

Abstract

A formal synthesis of (±)-roseophilin is described. Scandium(III)-catalyzed Nazarov cyclization of 2,5-disubstituted N-tosylpyrrole 19 gives a 5,5’-fused ketopyrrole, and ansa-bridge formation via π-allyl palladium macrocyclization gives 21.
Roseophilin (1), isolated from Streptomyces griseoviridis, was identified as a potent cytotoxic agent against K562 human erythroid leukemia cells (chronic myeloid leukemia model; IC50, 0.34 μM) and KB human epidermoid carcinoma cells (nasopharyngeal carcinoma model; IC50, 0.88 μM).1 Unexpectedly, the unnatural enantiomer of 1, ent-roseophilin, was shown to possess 2-10 fold greater potency than the naturally occuring enantiomer.2
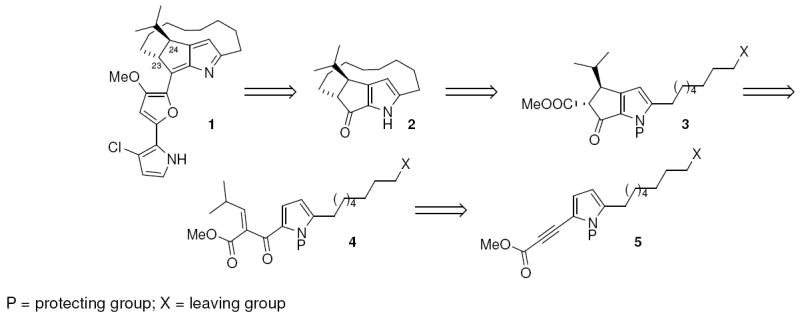
The structure of 1 contains an ansa-bridged macrocycle that is connected to a pyrrolylfuran moiety via an azafulvene subunit. Its unique chemical structure coupled with its poorly understood mechanism of cytotoxicity3 has led to the development of several synthetic approaches targeting 1.2,4 Herein, we disclose our approach to the formal synthesis of 1 via macrotricycle 2, using a strategy involving the catalytic Nazarov cyclization of polarized aryl vinyl ketones.5,6
Our retrosynthetic analysis of macrotricycle 2 is shown in Scheme 1. Execution of a late-stage macrocyclization to close the strained 13-membered ansa bridge was planned with some trepidation. In most of the published approaches toward this target, the final stages involved closure of either the cyclopentenyl ring or the pyrrole ring within an existing macrocyclic scaffold. In the few cases when a late-stage macrocyclization was performed on the fused 5,5’-fused ring system, bulky substituents on the side chain were needed to conformationally induce ring closure.4d,4h With greater confidence, we planned to prepare macrocyclization precursor 3 from Nazarov cyclization of pyrrolylvinyl ketone 4,6 which could in turn be assembled via [3+2] cycloaddition/chelotropic extrusion of the alkynyl ester 5, a sequence discovered by Padwa7 and investigated further in our laboratory.8
Scheme 1.
The synthesis began with ozonolytic desymmetrization9 of cyclohexene followed by Jones oxidation of the intermediate aldehyde to provide carboxylic acid 6 in excellent yield (Scheme 2). Under refluxing conditions in the presence of trifluoroacetic anhydride and carboxylic acid 6, N-tosylpyrrole was acylated selectively at the 2 position.10 Subsequently, the ketopyrrole formed was reductively deoxygenated under mild conditions using zinc iodide and sodium cyanoborohydride to provide ester 7 in good yield.11 Elaboration upon the alkyl side chain of ester 7 using the high yielding reduction/oxidation sequence of DIBAL-H and Swern conditions provides aldehyde 8. Under standard Horner-Wadsworth-Emmons conditions12 with methyl diethylphosphonoacetate, aldehyde 8 was converted to the α,β-unsaturated ester 9 in excellent yield.
Scheme 2.

Vilsmeier-Haack formylation13 of 9 provided pyrrolylcarboxaldehyde 10 which was subjected to the first step of the Corey-Fuchs transformation14 to arrive at gem-dibromoalkene 11 (Scheme 3). Reduction of the α,β-unsaturated ester moiety of 11 with DIBAL-H and subsequent silyl protection of the alcohol 12 yields gem-dibromoalkene 13. The rearrangement step of the Corey-Fuchs sequence provided alkyne 14 in good yield. Selective deprotonation of the acetylenic proton of 14 with lithium hexamethyldisilazide followed by addition of methyl chloroformate yielded alkynyl ester 15. One-pot formation of the alkynyl ester from 13 using organolithium reagents followed by quenching with methyl chloroformate was problematic due to orthometalation of the N-tosyl protecting group.15 Crude alkynyl ester 15 underwent a [1,3]-dipolar cycloaddition reaction with nitrone 16 at 80 °C in toluene (1.6 M) to provide isoxazoline 17 in excellent yield over 2 steps.
Scheme 3.

To unveil the Nazarov cyclization precursor, isoxazoline 17 was exposed to a slight excess of m-cpba at 0 °C, standard conditions for a chelotropic extrusion of this type.7,8 The desired 2-pyrrolyl vinyl ketone was obtained along with a byproduct resulting from epoxidation of the protected allylic alcohol. If the rate of N-oxidation is slightly faster than epoxidation, addition of a sacrificial alkene might minimize unwanted epoxidation of valuable substrate 17. Indeed, a protocol that involved alternating the addition of portions of cyclohexene and oxidant at -30 °C did protect 17 from overoxidation (Scheme 4). Further optimization of this reaction is required. With β-ketoester 18 in hand, we turned our attention to the Nazarov cyclization.
Scheme 4.

