

Abstract
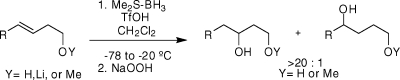
Metal-free homoallylic oxygen-directed intramolecular hydroboration is reported. Regioselectivities from 20:1 to 82:1 favoring the 1,3-dioxy-substituted products have been achieved using Me2S·BH3/TfOH followed by standard oxidative workup. Branching at the C5 position improves regioselectivity.
Despite repeated efforts over many years and several tantalizing empirical results that suggest oxygen-directed hydroboration (ODHB), definitive examples of this process have been rare.1–4 Evans’ metal-catalyzed reaction of catecholborane with several unsaturated alcohols, phosphinites, and carboxamides is the only method known to date with established synthetic potential for a range of substrates.(2) Another case of ODHB involving an α-methoxy-β,γ-unsaturated ester was encountered by Panek et al.(3) using Me2S·BH3. This example approaches the regioselectivity of the Evans result with a homoallylic alcohol (8:1 vs 11:1), but appears to be a special case reflecting unusual reactivity due to the presence of ester as well as methoxy groups in the starting material. The other historical examples reveal interesting perturbations of hydroboration stereoselectivity or regioselectivity by oxygen substituents,(4) but these reactions generally do not give useful product ratios. Our work reported below demonstrates a mechanistically distinct version of ODHB using metal-free conditions that afford unprecedented levels of regiocontrol.
The analogy of amine-directed hydroboration(5) suggested that alcohol borane complexes 6 (Scheme 1) might be accessible from activated boranes 3 or 4 and homoallylic alcohols 5. This would provide a basis for regiocontrol and intramolecular hydroboration if the potential complications from evolution of H2 and weak O−B complexation at the stage of 6 could be overcome. An SN2-like displacement of the leaving group X might then lead from 6 to the π-complex 7, which would afford hydroboration products via the labile O-protonated 1,2-oxaborolane 8 or related intermediates.
Scheme 1.

A number of conditions were tested in attempts to activate dimethylsulfide borane 1 or thioanisole borane 2(6) with I2 or TfOH at −78 °C in the presence of (E)-3-pentenol 5a. The resulting solution was warmed to −20 °C (5−19 h) and then quenched using standard oxidative workup (Table 1) for assay of the isomeric diols 9a and 10a. Both reactivity and regioselectivity were marginal in attempts to activate 1 with I2. A control experiment using 1 without I2 gave a 2.2:1 ratio of 9a and 10a at −20 °C, consistent with at least some HB involving unactivated Me2S·BH3 in entries 1 and 2. Switching to thioanisole borane 2 improved both reactivity and regioselectivity for the iodine activation (entries 3 and 4), but this required an inconvenient procedure using B2H6 gas to generate 2. Good selectivity was also observed by activating a solution of 5a and 1 with TfOH at −78 °C, probably because this procedure minimizes any chance of background reaction. However, product recovery was low (∼20%, entry 5).
Table 1. Reaction of 5a with 1 or 2 Activated in situa.
| entry | borane source | activator | time (h) | conversionb (%) | 9a:10ab |
|---|---|---|---|---|---|
| 1 | 1 | I2 | 10 | 40 | 4.3:1 |
| 2 | 1 | I2 | 19 | >90 | 4.7:1 |
| 3 | 2 | I2 | 5 | >90 | 7:1c |
| 4 | 2 | I2 | 5 | >90 | 18:1d |
| 5 | 1 | TfOH | 5 | >90 | >20:1 |
A 0.1 M solution of 5 and 2 equiv of 1 or 2 at −78 °C in CH2Cl2 was treated with 2 equiv of activator, warmed to −20 °C, stirred (time), and quenched with NaOOH/MeOH·H2O.
NMR assay.
Activated at −20 °C and warmed to −10 °C.
Warmed to −10 °C.
Preactivating the borane complex 1 with TfOH was found to be crucial for improved product recovery. With this modification, a series of homoallylic alcohols 5 gave useful yields and excellent regioselectivities (Table 2; preactivation at −78 °C and warming to −20 °C). In the highest yielding example, activation of 1 with TfOH followed by addition of 5e resulted in conversion on a time scale of hours at −20 °C. Diol 9e was obtained in 80% yield after oxidative workup (1.5 mmol scale), and NMR assay after derivatization(7) established 56:1 regioselectivity in favor of the 1,3-diol 9e over 10e (entry 5). Both the (E)- and (Z)-3-hexenols 5b and 5c (entries 2 and 3) reacted with >20:1 selectivity under these conditions, although the E-isomer gave the better result (37:1 9b:10b).(8) The highest regioselectivity was achieved in the conversion of 5f to 9f (entry 6), demonstrating that increased branching adjacent to C4 in 5f compared to 5d or 5e (entries 4 and 5) correlates with higher regioselectivity. In contrast to the successful TfOH experiments, attempted preactivation of 1 with iodine over the same temperature range (−78 to −20 °C) resulted in poor reactivity and low conversion.
Table 2. ODHB of Homoallylic Alcohols 5 with Me2S·BH3+ TfOHa.
| entry | Stg | R2 | time (h) | yield (%) | 9:10b |
|---|---|---|---|---|---|
| 1 | 5a | CH3 | 10 | 51c | >20:1 |
| 2 | 5b | C2H5 | 10 | 66c | 37:1 |
| 3 | 5c | Z-C2H5 | 5 | 51d | 28:1 |
| 4 | 5d | nC5H11 | 10 | 69 | >20:1 |
| 5 | 5e | cC6H11 | 5 | 80 | 56:1 |
| 6 | 5f | tC4H9 | 5 | 56 | 82:1 |
| 7 | 5g | Ph | 20 | >3 | N/D |
| 8 | 5h | Bn | 10 | 22 | N/D |
| 9 | 5i | CH2Bn | 5 | 59 | >20:1 |
Conditions: A 0.2 M solution of 1 (CH2Cl2) at −78 °C was treated with 1 equiv of TfOH, stirred 30 min, 0.2 M of 5 in CH2Cl2 added dropwise, and oxidative workup performed after (time) at −20 °C.
NMR assay.
Five equivalents of cyclohexene present.
Yield after derivatization.(7)
Surprisingly, phenyl substituents near the C=C subunit decreased reactivity using the TfOH preactivation conditions. Thus, the styrene 5g gave only trace conversion after 20 h at −20 °C, and the benzyl analogue 5h reacted very slowly compared to other aliphatic substrates (entry 8). On the other hand, 5i with a CH2CH2 spacer between the olefin and the phenyl group reacted normally, and good conversion was observed after 5 h (entry 9).
Entries 1 and 2 (Table 2) describe experiments where the ODHB reactions of 5a and 5b were conducted in the presence of 5 equiv of cyclohexene to confirm an intramolecular pathway. Selective consumption of the homoallylic alcohols was observed in both cases, while cyclohexanol was not detected after oxidative workup, as expected if the oxygen-directed internal pathway has a significant rate advantage. When these experiments were repeated in the absence of the cyclohexene, the yields of diols were somewhat lower (41% from 5a, 51% from 5b), suggesting that the cyclohexene additive serves in a protective role by scavenging residual triflic acid.(9)
Although the above experiments establish an intramolecular hydroboration process for the alcohol substrates, attempts to probe the sequence of events suggested in Scheme 1 using NMR spectroscopy provided only limited insight. When 1 was treated with TfOH at −78 °C, the 11B chemical shift of 1 (δ −20.6 ppm) was replaced by a major new signal (δ −2.0 ppm, t, J = 129 Hz) consistent with the activated borane 3 (X = OTf). Addition of substrate 5b at −78 °C and warming to −20 °C produced broad 11B signals (δ 7.5 to −8.0 ppm; tetravalent boron), but no signals were found from δ 50 to 70 ppm, the range estimated for the hypothetical trivalent ROBH2 (11)10,11 or oxaborolanes 12 (X = H or OTf).(12)
In an attempt to detect 11 or other intermediates, alcohol 5b was treated with BuLi (1.1 equiv) at −78 °C in DCM, followed by addition of the resulting alkoxide solution to preformed 3 (X= OTf, 2 equiv). The 1H and 11B NMR spectra were not definitive. However, quenching the solution after 5 h at −20 °C by oxidative workup with NaOH/MeOH/H2O2 gave the diol 9b (56%, >20:1 regioselectivity).(8) Using the best substrate 5e from Table 2, the lithium alkoxide procedure gave a 63:1 ratio of 9e:10e in 64% yield after derivatization.(7) Similar results were obtained in several other examples, indicating that the lithium alkoxides are competent substrates for directed hydroboration. The small difference in regioselectivity compared with Table 2, entry 5, implies that the lithium alkoxide from 5e need not react via the same regioselectivity-determining transition state as for the alcohol substrate, but a more convincing differentiation was desired. To this end, the methoxy ether 13 was subjected to the usual ODHB conditions using preformed 3 (X = OTf) at −78 °C (15 min) to −20 °C (10 h). After oxidative workup, the product was obtained as a 20:1 mixture of 14:15. In contrast to results with the alcohol substrates, the in situ procedure (TfOH added to 13 + 1 at −78 °C, 15 min; −20 °C, 10 h) gave a better result (61% yield, 50:1 14:15). By comparison, standard hydroboration using THF/borane (0 °C, 4 h, THF) afforded a 1.4:1 mixture of 14:15. These findings prove that ODHB can take place by mechanisms involving neither 6 nor the alkoxyborane 11, but do not rule out these intermediates for the alcohol substrates. If covalently bound B−O intermediates are involved in all three reactions (alcohol, Li alkoxide, and ether substrates), then each of the distinct species represented by structures 6, 16, and 17 may be reactive in ODHB.(13) The ether example via 17 would be mechanistically analogous to the amine borane reactions,(5) but the relative merits of 6 and 16 remain unknown.
Scheme 2.

