

Abstract
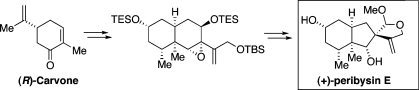
A convergent, stereocontrolled route to either antipode of the cell adhesion inhibitor, peribysin E, has been achieved from carvone. Highlights of the synthesis include a Diels−Alder reaction to generate a cis-decalin framework, followed by semipinacol-type ring contraction to secure the stereochemistry of the C7 quaternary center. Potential mechanistic pathways for the critical ring contraction were studied through deuterium incorporation studies. In addition, an optimized olefin isomerization/Saegusa oxidation protocol is described for the conversion of [4+2] cycloadducts of 2-(trialkylsilyloxy)-1,3-dienes to 1,6(2H,7H)-naphthalenediones, having stereochemical arrangements not accessible via conventional Robinson annulation protocols. Finally, the ability to independently prepare either enantiomer of peribysin E from the corresponding antipode of carvone led to a reassignment of the absolute configuration of peribysin E.
Introduction
An overarching objective of our laboratory is the synthesis and evaluation of small-molecule natural products (SMNPs) which possess biological profiles that may be of value in the treatment of cancer. High on our list of priorities in these pursuits are viable cancer chemotherapeutic agents which may function through a range of biological mechanisms.(1) In one setting, the drug is designed to attack the diseased cell itself, by targeting a component of the enzymatic or receptor machinery that is critical for the upkeep of neoplasticity.(2) Drugs which operate under this paradigm are generally termed cytotoxic or antiproliferative agents. Although such agents are widely used to great effect, there is potential benefit to be gained from an alternative approach, wherein the drug is optimized to inhibit some particular element of the support network required for tumor metastasis (i.e., cell adhesion,(3) cell migration,(4) angiogenesis(5)). These antimetastatic agents might be expected to exhibit lower toxicity than traditional cytotoxic agents, although, of course, target selectivity must still be considered. Because these types of agents would primarily serve to inhibit tumor metastasis, the optimal setting for success would be one in which the primary tumor had been resected, through surgery, radiation, or chemotherapy. The antimetastatic agent would be evaluated for its ability to halt progression of the disease, presumably through micrometastasis.
In the context described above, we took note of a newly isolated natural product, termed peribysin E (originally reported as 1(6)). Isolated by Yamada and co-workers from Periconia byssoides OUPS-N133, peribysin E was reported to exhibit potent cell adhesion inhibitory activity.7,8 In fact, peribysin E was claimed to be more strongly inhibitory than is herbimycin. In the hopes of gaining more insight into the antimetastatic potential of this particular cell adhesion inhibitor, we initiated a program directed toward the total synthesis of peribysin E. Furthermore, our synthetic route toward peribysin E would oblige us to confront mechanistic and stereochemical issues which are of considerable interest beyond the confines of this particular investigation. Indeed, a total synthesis of the properly configured peribysin E has been accomplished(9) and is described herein. The synthesis necessitated revision of the assignment of the absolute configuration of peribysin E. Furthermore, the work described below identified some issues of stereochemistry and mechanism not previously considered.
Results and Discussion
Synthetic Analysis of Peribysin E (1)
In designing a synthetic route toward peribysin E (1), we envisioned a late-stage intermediate of the type 6, confident that the final step of the synthesis, i.e., the closure of the C-ring, could be readily accomplished from aldehyde 6 (Figure 1). The defining feature of the proposed route would be a rearrangement step, with ring contraction, of an epoxide, 5, resulting in the formation of a five-membered ring incorporating the C7 quaternary center (i.e., 6). The relative configuration of the newly formed C7 locus would be controlled by inversion of the β-oriented epoxide (asterisks, 5) in the rearrangement step. If the migrating carbon (C8) were to exist as the free alcohol (5, R = H), aldehyde 6 would be obtained directly from the rearrangement. Alternatively, a protected alcohol might emerge from the rearrangement as an acetal. The latter would readily be converted to the target aldehyde. An analysis of the literature revealed a surprising paucity of examples involving comparable ring contractions of fused cyclohexene epoxides bearing internal oxygen nucleophiles, of the type 5→6. Actually, one could envision alternate and better-precedented reaction pathways for an epoxide such as 5.(10) Sensitive to the possibility of potentially complicating competitive pathways, we sought to design a system in which the desired ring contraction would be favored. Indeed, as will be shown, with considerable effort we were able to suppress nonproductive pathways, thereby achieving the desired rearrangement step with good efficiency.
Figure 1.