Initial attempts at cyclization of β-ketoester 18 were unsuccessful. The silyl protecting group was cleaved under the reaction conditions and the exposed alcohol appeared to inhibit catalyst turnover. Further heating resulted in decomposition. After exchanging the silyl protecting group for an acetyl protecting group, the Nazarov substrate 19 was heated in the presence of catalytic amounts of scandium(III) triflate and 1 equivalent of lithium perchlorate to provide Nazarov product 20 in good yield (Scheme 4).6b Furthermore, the reaction was relatively rapid with complete conversion observed in under 2 hours.
Initial attempts to cyclize structures similar to 3 (see Scheme 1) using mild base in a polar aprotic solvent16 were unsuccessful. Since intramolecular alkylation of the β-ketoester by SN2 displacement was not possible, alkylation with the appropriate π-allyl palladium species was explored.17 We sought to carry out the macrocyclization under dilute and controlled conditions using a suitable palladium(0) catalyst system (Table 1). Our initial attempt relied upon slow addition of the sodium enolate of 20 to a refluxing THF solution of tetrakis(triphenylphosphine)palladium (Entry 1).18 The reaction provided a 4:1 mixture of macrocycle 21 to a product resulting from β-hydride elimination (A). The two products were difficult to separate via flash chromatography. Fortunately, macrocycle 21 could be recrystallized and an X-ray crystal structure was obtained as shown in Figure 1. By preforming the Pd(0) catalyst using palladium acetate in the presence of dppp, we were pleased to find the overall yield increased to 73 % (Entry 2). The ratio was improved to 4:1 with the use of dppe (Entry 3). Decomposition was observed when dppb was employed (Entry 4).
Table 1.
Optimization of π-allyl Pd macrocyclization.a
 | ||||
|---|---|---|---|---|
| entry | catalyst | ligand (mol %) | Overall yield (%) | ratio 21:Ab |
| 1 | Pd(PPh3)4 | - | 56 | 4:1 |
| 2 | Pd(OAc)2 | dpppc (48 mol %) | 73 | 3:1 |
| 3 | Pd(OAc)2 | dpped (48 mol %) | 82 | 4:1 |
| 4 | Pd(OAc)2 | dppbe (48 mol %) | f | - |
Reaction conditions: 20 mol % catalyst, ligand, NaH (1.1 equiv), reflux, THF (0.001 M).
Based on 1H NMR integration.
dppp = 1,3-bis(diphenylphosphino)propane.
dppe = 1,2-bis(diphenylphosphino)-ethane.
dppb = 1,4-bis(diphenylphosphino)butane.
Decomposition was observed.
Figure 1.

ORTEP drawing of macrocycle 21.
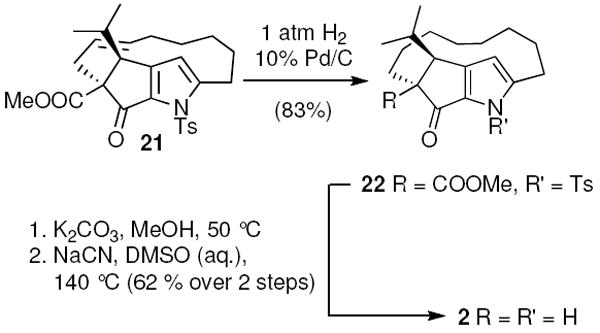
Functional group modification was required to complete the formal synthesis of 1 (Scheme 5). Hydrogenation followed by deprotection of the pyrrolyl nitrogen under basic conditions furnished macrocyclic β-ketoester 22. Krapcho dealkoxycarbonylation of 22 delivered 2 in good yield.
Scheme 5.
Future work will focus on developing a more efficient synthesis of Nazarov cyclization precursor 19, optimizing the macrocyclization and adapting the strategy to the asymmetric synthesis of 2.
Supplementary Material
Experimental procedures, characterization data, X-ray crystal structure coordinates and files for compound 21 in CIF fromat. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We are grateful to NSF (CAREER: CHE-0349045), NIH (NIGMS R01 GM079364), and the University of Rochester for generous financial support. We thank Prof. Robert K. Boeckman, Jr. (University of Rochester) and Prof. David W. C. MacMillan (Princeton University) for helpful discussions. We are also grateful to Dr. William Brennessel (University of Rochester) for solving the structure of 21 by X-ray crystallography, and Dr. Alice Bergmann (University of Buffalo) for carrying out high-resolution mass spectroscopy.
References
- 1.Hayakawa Y, Kawakami K, Seto H, Furihata K. Tetrahedron Lett. 1992;33:2701. [Google Scholar]
- 2.Boger DL, Hong J. J Am Chem Soc. 2001;123:8515. doi: 10.1021/ja011271s. [DOI] [PubMed] [Google Scholar]
- 3.(a) Furstner A, Grabowski EJ. ChemBioChem. 2001;2:706. doi: 10.1002/1439-7633(20010903)2:9<706::AID-CBIC706>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]; (b) Furstner A, Reinecke K, Prinz H, