) under argon atmosphere, according to the Yoshizawa procedure.(16) Then, 9a−h were alkylated with 4-fluorobenzylbromide in alkaline medium (K2CO3) to give the N-1 substituted quinolones 10a−h. These compounds were condensed with diethyl oxalate in the presence of sodium ethoxide to provide ethyl esters 5a−h, which were in turn hydrolyzed with 6 N NaOH to afford the corresponding acids 6a−h (Scheme 1).
Scheme 1.

Reagents and conditions: (a) ethyl orthoformate, ethyl acetoacetate, Dowtherm A, 95−254 °C, 8 h; (b) 4-fluorobenzyl bromide, K2CO3, DMF, 100 °C, 1 h; (c) diethyl oxalate, C2H5ONa, THF, room temp, 2 h; (d) 1 N NaOH, THF/CH3OH, room temp, 40 min.
Scheme 3.

Reagents and conditions: (a) pyrrolidine, Et3N, DMF, microwave, 100 W, 153 °C, 10 min; (b) diethyl oxalate, C2H5ONa, THF, room temp, 2 h; (c) 1 N NaOH, THF/CH3OH, room temp, 40 min.
Synthesis of 1-unsubstituted derivatives 7a,b and 8a,b was achieved in a similar fashion by coupling of 4-quinolinones 9e,f with diethyl oxalate in the presence of the sodium ethoxide, followed by alkaline hydrolysis of the resulting ethyl esters 7a,b to provide acids 8a,b (Scheme 2).
Scheme 2.

Reagents and conditions: (a) diethyl oxalate, C2H5ONa, THF, room temp, 2 h; (b) 1 N NaOH, THF/CH3OH, room temp, 40 min.
Derivatives 5i and 6i were synthesized as described in Scheme 3. Pyrrolidinyl derivative 10i was obtained in few minutes with good yields by substitution of fluorine atom of 10c with pyrrolidine in the presence of NEt3, under microwave irradiation. 10i was then condensed with ethyl oxalate, and the ester 5i that formed was hydrolyzed to afford the required acid 6i. Chemical, physical, and analytical data of intermediates 9a−h and 10a−i are reported in Table 1, while spectroscopic data are shown in Supporting Information. Data for final products 5a−i, 6a−i, 7a,b, and 8a,b are listed in .
Table 1. Chemical, Physical, and Analytical Data of Derivatives 9a−h and 10a−i.
| compd | R6 | R7 | R8 | mp (°C) | recryst solventa | yield (%) | analysis |
|---|---|---|---|---|---|---|---|
| 9ab | H | H | H | 242−244 | a | 56 | C, H, N (C11H9NO2) |
| 9b | F | H | H | >300 | b | 29 | C, H, N, F (C11H8FNO2) |
| 9c | H | F | H | 270−271 | c | 41 | C, H, N, F (C11H8FNO2) |
| 9d | H | H | F | 268−270 | b | 40 | C, H, N, F (C11H8FNO2) |
| 9eb | Cl | H | H | >300 | b | 29 | C, H, N, Cl (C11H8ClNO2) |
| 9fb | H | Cl | H | >300 | b | 96 | C, H, N, Cl (C11H8ClNO2) |
| 9gb | H | H | Cl | >300 | b | 55 | C, H, N, Cl (C11H8ClNO2) |
| 9h | Cl | Cl | H | 114−115 | d | 19 | C, H, N, Cl (C11H7Cl2NO2) |
| 10a | H | H | H | 213−214 | d | 65 | C, H, N, F (C18H14FNO2) |
| 10b | F | H | H | 234−235 | e | 94 | C, H, N, F (C18H13F2NO2) |
| 10c | H | F | H | 186−187 | e | 100 | C, H, N, F (C18H13F2NO2) |
| 10d | H | H | F | 193−194 | f | 78 | C, H, N, F (C18H13F2NO2) |
| 10e | Cl | H | H | 214−216 | e | 92 | C, H, N, Cl, F (C18H13ClFNO2) |
| 10f | H | Cl | H | 185−187 | e | 73 | C, H, N, Cl, F (C18H13ClFNO2) |
| 10g | H | H | Cl | 215−217 | g | 92 | C, H, N, Cl, F (C18H13ClFNO2) |
| 10h | Cl | Cl | H | 235−237 | d | 49 | C, H, N, Cl, F (C18H12Cl2FNO2) |
| 10i | H | 1-pyrrolidinyl | H | 175−177 | e | 56 | C, H, N, F (C22H21FN2O2) |
Recrystallization solvents: (a) aceton, (b) ethanol, (c) isopropanol, (d) toluene, (e) toluene/cyclohexane, (f) benzene/cyclohexane, (g) benzene.
Reference (17).
Evaluation of Biological Activities
In Vitro Assays
Derivatives 5a−i, 6a−i, 7a,b, and 8a,b were tested in vitro for ST inhibition in the presence of magnesium (Mg2+) using a recently described high throughput electrochemiluminescent (HTECL) assay.(14) IC50 values were generated from duplicate experiments (Table 2). Compounds 5a−i, 6a−i, 7a,b, and 8a,b were also tested for ST and 3′-P using gel-based assays carried out in the presence of Mg2+ (Figure 3A). IC50 values were calculated using dose response curves (Figure 3B) and are summarized in Table 2. The newly synthesized DKAs exhibited high potency against IN with high selectivity against ST, thus confirming the hypothesis that the removal of the diketo acid branch in the 6-position of the quinolinone ring of 4 can lead to ST-selective inhibitors (6a−i showed 3′-P/ST ratios ranging from 45 to 667). The acid derivatives 6a−i were more potent than the corresponding esters 5a−i (Figure 3A and Figure 3B, compare compounds 6h and 5h), and the 1-p-F-benzyl substituted quinolinones (5e,f and 6e,f) were markedly more active than their unsubstituted counterparts (7a,b and 8a,b). This trend is consistent with the results obtained previously on the bifunctional DKA (BDKA) series.(14) In particular, the relevant role played by the p-F-benzyl moiety at the 1-position of the quinolinone ring was previously noted for the DKA derivatives.12,18 The newly synthesized acid derivatives 6a−i were potent inhibitors, showing IC50 values in the range of 18−40 nM for the ST step (Table 2, HTECL assay). The most active compound of this series is 6d with a ST IC50 value of 18 nM, which is comparable to reference drugs 2 and 3. Removal of the diketo acid chain from the 6-position of our BDKA lead molecule (compound 4) (14) led to compound 6a that retains the potent anti-IN activity of the parent derivative with an IC50 value of 40 nM for ST (Table 2, HTECL assay). However, 6a is a more ST selective inhibitor than 4.(14) Introduction of a halogen (F or Cl) in the 6-, 7-, or 8-position of 6a led to compounds 6b−g, which show similar anti-IN activities (IC50 = 18−30 nM, Table 2, HTECL assay) as their unsubstituted counterpart 6a. Interestingly, the anti-IN activities appear to be influenced by the position of the halogen. Indeed, inhibitory activities decrease in the following order: 8-F(Cl) ∼ 7-F(Cl) > 6-F(Cl) and 7-Cl > 8-Cl > 6-Cl. The introduction of a second chlorine atom in the structure of the potent inhibitor 6f led to 6h, which is approximately 1.5 times less potent than the parent counterpart. Similarly, replacement of the chlorine atom of 6f with a pyrrolidine ring led to 6i, which is approximately 1.5 times less potent than the parent compound. In conclusion, the activities of compounds with a substituent in the 7-position decrease in the following order: Cl > F > 1-pyrrolidinyl > H (Table 2, HTECL assay).
Table 2. Cytotoxicity, Antiviral, and Anti-Integrase Activities of Derivatives 5a−i, 6a−i, 7a,b, and 8a,b.

| anti-IN activity, IC50a |
antiviral activity |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| compd | R6 | R7 | R8 | X | Mg2+ b | Mg2+ c | Mg2+ c | CC50d | EC50e | SIf |
| 5a | H | H | H | C2H5 | 0.67 | 0.79, 0.44 | 135 | 99 | 34.8 | 2.8 |
| 5b | F | H | H | C2H5 | 0.58 | 0.23, 0.44 | >333 | >200 | >50 | |
| 5c | H | F | H | C2H5 | 1.3 | 1.2, 2.9 | >333 | >200 | >50 | |
| 5d | H | H | F | C2H5 | 26 | 5.2 | >333 | >200 | >50 | |
| 5e | Cl | H | H | C2H5 | 21 | 3.0 | >333 | ntg | ntg | |
| 5f | H | Cl | H | C2H5 | 14 | 3.6 | >333 | ntg | ntg | |
| 5g | H | H | Cl | C2H5 | 32 | 5.2 | >333 | >200 | >50 | |
| 5h | Cl | Cl | H | C2H5 | 3.9 | 1.8 | 135 | 72 | >50 | |
| 5i | H | Pyh | H | C2H5 | 2.3 | 2.7 | 110 | >200 | 4.1 | 48.8 |
| 6a | H | H | H | H | 0.040 | 0.06, 0.16 | 14 | >200 | 1.17 | >171 |
| 6b | F | H | H | H | 0.030 | 0.14 | 2.3 | >200 | >50 | |
| 6c | H | F | H | H | 0.020 | 0.050 | 4.4 | >200 | >50 | |
| 6d | H | H | F | H | 0.018 | 0.16 | 16 | >200 | >50 | |
| 6e | Cl | H | H | H | 0.028 | 0.40 | 6.4 | >200 | 46.1 | 4.3 |
| 6f | H | Cl | H | H | 0.019 | 0.11 | 4.0 | >200 | >50 | |
| 6g | H | H | Cl | H | 0.023 | 0.51 | 44 | >200 | >50 | |
| 6h | Cl | Cl | H | H | 0.033 | 0.075 | 11, 34 | 156 | 3.2 | 62.5 |
| 6i | H | Pyh | H | H | 0.028 | 0.18 | 14, 9.0 | >200 | 0.17 | >1176 |
| 7a | Cl | H | C2H5 | 3.3 | 2.1 | 120 | ntg | ntg | ||
| 7b | H | Cl | C2H5 | >111 | 224 | >333 | ntg | ntg | ||
| 8a | Cl | H | H | >111 | 56 | 166 | ntg | ntg | ||
| 8b | H | Cl | H | 37 | 17 | >333 | ntg | ntg | ||
| 1i | 2.1 | |||||||||
| 2i | 0.05 | 1.0 | ||||||||
| 3i | 0.01 | 0.39j | ||||||||
| 4 | 0.016 | 0.017 | 0.44 | >200 | 4.29 | >47 | ||||
Inhibitory concentration 50% (μM) determined from dose response curves.
Experiments performed in duplicate using the high throughput electrochemiluminescent (HTECL) assay (ST assay in the presence of Mg2+).
Experiments performed in a gel-based assay in the presence of Mg2+ (see Figure 3).
Cytotoxic concentration 50% (μM).
Effective concentration 50% (μM).
Selectivity index = CC50/EC50.
nt: not tested.
Py = 1-pyrrolidinyl.
This data is referred to EC95.
Figure 3.

Comparison of HIV-1 integrase inhibition by compound 6h and 5h in a gel-based assay and in the presence of Mg2+. (A) Phosphorimager image showing a representative experiment. 21, 19, and STP correspond to the DNA substrate, the 3′-P product, and the ST products, respectively. (B) HIV-1 integrase inhibition curves for 3′-P and ST (derived from densitometric analysis of the gels presented in part A).