Synthetic strategy toward peribysin E (1).
As outlined in Figure 1, we envisioned preparing the key rearrangement precursor, 5, through a multistep process commencing with diastereoselective Diels−Alder cycloaddition of a diene of the type 3 and an optically active dienophile such as 2. It was anticipated that perhaps the R′ dienophilic substituent would shield the β-face of the dienophile, thus directing cycloaddition to the desired α-face. In this way, the cis-relationship of the C5 angular methyl group and the C2 substituent would have been established.
Construction of Enone 10: Unexpected Isomerization of Silyl Enol Ether 9
We originally selected as the dienophilic coupling partner commercially available (S)-carvone(11) (7, Scheme 1). The expectation was that the resident isopropenyl moiety would favorably direct the stereofacial sense of the Diels−Alder cycloaddition. At the appropriate time, this functionality would be readily converted to a hydroxy group with retention of configuration. As hoped, Lewis acid-catalyzed cycloaddition of 7 with diene 8 proceeded to afford intermediate 9,(12) which was well disposed to undergo subsequent Saegusa oxidation, thereby providing the desired enone 10 in overall 63% yield and high cis-selectivity.
Scheme 1.

Reagents and conditions: (a) EtAlCl2, toluene, 0 °C to rt, 4 h; (b) Pd(OAc)2, CH3CN, rt, 4 h, 63% over two steps, 19:1 (cis:trans); (c) 100 °C, toluene, 50%, 2:3 (12:13).
It was in the course of optimizing this sequence that we took note of an interesting and unanticipated isomerization event. Though it is not directly relevant to the synthesis of the target structure at hand, we felt it to be worthy of further study, since it charts some new directions for stereoselection. Thus, we had found that when intermediate 9 was heated following workup, the silyl enol ether had isomerized to compound 11. Subsequent Saegusa oxidation provided a mixture of enone 12 (20%) and the nonoxidized hydrolysis product, ketone 13 (30%).
We note that Δ4,3-keto octalone systems of the general type 15 are typically synthesized through Robinson annulation.(13) However, as is well precedented, this approach tends to result in an octalone bearing a trans relationship between the angular substituents and that at C6 (proto-steroid numbering).(14) Thus, a protocol which would allow access to the cis-series related system, as encompassed in octalone 12, would constitute a new capability for stereocontrol. As such, we sought to investigate the fortuitously discovered isomerization−oxidation sequence in more detail, in the hopes of optimizing conditions to maximize the formation of 12.
We hoped that a more robust silicon group would tend to suppress nonoxidative hydrolysis of the silyl enol ether, minimizing formation of ketone type 13. Accordingly, we replaced the trimethylsilyl (TMS) group on the diene with a triethylsilyl (TES) group (16, Scheme 2). Cycloaddition with S-carvone (7) yielded adduct 17 along with a small amount of isomerized silyl enol ether 18 (17:18 = 11:1). Upon silica gel purification, facile olefin isomerization occurred to provide 18 as the major product. Alternatively, simply stirring 17 with silica gel in CH2Cl2 afforded 92% of the isomerized adduct 18, along with 8% of the original silyl enol ether, 17. Saegusa oxidation of 18 provided the corresponding target enone, 12, in 81% yield.
Scheme 2.