Pending the detection of hypothetical intermediates such as 11, 6, 16, or 17, we cannot comment further on the mechanism of ODHB. However, the preparative results show that the oxygen-directed intramolecular hydroboration is feasible for homoallylic alcohols, alkoxides, and ethers using 3 as an activated reagent equivalent to TfOBH2.(14)
Acknowledgments
This work was supported by NIH (GM067146; CBI training Grant GM08597).
Supporting Information Available
Experimental procedures and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hoveyda A. H.; Evans D. A.; Fu G. C. Chem. Rev. 1993, 93, 1307. [Google Scholar]
- a Evans D. A.; Muci A. R.; Stürmer R. J. Org. Chem. 1993, 58, 5307. [Google Scholar]; b Evans D. A.; Fu G. C.; Hoveyda A. H. J. Am. Chem. Soc. 1988, 110, 6917. [Google Scholar]; c Evans D. A.; Fu G. J. Am. Chem. Soc. 1991, 113, 4042. [Google Scholar]
- Panek J. S.; Xu F. J. Org. Chem. 1992, 25, 215. [Google Scholar]
- a Schulte-Elte K. H.; Ohloff G. Helv. Chim. Acta 1967, 50, 153. [DOI] [PubMed] [Google Scholar]; b Heathcock C. H.; Jarvi E. T.; Rosen T. Tetrahedron Lett. 1984, 25, 243. [Google Scholar]; c Smith A. B.; Yokoyama Y.; Huryn D. M.; Dunlap N. K. Tetrahedron Lett. 1987, 28, 3659. [Google Scholar]; d Zweifel G.; Najafi M. R.; Rajagopalan S. Tetrahedron Lett. 1988, 29, 1895. [Google Scholar]; e Suzuki K.; Miyazawa M.; Shimazaki M.; Tsuchihashi G. Tetrahedron 1988, 44, 4061. [Google Scholar]; f Jung M. E.; Karama U. Tetrahedron Lett. 1999, 40, 7907. [Google Scholar]
- a Scheideman M.; Shapland P.; Vedejs E. J. Am. Chem. Soc. 2003, 125, 10502. [DOI] [PubMed] [Google Scholar]; b Scheideman M.; Wang G.; Vedejs E. J. Am. Chem. Soc. doi: 10.1021/ja0774663. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidlewicz M.; Kanth J. V. B.; Brown H. C. J. Org. Chem. 2000, 65, 6697. [DOI] [PubMed] [Google Scholar]
- 1H NMR assay of 9+10 was best done after diaroylation with 2-CF3C6H4COCl (resolved methine H’s for all diol pairs except 9h/10h).
- Diols 9b, 9c, and 9f were difficult to purify due to water solubility and co-elution with Me2SO2 formed from Me2S during oxidative workup.
- 1-Phenethyltetrahydrofuran was isolated in 6% yield using ODHB from 5i. Analogous (volatile) byproducts from TfOH-induced cyclization were detected in ODHB experiments with some of the other alcohols 5. The corresponding Li alkoxides give no cyclic ethers, but conversion was lower using 2 equiv of 3 (X = OTf) and the procedure more tedious.
- Alkoxyboranes similar to 11 are unknown. For detection of ROBHThx, see:Cha J. S.; Seo W. W.; Kim J. M.; Kwon O. O.. Bull. Korean Chem. Soc. 1996, 17, 892. Activation of 1 afforded 92% of the expected H2 within 30 min at −78 °C and an additional 0.15 equiv of H2 vs the starting TfOH upon addition of 5b and warming to −20 °C, but further hydrogen evolution was too slow to measure. In a control experiment, reaction of 1 with EtOH in DCM at −20 °C gave 5−7% H2 within 20 min, and very slow H2 evolution thereafter (9% total after 1.5 h). [Google Scholar]
- Nöth H.; Wrackmeyer B. In Nuclear Magnetic Resonance Spectroscopy of Boron Compounds; Diehl P., Fluck E., Kosfeld R., Eds.; Springer-Verlag: Berlin: 1978; Vol. 14 (see Tables I, XIV, IL on pages 115, 141, and 253). [Google Scholar]
- Wrackmeyer B. In Annual Reports on NMR Spectroscopy; Webb G. A., Ed.; Harcourt Brace Jovanovich: London, 1988, Vol. 20, pp 61−203see Tables 10, 14, and 19 on pages 89, 92, and 105. [Google Scholar]
- The 1H NMR spectrum after mixing 3 (X = OTf) and 5b at −78 °C and warming to −20 °C (5 min) revealed a new signal at δ = 12.5 ppm, consistent with the O−H subunit of 6. This signal disappeared over 15 min at −20 °C, but the olefinic signals were still present. Activation of n-Bu4NBH4 in CH2Cl2 at −78 °C with TfOH (2 equiv), addition of 5d, and warming to −20 °C gave >20:1 9d:10d (60%) after oxidative workup, suggesting that Me2S plays no major role.
- Allylic alcohols such as (E)-2-octenol are not reactive under the standard conditions of Table 2. Bis-homoallylic alcohols are reactive, but give lower regioselectivity (5:1 ratio favoring the 1,4-diol from (E)-hex-4-enol). Homoallylic alcohols containing alkyl groups at C2 react with ≤1.5:1 dr.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.