Compounds 5a−i and 6a−i also inhibited integrase with similar potency in the presence of Mg2+ or Mn2+ (data not shown). The activity in the presence of Mg2+ in addition to the selectivity for ST represents a trademark of the most potent DKAs active against HIV replication in cells.(19)
Cell-Based Assays
Cytotoxicity and antiviral activities of compounds 5a−i, 6a-i, 7a,b, and 8a,b are presented in Table 2. Among the tested compounds, derivatives 5a,i, and 6a,e,h,i show EC50 < 50 μM when tested against HIV-1-infected H9/HTLVIIIB cells. In particular, 5i and 6a,h,i are active in the micromolar range, with SI ranging from 48.8 to >1176. The most potent derivative of this series is 6i (EC50 = 0.17 μM and SI > 1176). Compound 6i is in fact more potent than reference derivatives 2−4. In general, all the active compounds were characterized by low cytotoxicity against human histiocytic lymphoma (U937) cell line, showing CC50 values in the range of 99 to >200 μM.
Conclusions
We designed, synthesized, and tested a series of novel quinolinonyl diketo acid derivatives in both enzyme- and cell-based assays as anti-HIV-1 agents to selectively target the ST step of integration. We were able to generate such new compounds, which are potent ST-selective IN inhibitors and active against HIV-1 replication in acutely infected cells. These compounds could be useful tools to study the mechanism of action of HIV-1 IN.
Experimental Section
Chemistry. General
Melting points were determined on a Buchi 530 melting point apparatus and are uncorrected. Infrared (IR) spectra were recorded on a Perkin-Elmer Spectrum-one spectrophotometer. 1H NMR spectra were recorded at 400 MHz on a Bruker AC 400 Ultrashield spectrometer. Merck silica gel 60 F254 plates were used for analytical TLC. Developed plates were visualized by UV light. Column chromatographies were performed on silica gel Merck 70−230 mesh. Solvents were reagent grade and, when necessary, were purified and dried by standard methods. Concentration of solutions after reactions and extractions involved the use of a rotatory evaporator (Büchi) operating at a reduced pressure of approximately 20 Torr. Organic solutions were dried over anhydrous sodium sulfate. Analytical results agreed to within ±0.40% of the theoretical values.
Microwave Irradiation Experiments
Microwave reactions were conducted using a CEM Discover synthesis unit (CEM Corp., Matthews, NC). The machine consists of a continuous focused microwave-power delivery system with operator-selectable power output from 0 to 300 W. The temperature of the contents of the vessel was monitored using a calibrated infrared temperature control mounted under the reaction vessel. All experiments were performed using a stirring option whereby the contents of the vessel are stirred by means of a rotating magnetic plate located below the floor of the microwave cavity and a Teflon-coated magnetic stir bar in the vessel.
General Procedure for the Synthesis of 3-Acetyl-4(1H)-quinolinones 9a−h
Ethyl orthoformate (4.0 g, 27 mmol), ethyl acetoacetate (3.5 g, 27 mmol), the appropriate substituted aniline (27 mmol), and Dowtherm A (5.6 mL) were charged in a three-necked flask equipped with a water separator. This mixture was stirred under argon atmosphere, while the temperature was increased to 95 °C in 1 h, then gradually up to 162 °C in a further hour. Then the mixture was stirred at this temperature for 6 h. After this time, the resulting solution was added in portions during 3 h into 42 mL of Dowtherm A stirred in a three-necked flask equipped with thermometer and water separator and heated at 253−254 °C. After addition the mixture was heated at the same temperature for 2 h. Then the mixture was cooled at 90 °C, treated with isopropanol (10 mL), cooled at 30 °C, filtered, and washed with isopropanol and light petroleum ether in turn to give pure derivatives 9a−h. Chemical, physical, and analytical data of derivatives 9a−h are reported in Table 1. For spectroscopic data see Supporting Information.
General Procedure for the Synthesis of 3-Acetyl-1-(4-fluorophenyl)methyl-4(1H)-quinolinones 10a−h
A mixture of the proper 9a−h derivative (1.1 mmol), 4-fluorophenylmethyl bromide (610 mg, 3.3 mmol), and anhydrous K2CO3 (210 mg, 1.5 mmol) in dry DMF (10 mL) was stirred at 100 °C for 1 h. After the mixture was cooled, water was added (40 mL) and the precipitate that formed was filtered, washed with water and light petroleum ether in turn, and then dried under IR lamp to provide pure derivatives 10a−h. Chemical, physical, and analytical data of derivatives 10a−h are reported in Table 1. For spectroscopic data see Supporting Information.
3-Acetyl-1-(4-fluorophenyl)methyl-7-(pyrrolidin-1-yl)-4(1H)-quinolinone (10i)
A mixture of 10c (2.7 g, 8.5 mmol), pyrrolidine (1.8 g, 25.6 mmol), and NEt3 (0.7 g, 7.7 mmol) in dry DMF (40 mL) was irradiated with microwave at 153 °C for 10 min (applied potency 100 W), in an open vessel equipped with a condenser. After cooling, the reaction mixture was diluted with water (100 mL) and treated with 1 N HCl until pH 7. The solid that formed was collected by filtration. Purification of crude product was performed by chromatography on silica gel column (chloroform/ethyl acetate 1:1 as eluent) to give 1.7 g (56%) of pure 10i. Chemical, physical, and analytical data of derivatives 10i are reported in Table 1. For spectroscopic data see Supporting Information.
General Procedure for the Synthesis of Diketo Esters 5a−i and 7a,b
Sodium ethoxide (390 mg, 5.5 mmol) was added into a well stirred mixture of the appropriate acetyl derivative 9a−h or 10a−i (2.7 mmol) and diethyl oxalate (790 mg, 5.4 mmol) in anhydrous THF (2.7 mL) under nitrogen atmosphere. The mixture was stirred at room temperature for 2 h, then was poured into n-hexane (50 mL). The collected precipitate was vigorously stirred for 30 min in 1 N HCl (50 mL). The yellow solid that formed was filtered, washed with water, and dried under IR lamp to afford the pure diketo esters 5a−i and 7a,b. Yield (%), melting point (°C), recrystallization solvent, IR, 1H NMR, and analytical data are reported for each of the following compounds.