Reagents and conditions: (a) EtAlCl2, toluene, 0 °C to rt, 4 h, 11:1 (17:18); (b) silica gel, CH2Cl2, rt, 1 h, 70% over two steps, 1:11 (17:18); (c) Pd(OAc)2, DMSO, 0 °C to rt, 12 h, 81%.
The chemistry described above, admittedly in a preliminary setting, points to a new idea for preparing cis-substituted decalin systems such as 12 (see asterisks) through recourse to a Diels−Alder isomerization−oxidation sequence. This protocol, discovered by chance, does not have direct implications for the synthesis of peribysin E. However, if generalizable, it should prove to be of great value in the synthesis of certain types of octalones bearing a cis-relationship between C7 and C10 (proto-steroid numbering); see asterisks in 12.
Construction of Intermediate 25
We now return to the synthesis of peribysin E. With an efficient route to enone 10, the next objective envisioned replacement of the ketonic function at C4 with a methyl group. First, the conjugated C8 ketone was temporarily masked as a dithiane, through selective ketalization(15) (10→19, Scheme 3). Wittig−Levine-type methoxymethenylation(16) of the free C4 ketone afforded 20. The latter, following hydrolysis, emerged as 21. Its aldehyde moiety was converted to a methyl group through a well-established sequence involving reduction, followed by mesylation and a second hydride-mediated reduction.(17) Finally, cleavage of the dithiane function served to restore the C8 ketone (see intermediate 23).
Scheme 3.

Reagents and conditions: (a) 1,2-ethanedithiol, MeOH, BF3·OEt2, 0 °C, 30 h, 83%; (b) Ph3PCH2OCH3Cl, KN(SiMe3)2, THF, −30 to 0 °C to rt, 24 h; (c) 4 N HCl, MeOH, THF, 0 °C to rt, 36 h, 89% over two steps, 13:1 β:α; (d) NaBH4, MeOH, THF, 0 °C to rt, 2 h, 90%; (e) MsCl, Et3N, CH2Cl2, 0 °C to rt, 1.5 h; (f) LiBHEt3, THF, 0 °C to rt, 24 h, 71% over two steps; (g) (CF3CO2)2IPh, MeOH, H2O, CH2Cl2, rt, 15 min, 87%.
With 23 in hand, we directed our attention to the conversion of the C2 isopropenyl group to the alcohol group ultimately required at C2. As shown in Scheme 4, the terminal methylene moiety was converted, by concurrent Johnson−Lemieux oxidation,(18) to a ketone (see 24). Finally, Baeyer−Villiger oxidation of 24 served to provide an acetate formulated as 25, given the expected retention of configuration in the migration step (see red asterisks at C2 in 24 and 25).
Scheme 4.

Reagents and condition: (a) OsO4, H2O2, NaIO4, 2,6-lutidine, dioxane, rt, 4 h, 85%; (b) mCPBA, CH2Cl2, 0 °C to rt, 24 h, 45% (80% BORSM).
Elaboration of the cis-Decalin Framework: Iodination and Suzuki Cross-Coupling
We next focused on the installation of a side chain at C7 (see blue asterisk, 25, Scheme 4). We hoped to exploit a Suzuki cross-coupling toward that end. Our initial attempts to install a C7 iodo group following the Johnson protocol (I2, pyridine)(19) were largely unsuccessful (∼20% conversion). We surmised that the rather low reaction yield could well be attributable to steric interference from the C5 quaternary center, which might be blocking the pyridine nucleophile from engaging in the required conjugate addition. We hoped to circumvent this problem through the use of TMSN3, with the expectation that the TMS group would serve as the activator for conjugate addition by the smaller azide (i.e., 26→27, Scheme 5). In the event, we were pleased to observe that addition of TMSN3, followed by iodine, did provide intermediate 28 which, following azide elimination, suffered conversion to α-iodoenone 29.(20) This reaction proceeded in 71% yield. Intermediate 29 reacted readily with vinyl boronic ester 30(21) in a successful Suzuki cross-coupling reaction to afford the fully elaborated cis-decalin system, 31.
Scheme 5.