4-[1-(4-Fluorophenyl)methyl-4(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid Ethyl Ester (5a)
82%; 175−177 °C; ethanol; IR ν 3400 (OH), 1721 (C=O ester), 1661, 1635, and 1607 (C=O ketone) cm−1. 1H NMR (CDCl3) δ 1.44 (t, 3H, CH3), 4.42 (q, 2H, CH2CH3), 5.48 (s, 2H, CH2), 7.06−7.26 (m, 4H, benzene H), 7.37−7.65 (m, 3H, quinolinone C6−H, C7−H, and C8−H), 8.16 (s, 1H, butenoate C3−H), 8.56 (m, 1H, quinolinone C5−H), 8.77 (s, 1H, quinolinone C2−H), 15.50 (bs, 1H, OH). Anal. (C22H18FNO5) C, H, N, F.
4-[6-Fluoro-1-(4-fluorophenyl)methyl-4(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid Ethyl Ester (5b)
92%; 152−153 °C; toluene/cyclohexane; IR ν 1740 (C=O ester), 1650 and 1624 (C=O ketone) cm−1; 1H NMR (DMSO-d6) δ 1.29 (t, 3H, CH3), 4.30 (q, 2H, CH2CH3), 5.78 (s, 2H, CH2), 7.16−7.20 (m, 2H, benzene H), 7.29−7.37 (m, 2H, benzene H), 7.66 (m, 1H, quinolinone C7−H), 7.81 (m, 1H, quinolinone C8−H), 7.97 (m, 1H, quinolinone C5−H), 8.00 (s, 1H, butenoate C3−H), 9.13 (s, 1H, quinolinone C2−H), 15.33 (bs, 1H, OH). Anal. (C22H17F2NO5) C, H, N, F.
4-[7-Fluoro-1-(4-fluorophenyl)methyl-4(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid Ethyl Ester (5c)
84%; 151−152 °C; benzene/cyclohexane; IR ν 1726 (C=O ester), 1644 (C=O ketone) cm−1. 1H NMR (DMSO-d6) δ 1.30 (t, 3H, CH3), 4.30 (q, 2H, CH2CH3), 5.72 (s, 2H, CH2), 7.17−7.21 (m, 2H, benzene H), 7.31−7.40 (m, 3H, benzene H and quinolinone C6−H), 7.62 (m, 1H, quinolinone C8−H), 7.97 (s, 1H, butenoate C3−H), 8.36 (m, 1H, quinolinone C5−H), 9.10 (s, 1H, quinolinone C2−H), 14.00 (bs, 1H, OH). Anal. (C22H17F2NO5) C, H, N, F.
4-[8-Fluoro-1-(4-fluorophenyl)methyl-4(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid Ethyl Ester (5d)
83%; 179−180 °C; benzene/cyclohexane; IR ν 3300 (OH), 1744 (C=O ester), 1648 and 1604 (C=O ketone) cm−1. 1H NMR (DMSO-d6) δ 1.27 (t, 3H, CH3), 4.30 (q, 2H, CH2CH3), 5.78 (s, 2H, CH2), 7.14−7.24 (m, 4H, benzene H), 7.48−7.53 (m, 1H, quinolinone C6−H), 7.59−7.69 (m, 1H, quinolinone C7−H), 7.97 (s, 1H, butenoate C3−H), 8.17−8.19 (m, 1H, quinolinone C5−H), 9.01 (s, 1H, quinolinone C2−H), 15.50 (bs, 1H, OH). Anal. (C22H17F2NO5) C, H, N, F.
4-[6-Chloro-1-(4-fluorophenyl)methyl-4(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid Ethyl Ester (5e)
34%; 131−133 °C; washed with cyclohexane; IR ν 3400 (OH), 1722 (C=O ester), 1700 and 1628 (C=O ketone) cm−1. 1H NMR (DMSO-d6) δ 1.33 (t, 3H, CH3), 4.33 (q, 2H, CH2CH3), 5.71 (s, 2H, CH2), 7.14−7.34 (m, 4H, benzene H), 7.71−7.77 (m, 2H, quinolinone C7−H and C8−H), 7.91 (s, 1H, butenoate C3−H), 8.25 (m, 1H, quinolinone C5−H), 9.09 (s, 1H, quinolinone C2−H), 12.70 (bs, 1H, OH). Anal. (C22H17ClFNO5) C, H, N, Cl, F.
4-[7-Chloro-1-(4-fluorophenyl)methyl-4(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid Ethyl Ester (5f)
76%; 173−175 °C; washed with isopropanol; IR ν 3300 (OH), 1727 (C=O ester), 1647 (C=O ketone) cm−1. 1H NMR (DMSO-d6) δ 1.30 (t, 3H, CH3), 4.32 (q, 2H, CH2CH3), 5.76 (s, 2H, CH2), 7.16−7.25 (m, 2H, benzene H), 7.36−7.38 (m, 2H, benzene H), 7.54 (m, 1H, quinolinone C6−H), 7.88 (m, 1H, quinolinone C8−H), 7.96 (s, 1H, butenoate C3−H), 8.29 (m, 1H, quinolinone C5−H), 9.08 (s, 1H, quinolinone C2−H), 12.70 (bs, 1H, OH). Anal. (C22H17ClFNO5) C, H, N, Cl, F.
4-[8-Chloro-1-(4-fluorophenyl)methyl-4(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid Ethyl Ester (5g)
98%; 164−165 °C; toluene; IR ν 3400 (OH), 1749 (C=O ester), 1644 (C=O ketone) cm−1. 1H NMR (DMSO-d6) δ 1.29 (t, 3H, CH3), 4.30 (q, 2H, CH2CH3), 6.09 (s, 2H, CH2), 7.14−7.16 (m, 4H, benzene H), 7.15 (m, 1H, quinolinone C6−H), 7.82 (m, 1H, quinolinone C7−H), 7.91 (s, 1H, butenoate C3−H), 8.34 (m, 1H, quinolinone C5−H), 8.81 (s, 1H, quinolinone C2−H), 15.25 (bs, 1H, OH). Anal. (C22H17ClFNO5) C, H, N, Cl, F.
4-[6,7-Dichloro-1-(4-fluorophenyl)methyl-4(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid Ethyl Ester (5h)
98%; 144−145 °C; toluene/cyclohexane; IR ν 1727 (C=O ester), 1638 (C=O ketone) cm−1. 1H NMR (DMSO-d6) δ 1.35 (t, 3H, CH3), 4.38 (q, 2H, CH2CH3), 5.82 (s, 2H, CH2), 7.18−7.30 (m, 2H, benzene H), 7.43−7.46 (m, 2H, benzene H), 7.98 (s, 1H, butenoate C3−H), 8.14 (s, 1H, quinolinone C8−H), 8.41 (s, 1H, quinolinone C5−H), 9.14 (s, 1H, quinolinone C2−H), 16.00 (bs, 1H, OH). Anal. (C22H16Cl2FNO5) C, H, N, Cl, F.
4-[1-(4-Fluorophenyl)methyl-7-(pyrrolidin-1-yl)-4(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid Ethyl Ester (5i)
82%; 170 °C (dec); methanol; IR ν 3300 (OH), 1735 (C=O ester), 1621 (C=O ketone) cm−1. 1H NMR (DMSO-d6) δ 1.31 (t, 3H, CH3), 1.98−2.10 (m, 4H, pyrrolidine H), 3.32−3.36 (m, 1H, pyrrolidine H), 4.30 (q, 2H, CH2CH3), 5.64 (s, 2H, CH2), 6.37−6.39 (m, 1H, quinolinone C8−H), 6.77−6.79 (m, 1H, quinolinone C6−H), 7.29−7.32 (m, 2H, benzene H), 7.45−7.48 (m, 2H, benzene H), 8.12−8.16 (m, 2H, quinolinone C5−H and butenoate C3−H), 8.79 (s, 1H, quinolinone C2−H), 13.50 (bs, 1H, OH). Anal. (C26H25FN2O5) C, H, N, Cl, F.
4-(6-Chloro-4(1H)-quinolinon-3-yl)-2-hydroxy-4-oxo-2-butenoic Acid Ethyl Ester (7a)
57%; 209−211 °C; ethanol; IR ν 3300 (NH and OH), 1734 (C=O ester), 1684 (C=O ketone) cm−1. 1H NMR (DMSO-d6) δ 1.31 (t, 3H, CH3), 4.25 (q, 2H, CH2), 7.40−8.00 (m, 3H, quinolinone C7−H, C8−H and butenoate C3−H), 8.20 (m, 1H, quinolinone C5−H), 8.50 (s, 1H, quinolinone C2−H), 12.75 (bs, 2H, NH and OH). Anal. (C15H12ClNO5) C, H, N, Cl.
4-(7-Chloro-4(1H)-quinolinon-3-yl)-2-hydroxy-4-oxo-2-butenoic Acid Ethyl Ester (7b)
77%; 187−189 °C; washed with isopropanol; IR ν 3200, 3064 (NH and OH), 1726 (C=O ester), 1700 and 1624 (C=O ketone) cm−1. 1H NMR (DMSO-d6) δ 1.30 (t, 3H, CH3), 4.30 (q, 2H, CH2), 7.47 (m, 1H, quinolinone C6−H), 7.68 (s, 1H, quinolinone C8−H), 7.94 (s 1H, butenoate C3−H), 8.18 (m, 1H, quinolinone C5−H), 8.77 (s, 1H, quinolinone C2−H), 13.00 (bs, 1H, NH and OH). Anal. (C15H12ClNO5) C, H, N, Cl.
General Procedure for the Synthesis of Diketo Acids 6a−i and 8a,b
A mixture of 1 N NaOH (6.5 mL) and the appropriate ester 5a−i or 7a,b (1.3 mmol) in 1:1 THF/methanol (12 mL) was stirred at room temperature for 40 min and then poured onto crushed ice. The aqueous layer was separated and treated with 1 N HCl until pH 3 was reached, and the yellow solid that formed was collected by filtration, then washed with water, hot dry ethanol, and light petroleum ether to afford pure acids 6a−i and 8a,b. Yield (%), melting point (°C), recrystallization solvent, IR, 1H NMR, and analytical data are reported for each of the following compounds.
4-[1-(4-fluorophenyl)methyl-4(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid (6a)
50%; 207−209 °C; washed with anhydrous ethanol; IR ν 3400 (OH), 1732 (C=O acid), 1619 (C=O ketone) cm−1. 1H NMR (CDCl3) δ 5.79 (s, 2H, CH2), 7.22−7.38 (m, 4H, benzene H), 7.53−8.34 (m, 5H, quinolinone C6−H, C7−H, C8−H and butenoate C3−H), 9.16 (s, 1H, quinolinone C2−H), 15.50 (bs, 2H, OH). Anal. (C20H15FNO5) C, H, N, F.
4-[6-Fluoro-1-(4-fluorophenyl)methyl-4(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid (6b)
95%; 183−184 °C; washed with anhydrous ethanol; IR ν 3100 (OH), 1730 (C=O acid), 1613 (C=O ketone) cm−1. 1H NMR (DMSO-d6) δ 5.76 (s, 2H, CH2), 7.16−7.20 (m, 2H, benzene H), 7.33−7.36 (m, 2H, benzene H), 7.65 (m, 1H, quinolinone C7−H), 7.80 (m, 1H, quinolinone C8−H), 7.94−7.97 (m, 2H, quinolinone C5−H and butenoate C3−H), 9.10 (s, 1H, quinolinone C2−H), 14.67 (bs, 2H, OH). Anal. (C20H13F2NO5) C, H, N, F.