Reagents and conditions: (a) TMSN3, I2, pyridine, CH2Cl2, 71% (100% BORSM); (b) Pd(PhCN)2Cl2, THF:H2O, Ag2O, Ph3As, rt, 4 h, 89%.
“Nucleophilic epoxidation” (22a) of 31 proceeded with high diastereoselectivity to generate 32, presumably through axial attack of the hydrogen peroxide at the β-carbon of the enone (Scheme 6).(22) Subsequent reduction of the ketone was achieved through delivery of the hydride from the convex face of the decalin system, thereby affording epoxy alcohol 33.(23)
Scheme 6.

Reagents and conditions: (a) H2O2, NaOH, MeOH, 0 °C, 48 h, (85% BORSM); (b) NaBH4, MeOH, THF, 0 °C, 45 min, 91%, 7:1.
Modalities for Accomplishing Ring Contraction: Mechanistic Evaluations
With epoxy alcohol 33 in hand, we were now prepared to evaluate the viability of the critical ring-contracting rearrangement. During the planning stages of the synthesis, some potential complications in the proposed rearrangement had not gone unnoticed. In the conformations of the cis-decalin framework (cf. 33a and 33b, Figure 2), the epoxide appears to lack the optimal stereoelectronic alignments for concerted base-catalyzed rearrangement. The optimal setting would be one in which the migrating C8−C9 bond would be trans-anticoplanar to the epoxide C−O bond. Indeed, as shown in Scheme 7, initial attempts to effect ring contraction through base-mediated quasi-Favorski rearrangement(24) (33→34) or through benzylic acid type rearrangement(25) (32→36) were unsuccessful.
Figure 2.

Chair conformations of epoxy diol 36.
Scheme 7.

Reagents and conditions: (a) KH, THF, 0 °C to rt, 1 h, decomposition; (b) NaOH, MeOH, 0 °C to rt, 2 h, multiple products.
In light of these initial difficulties, we wondered whether a Lewis acid-catalyzed semipinacol rearrangement—operating through the agency of a more reactive allylic cationoid species—would be more suitable for our purposes. In the event, we were pleased to find that exposure of epoxide 33 to YbOTf3 did give rise to a 2:1 ratio of the desired aldehyde 34 and ketone 37, respectively (Scheme 8). The ketone 37 was found to be unstable and underwent ready dehydration, giving rise to the corresponding enone 38 wherein the required aldehyde had been generated via migration of the C8−C9 bond. Two distinct mechanistic interpretations could be offered to account for the competitive formation of the undesired 37. The first (path a, Scheme 8) was of a type previously proposed by Edwards and Maitland(26) in the context of the synthesis of cytochalasin N. Applied to the case at hand, it envisions proton elimination from epoxide 33 to afford enol 39. Following tautomerization, ketone 37 would emerge. In an alternative pathway (path b), the hydride embedded on C8 would migrate directly, thereby providing the observed ketone byproduct.
Scheme 8.

Reagents and conditions: (a) YbOTf3, THF, 0 °C to rt, 24 h, 50%, 2:1.
The alternate mechanistic pathways suggested above might be distinguishable by suitable deuterium labeling experiments. It is known that there are smaller deuterium isotope effects for hydride migration reactions compared with more common proton transfer steps.(27) We thus prepared deuterated epoxy alcohol 40 (Scheme 9). We reasoned that if the reaction proceeded through a proton-transfer pathway (see path a), a considerable isotope effect would be expected.(28) By contrast, if the hydride migration pathway (path b) were operative, the isotope effect would be quite small.(29) In the event, treatment of 40 withYbOTf3 gave rise to only a minimal change in the ratio (3:1) of aldehyde 41 and ketone 42. Given the observed product distribution, one would presume that the undesired ketone side product 37 is primarily generated through a hydride migration pathway (path b).
Scheme 9.

Reagents and conditions: (a) NaBD4, MeOH:THF, 0 °C, 30 min, 91%, 6:1; (b) YbOTf3, THF, 0 °C to rt, 24 h, 39%, 3:1.
Armed with this added mechanistic insight, we now returned to the total synthesis of peribysin E. With aldehyde 34 in hand, we had confirmed the viability of the semipinacol rearrangement in effecting the critical ring contraction step. However, for efficiency purposes, we sought a means by which to suppress the undesired hydride migration pathway. In an effort to improve the product distribution of the pinacol rearrangement (33→34), we first screened a range of Lewis acids. Unfortunately, all Lewis acids examined led to recovery of starting material or decomposition. Postulating that the resident free hydroxyl groups might be incompatible with mediation by Lewis acids, we prepared modified intermediate 43, in which both free hydroxyls were protected with TES groups (Scheme 10). Happily, upon exposure of 43 to TiCl4, the desired ring contracted rearrangement product 34 was formed as the principal product, with the required configuration at the newly formed C7 quaternary center.(30) Only a 5% yield of the β-hydroxyketone (37) was observed under optimized conditions.(31) Treatment of 34 with HCl−methanol afforded the target system, peribysin E. We think that these findings are illustrative of the connectivity between mechanistic thinking and process optimization.
Scheme 10.