4-[7-Fluoro-1-(4-fluorophenyl)methyl-4(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid (6c)
95%; 162−163 °C; washed with anhydrous ethanol; IR ν 3300 (OH), 1719 (C=O acid), 1635 (C=O ketone) cm−1. 1H NMR (DMSO-d6) δ 5.70 (s, 2H, CH2), 7.14−7.21 (m, 2H, benzene H), 7.31−7.37 (m, 3H, benzene H and quinolinone C6−H), 7.60 (m, 1H, quinolinone C8−H), 7.93 (s, 1H, butenoate C3−H), 8.34 (m, 1H, quinolinone C5−H), 9.05 (s, 1H, quinolinone C2−H), 14.60 (bs, 2H, OH). Anal. (C20H13F2NO5) C, H, N, F.
4-[8-Fluoro-1-(4-fluorophenyl)methyl-4(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid (6d)
78%; 180−181 °C; washed with hot anhydrous ethanol; IR ν 3300 (OH), 1732 (C=O acid), 1634 and 1602 (C=O ketone) cm−1. 1H NMR (DMSO-d6) δ 5.86 (s, 2H, CH2), 7.12−7.23 (m, 4H, benzene H), 7.45−7.50 (m, 1H, quinolinone C6−H), 7.57−7.62 (m, 1H, quinolinone C7−H), 7.70 (s, 1H, butenoate C3−H), 8.16−8.18 (m, 1H, quinolinone C5−H), 8.96 (s, 1H, quinolinone C2−H), 14.30 (bs, 2H, OH). Anal. (C20H13F2NO5) C, H, N, F.
4-[6-Chloro-1-(4-fluorophenyl)methyl-4(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid (6e)
71%; 194−197 °C; DMF/water; IR ν 3385 (OH), 1710 (C=O acid), 1663 and 1622 (C=O ketone) cm−1. 1H NMR (DMSO-d6) δ 5.72 (s, 2H, CH2), 7.18−7.32 (m, 4H, benzene H), 7.75−7.85 (m, 2H, quinolinone C7−H and C8−H), 8.22 (m, 1H, C5−H quinolinone), 7.90 (s, 1H, butenoate C3−H), 9.08 (s, 1H, quinolinone C2−H), 13.00 (bs, 2H, OH). Anal. (C20H12ClFNO5) C, H, N, Cl, F.
4-[7-Chloro-1-(4-fluorophenyl)methyl-4(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid (6f)
64%; 188−190 °C; washed with isopropanol; IR ν 3400 (OH), 1734 (C=O acid), 1646 (C=O ketone) cm−1. 1H NMR (DMSO-d6) δ 5.75 (s, 2H, CH2), 7.21−7.36 (m, 4H, benzene H), 7.55 (m, 1H, quinolinone C6−H), 7.83−7.85 (m, 2H, quinolinone C8−H and butenoate C3−H), 8.30 (m, 1H, quinolinone C5−H), 9.07 (s, 1H, quinolinone C2−H), 13.00 (bs, 2H, OH). Anal. (C20H12ClFNO5) C, H, N, Cl, F.
4-[8-Chloro-1-(4-fluorophenyl)methyl-4(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid (6g)
82%; 175−177 °C; washed with anhydrous ethanol; IR ν 3300 (OH), 1742 (C=O acid), 1637 and 1603 (C=O ketone) cm−1. 1H NMR (DMSO-d6) δ 6.08 (s, 2H, CH2), 7.00−7.30 (m, 4H, benzene H), 7.44−7.46 (m, 1H, quinolinone C6−H), 7.81−7.83 (m, 2H, butenoate C3−H and quinolinone C7−H), 8.33−8.35 (m, 1H, quinolinone C5−H), 8.89 (s, 1H, quinolinone C2−H), 14.70 (bs, 2H, OH). Anal. (C20H13ClFNO5) C, H, N, Cl, F.
4-[6,7-Dichloro-1-(4-fluorophenyl)methyl-4(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid (6h)
86%; 175−177 °C; washed with isopropanol; IR ν 3300 (OH), 1722 (C=O acid), 1630 (C=O ketone) cm−1. 1H NMR (DMSO-d6) δ 5.75 (s, 2H, CH2), 7.17−7.22 (m, 2H, benzene H), 7.32−7.39 (m, 2H, benzene H), 7.88 (s, 1H, quinolinone C8−H), 8.06 (s, 1H, butenoate C3−H), 8.32 (s, 1H, quinolinone C5−H), 9.05 (s, 1H, quinolinone C2−H), 16.00 (bs, 2H, OH). Anal. (C20H12Cl2FNO5) C, H, N, Cl, F.
4-[1-(4-Fluorophenyl)methyl-7-(pyrrolidin-1-yl)-4(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid (6i)
87%; 197−199 °C; washed with isopropanol; IR ν 3300 (OH), 1718 (C=O acid), 1614 (C=O ketone) cm−1. 1H NMR (DMSO-d6) δ 2.06−2.13 (m, 4H, pyrrolidine H), 3.26−3.30 (m, 4H, pyrrolidine H), 5.77 (s, 2H, CH2), 6.42−6.45 (m, 1H, quinolinone C8−H), 6.81−6.85 (m, 1H, quinolinone C6−H), 7.18−7.25 (m, 2H, benzene H), 7.49−7.52 (m, 2H, benzene H), 8.02 (s, 1H, butenoate C3−H), 8.15−8.17 (m, 1H, quinolinone C5−H), 9.09 (s, 1H, quinolinone C2−H), 14.00 (bs, 2H, OH). Anal. (C24H21FN2O5) C, H, N, F.
4-(6-Chloro-4(1H)-quinolinon-3-yl)-2-hydroxy-4-oxo-2-butenoic Acid (8a)
50%; 220 °C (dec); toluene/cyclohexane; IR ν 3400, 3200 (NH and OH), 1718 (C=O acid), 1640 and 1611 (C=O ketone) cm−1. 1H NMR (DMSO-d6) δ 7.72−7.75 (m, 2H, quinolinone C7−H and C8−H), 8.13 (s, 1H, butenoate C3−H), 8.53 (m, 1H, quinolinone C5−H), 8.80 (s, 1H, quinolinone C2−H), 13.00 (bs, 3H, NH and OH). Anal. (C13H8ClNO5) C, H, N, Cl.
4-(7-Chloro-4(1H)-quinolinon-3-yl)-2-hydroxy-4-oxo-2-butenoic Acid (8b)
52%; 200 °C (dec); washed with isopropanol; IR ν 3400, 3200 (NH and OH), 1740 (C=O acid), 1640 and 1616 (C=O ketone) cm−1. 1H NMR (DMSO-d6) δ 7.47 (m, 1H, quinolinone C6−H) 7.69 (m, 1H, quinolinone C8−H), 7.92 (s, 1H, butenoate C3−H), 8.21 (m, 1H, quinolinone C5−H), 8.88 (s, 1H, quinolinone C2−H), 12.75 (bs, 3H, NH and OH). Anal. (C13H8ClNO5) C, H, N, Cl.
Biological Assays. Integrase Assays
Compounds 5a−i, 6a−i, 7a,b, and 8a,b were tested for their ability to inhibit HIV-1 integrase in vitro using a gel-based assay in addition to a 96-well plate-based high throughput electrochemiluminescent assay.(14)
In the gel assay, a 5′-end-labeled 21-mer double-stranded DNA oligonucleotide corresponding to the last 21 bases of the U5 viral LTR is used to follow both 3′-P and ST (for review see ref (4)). Briefly, a DNA−enzyme complex is preformed by mixing the drug at the desired concentration with 400 nM recombinant HIV-1 integrase in a buffer containing 50 mM MOPS, pH 7.2, 7.5 mM MnCl2, and 14.3 mM β-mercaptoethanol. The integration reaction is then initiated by the addition of 20 nM 5′-labeled double-stranded DNA template and continued in a total volume of 10 μL for 60 min at 37 °C. The reaction samples are stopped by adding the same volume of electrophoresis denaturing dye and loaded on 20% 19/1 acrylamide denaturing gel. Gels were exposed overnight and analyzed using a Molecular Dynamics phosphorimager (Sunnyvale, CA).
The 96-well plate-based high throughput electrochemiluminescent assay was performed using a BioVeris M series analyzer (Gaithersburg, MD) as described previously.(14) Briefly, DNA substrates were obtained from BioVeris and used according to the manufacturer’s recommendations. Donor DNA is incubated for 30 min at 37 °C in the presence of 250 nM recombinant HIV-1 integrase. After addition of the drug, the integration reaction is initiated by addition of target DNA. Reaction is carried out for 60 min at 37 °C and then read on the BioVeris M series analyzer.
Anti-HIV Assays in Cultured Cell Lines
The anti-HIV drug testing was performed in 96-well plates with a defined, previously titered inoculum of a laboratory strain (HTLV-IIIB) to minimize the inoculum effect.
In brief, all compounds were dissolved in dimethyl sulfoxide and diluted in cell culture medium at concentrations ranging from 0.1 to 50 μM. Exponentially growing human T lymphocytes (H9 cell line) were added at 5000 cells/well. After the infectivity of a virus stock (HTLV-IIIB) is quantified, an aliquot containing 1 × 105 50% tissue culture infectious dose (TCID50) per 5000 H9 cells per well is used as inoculum in each set of in vitro infections of H9 cells. Uninfected cells with the compound served as a toxicity control, and infected and uninfected cells without the compound served as basic controls. Culture were incubated at 37 °C in a 5% CO2 atmosphere for 4 days. Supernatant fluid of infected wells (in the absence of drug and at each of a number of drug concentrations) is harvested. HIV p24 antigen is quantified, and the 50% inhibitory concentration (IC50) of drug is determined using the median effect equation.
Cytotoxicity Assays
Cytotoxicity of test compounds has been evaluated on human histiocytic lymphoma (U937) cell line obtained from American Type Culture Collection (ATCC, Rockville, MD). Cells were plated in a 96 well-plate at a concentration of 5 × 103/mL in RPMI-1640 without phenol red, supplemented with 20% fetal calf serum and antibiotics. Two hours after plating, compounds were added at different concentrations ranging from 1 to 200 μM and cells were incubated at 37 °C for 24 h. Then cells were incubated at 37 °C for 3 h with 1 mg/mL of MTT (Sigma-Aldrich, Milan, Italy). After incubation, the remaining water insoluble formazan was solubilized in absolute isopropanol containing 0.1 N HCl. Absorbance of converted dye was measured in an ELISA plate reader at a wavelength of 570 nm. The cytotoxicity of the compounds was calculated as the percentage reduction of the viable cells compared with the drug-free control culture. The drug concentration required to reduce the cell viability by 50% has been called IC50.
Acknowledgments
This project was supported by the Ministero della Sanità, Istituto Superiore di Sanità, “Programma Nazionale di ricerca sull’AIDS” (Grant No. 30G.9), by the Italian MIUR (PRIN 2006, Grant No. 2006030809), and by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
Supporting Information Available
Spectroscopic data of compounds 9a−h and 10a−i; elemental analysis data for derivatives 5a−i, 6a−i, 7a,b, and 8a,b. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Footnotes
Abbreviations: HIV-1, human immunodeficiency virus type 1; RTI, reverse transcriptase inhibitors; PRI, protease inhibitors; HAART, highly active antiretroviral therapy; IN, integrase; 3′-P, 3′-processing; ST, strand transfer; DKA, aryl diketo acid; 5CITEP, 1-(5-chloroindol-3-yl)-3-hydroxy-3-(2H-tetrazol-5-yl)propenone; HTECL, high throughput electrochemiluminescent; BDKA, bifunctional diketo acid; SAR, structure−activity relationship.