Reagents and conditions: (a) TESCl, imidazole, DMF, rt, 12 h, 93%; (b) TiCl4, CH2Cl2,−78 °C, 5 min, 50%, 10:1 (34:37); (c) HCl, MeOH, 0 °C, 1 h, 80%.
Reassignment of the Absolute Configuration of Peribysin E
The proton and carbon spectra of fully synthetic peribysin E were identical to the spectra of naturally derived natural product provided by Professor Yamada, though the spectra of the synthetic material were cleaner. We noted, however, a very significant discrepancy in the optical rotation measurements. The synthetic material had a specific rotation of [α]25D = −52.17 (c = 0.11, EtOH), while the natural peribysin E is reported to possess a specific rotation of [α]25D = −262.2 (c = 0.11, EtOH). Though both samples exhibited the same sign of rotation, the discrepancy in the magnitude of rotation was so large as to raise a serious concern as to the matter of identity. In order to further clarify this unexpected finding, we prepared the diacetate of our synthetic peribysin E and determined the rotation to be [α]25D = −34.78 (c = 0.069, EtOH). By contrast, the naturally derived peribysin E diacetate was reported to have a specific rotation of [α]25D = +35.00 (c = 0.069, EtOH). On the basis of this information, we could only conclude that the original assignment of absolute configuration for peribysin E had, in fact, been incorrect. We had thus synthesized ent-peribysin E (1, Scheme 10).
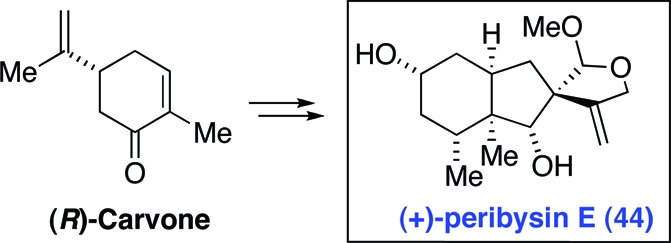
Finally, in order to provide full clarity to the situation, we repeated all of the steps of the synthesis, starting from (R)-carvone. We ultimately reached peribysin E, with the absolute configuration 44 (Scheme 11). Indeed, the optical rotation of the fully synthetic diacetate of 44, [α]25D = +37.49 (c = 0.069, EtOH), was found to be consistent with that reported for the naturally derived material. In light of the validity of the assignments of absolute configurations of carvone enantiomers,(11) and our full confidence in the stereochemical logic of the syntheses, we now conclude that peribysin E is properly depicted as 44, and that our original target structure (1) corresponds to ent-peribysin E.
Scheme 11. Synthesis of (+)-Peribysin E (44).
Preliminary in vitro biological evaluations were conducted with synthetic peribysin E (44) and ent-peribysin E (1). In an adhesion assay of human leukemia HL60 cells to human umbilical vein endothelial cells (HUVECs), a dose-dependent effect was observed for peribysin E (44), first detectable at 4 μM and reaching a maximum at 100 μM. At concentrations above 100 μM, the HUVECs began to detach from the bottom of the well in response to the drug treatment, thus rendering specific HL60 adhesive evaluation unfeasible. In contrast, ent-peribysin E (1) exhibited minor effects on HL60 adhesion at concentrations above 50 μM. We are thus able to conclude that the anti-adhesive property of peribysin E is apparently enantiospecific.
Summary
In conclusion, we have achieved the enantioselective total syntheses of both (+)- and (−)-peribysin E, commencing with the corresponding enantiomers of carvone. A chance observation as to the isomerization of 17→18 opens up new possibilities for stereocontrol in octalone synthesis. A critical semipinacol type ring contraction allowed for the stereocontrolled generation of a 5,6-spiro system from a Diels−Alder-derived cis-decalin. It is expected that the quantities of peribysin E now available through total synthesis will enable a more thorough evaluation of its promising cell-adhesion inhibitory properties. Such investigations are currently being pursued as part of our long-standing program in the area of oncologically relevant SMNPs through biomechanistically relevant pathways.
Acknowledgments
This work was supported by the National Institutes of Health (CA28824 to S.J.D., HL61401 and CA062948 to A.R.A.). We thank Dr. Louis Todaro (Hunter College) and Daniela Buccella (Columbia University, NSF grant CHE-0619638) for X-ray structure analyses. We thank Professor T. Yamada (Osaka University of Pharmaceutical Sciences) for furnishing us with the 1H and 13C NMR spectra of peribysin E.
Supporting Information Available
Experimental procedures and spectroscopic and analytical data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Mayer A. M. S.; Gustafson K. R. Eur. J. Cancer 2006, 42, 2241–2270. [DOI] [PubMed] [Google Scholar]
- a Cox M. B.; Miller C. A. III. Mol. Pharmacol. 2003, 64, 1549–1556. [DOI] [PubMed] [Google Scholar]; b Baselga J. Science 2006, 312, 1175–1178. [DOI] [PubMed] [Google Scholar]