Supplementary Material
References
- Richman D. D. HIV chemotherapy. Nature 2001, 410, 995–1001. [DOI] [PubMed] [Google Scholar]
- Cohen J. Therapies. Confronting the limits of success. Science 2002, 296, 2320–2324. [DOI] [PubMed] [Google Scholar]
- Fesen M. R.; Kohn K. W.; Leteurtre F.; Pommier Y. Inhibitors of human immunodeficiency virus integrase. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 2399–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y.; Johnson A. A.; Marchand C. Integrase inhibitors to treat HIV/AIDS. Nat. Rev. Drug Discovery 2005, 4, 236−248. [DOI] [PubMed] [Google Scholar]
- Dayam R.; Gundla R.; Al-Mawsawi L. Q.; Neamati N. HIV-1 integrase inhibitors: 2005−2006 update. Med. Res. Rev. 2008, 28, 118–154. [DOI] [PubMed] [Google Scholar]
- Evering T. H.; Markowitz M. Raltegravir (MK-0518): an integrase inhibitor for the treatment of HIV-1. Drugs Today 2007, 43, 865–877. [DOI] [PubMed] [Google Scholar]
- Fesen M. R.; Pommier Y.; Leteurtre F.; Hiroguchi S.; Yung J.; Kohn K. W. Inhibition of HIV-1 integrase by flavones, caffeic acid phenethyl ester (CAPE) and related compounds. Biochem. Pharmacol. 1994, 48, 595–608. [DOI] [PubMed] [Google Scholar]
- Cushman M.; Sherman P. Inhibition of HIV-1 integration protein by aurintricarboxylic acid monomers, monomer analogs, and polymer fractions. Biochem. Biophys. Res. Commun. 1992, 185, 85–90. [DOI] [PubMed] [Google Scholar]
- Carteau S.; Mouscadet J.-F.; Goulaouic H.; Subra F.; Auclair C. Inhibitory effect of the polyanionic drug suramin on the in vitro HIV DNA integration reaction. Arch. Biochem. Biophys. 1993, 305, 606–610. [DOI] [PubMed] [Google Scholar]
- Pluymers W.; Neamati N.; Pannecouque C.; Fikkert V.; Marchand C.; Burke T. R. Jr.; Pommier Y.; Schols D.; De Clercq E.; Debyser Z.; Witvrouw M. Viral entry as the primary target for the anti-HIV activity of chicoric acid and its tetra-acetyl esters. Mol. Pharmacol. 2000, 58, 641–648. [DOI] [PubMed] [Google Scholar]
- Goldgur Y.; Craigie R.; Cohen G. H.; Fujiwara T.; Yoshinaga T.; Fujishita T.; Sugimoto H.; Endo T.; Murai H.; Davies D. R. Structure of the HIV-1 integrase catalytic domain complexed with an inhibitor: a platform for antiviral drug design. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 13040–13043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazuda D. J.; Felock P.; Witmer M.; Wolfe A.; Stillmock K.; Grobler J. A.; Espeseth A.; Gabryelski L.; Schleif W.; Blau C.; Miller M. D. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science 2000, 287, 646–650. [DOI] [PubMed] [Google Scholar]
- Zhuang L.; Wai J. S.; Embrey M. W.; Fisher T. E.; Egbertson M. S.; Payne L. S. Jr.; Vacca J. P.; Hazuda D. J.; Felock P. J.; Wolfe A. L.; Stillmock K. A.; Witmer M. V.; Moyer G.; Schleif W. A.; Gabryelski L. J.; Leonard Y. M. Jr.; Michelson S. R.; Young S. D. Design and synthesis of 8-hydroxy-[1,6]naphthyridines as novel inhibitors of HIV-1 integrase in vitro and in infected cells. J. Med. Chem. 2003, 46, 453–456. [DOI] [PubMed] [Google Scholar]
- Di Santo R.; Costi R.; Roux A.; Artico M.; Lavecchia A.; Marinelli L.; Novellino E.; Palmisano L.; Andreotti M.; Amici R.; Galluzzo C. M.; Nencioni L.; Palamara A.; Pommier Y.; Marchand C. Novel bifunctional quinolonyl diketo acid derivatives as HIV-1 integrase inhibitors: design, synthesis, biological activities, and mechanism of action. J. Med. Chem. 2006, 49, 1939–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison A. A.; Marchand C.; Patil S. S.; Costi R.; Di Santo R.; Burke T. R. Jr.; Pommier Y. Probing HIV-1 integrase inhibitor binding sites with position-specific integrase-DNA cross-linking assays. Mol. Pharmacol. 2007, 71, 893–901. [DOI] [PubMed] [Google Scholar]
- Yoshizawa H.Process for Producing 5,7-Dichloro-4-hydroxy Quinoline. PCT Int. Appl. WO 9523787, 1995; CAN 124: 29619, 1995.
- Mapara R. K.; Desai C. Synthesis of 4-hydroxyquinolines. J. Indian Chem. Soc. 1954, 31, 951–956. [Google Scholar]
- Sato M.; Motomura T.; Aramaki H.; Matsuda T.; Yamashita M.; Ito Y.; Kawakami H.; Matsuzaki Y.; Watanabe W.; Yamataka K.; Ikeda S.; Kodama E.; Matsuoka M.; Shinkai H. Novel HIV-1 integrase inhibitors derived from quinolone antibiotics. J. Med. Chem. 2006, 49, 1506–1508. [DOI] [PubMed] [Google Scholar]
- Grobler J. A.; Stillmock K.; Hu B.; Witmer M.; Felock P.; Espeseth A. S.; Wolfe A.; Egbertson M.; Bourgeois M.; Melamed J.; Wai J. S.; Young S.; Vacca J.; Hazuda D. J. Diketo acid inhibitor mechanism and HIV-1 integrase: implications for metal binding in the active site of phosphotransferase enzymes. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 6661–6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.