- Musza L. L.; Speight P.; McElhiney S.; Barrow C. J.; Gillum A. M.; Cooper R.; Killar L. M. J. Nat. Prod 1994, 57, 1498–1502. [DOI] [PubMed] [Google Scholar]
- Sawyer T. K. Expert Opin. Investig. Drugs 2004, 13, 1–19. [DOI] [PubMed] [Google Scholar]
- Ingber D.; Fugita T.; Kishimoto S.; Sudo K.; Kanamaru T.; Brem H.; Folkman J. Nature 1990, 348, 555–557. [DOI] [PubMed] [Google Scholar]
- The structure including absolute as well as relative stereochemistry was assigned as shown in structure 1.
- Yamada T.; Doi M.; Miura A.; Harada W.; Hiramura M.; Minoura K.; Tanaka R.; Numata A. J. Antibiot. 2005, 58, 185–191. [DOI] [PubMed] [Google Scholar]
- Yamada T.; Iritani M.; Doi M.; Minoura K.; Ito T.; Numata A. J. Chem. Soc., Perkin Trans. 2001, 1, 3046–3053. [Google Scholar]
- Angeles A. R.; Dorn D. C.; Kou C. A.; Moore M. A. S.; Danishefsky S. J. Angew. Chem., Int. Ed. 2007, 46, 1451–1454. [DOI] [PubMed] [Google Scholar]
- a Kunisch F.; Hobert K.; Welzel P. Tetrahedron Lett. 1985, 26, 6039–6042. [Google Scholar]; b Maruoka L.; Ooi T.; Nagahara S.; Yamamoto H. J. Am. Chem. Soc. 1989, 111, 6431–6432. [Google Scholar]
- a Preite M. D.; Gonzalez-Sierra M.; Zinczuk J.; Ruveda E. Tetrahedron: Asymmetry 1994, 5, 503–506. [Google Scholar]; b Kawai Y.; Hayashi M.; Tokitoh N. Tetrahedron 2005, 61, 5049–5055. [Google Scholar]
- Haaksma A. A.; Jansen B. J. M.; de Groot A. Tetrahedron 1992, 48, 3121–3130. [Google Scholar]
- Rapson W. S.; Robinson R. J. Chem. Soc. 1935, 1285–1288. [Google Scholar]
- Heathcock C. H.; Graham S. L.; Pirrung M. C.; Flavac F.; White C. T. In The Total Synthesis of Natural Products; ApSimon J. E., Ed.; Wiley: New York, 1983; Vol. 5, p 129. [Google Scholar]
- Harayama T.; Cho H.; Inubushi Y. Chem. Pharm. Bull. 1978, 26, 1201–1214. [Google Scholar]
- Levine S. G. J. Am. Chem. Soc. 1958, 80, 6150–6151. [Google Scholar]
- For related transformations in the AB trans series, see:; a Paquette L. A.; Wang T.-Z.; Philippo C. M. G.; Wang S. J. Am. Chem. Soc. 1994, 116, 3367–3374. [Google Scholar]
- Pappo R.; Allen D. S. Jr.; Lemieux R. U.; Johnson W. S. J. Org. Chem. 1956, 21, 478–479. [Google Scholar]
- Johnson C. R.; Adams J. P.; Braun M. P.; Senanayake C. B. W. Tetrahedron Lett. 1992, 33, 917–918. [Google Scholar]
- Sha C.-K.; Huang S.-J. Tetrahedron Lett. 1995, 36, 6927–6928. [Google Scholar]
- Takahashi K.; Ishiyama T.; Miyaura N. J. Organomet. Chem. 2001, 625, 47–53. [Google Scholar]
- a Plattner P. A.; Heusser H.; Kulkarni A. B. Helv. Chim. Acta 1948, 31, 1822–1831. [DOI] [PubMed] [Google Scholar]; b The very high level of stereoselection in the nucleophilic epoxidation presumably reflects the mutually compatible tendencies for attack by the hydroperoxide anion from the convex face along an axial trajectory (cf.Wu Y-D.; Houk K. N.; Trost B. M. J. Am. Chem. Soc. 1987, 109, 5560–5561. [Google Scholar]; ). The presence of cis-1,3-related substituents in the B-ring of 31 may well serve to anchor the conformation of the system such that attack from the β-face corresponds to axial attack.
- The relative stereochemistry of 33 was established by crystallographic determination on a derived triol (Supporting Information).
- Harmata M.; Rashatasakhon P. Org. Lett. 2001, 3, 2533–2535. [DOI] [PubMed] [Google Scholar]
- Gill G. B. In Comprehensive Organic Synthesis; Trost B. M., Fleming I., Ed.; Pergamon: Oxford, 1991; pp 821−838 [Google Scholar]
- Edwards R. L.; Maitland D. J.; Whalley A. J. S. J. Chem. Soc., Perkin Trans. 1 1989, 57–65. [Google Scholar]
- Experimentally determined deuterium isotope effects for 1,2-hydride shifts lie in the range of 1.2−3.0:; a Myhre P. C.; Brown K. S. J. Am. Chem. Soc. 1969, 91, 5639–5641. [Google Scholar]; b Myhre P. C.; Evans E. J. Am. Chem. Soc. 1969, 91, 5641–5644. [Google Scholar]; c Karabatsos G. J.; His N.; Meyerson S. J. Am. Chem. Soc. 1970, 92, 621–626. [Google Scholar]; d Karabatsos G. J.; Mount R. A.; Rickter D. O.; Meyerson S. J. Am. Chem. Soc. 1970, 92, 1248–1253. [Google Scholar]
- Salomon R. G.; Miller D. B.; Zagorski M. G.; Coughlin D. J. J. Am. Chem. Soc. 1984, 106, 6049–6060. [Google Scholar]
- Coxon J. M.; Thorpe A. J. J. Org. Chem. 2000, 65, 8421–8429. [DOI] [PubMed] [Google Scholar]
- Maruoka K.; Hasegawa M.; Yamamoto H. J. Am. Chem. Soc. 1986, 108, 3827–3829. [DOI] [PubMed] [Google Scholar]
- M E.; Jung A.; van den Heuvel A. G.; Leach K. N.; Houk Org. Lett. 2003, 5, 3375–3378. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.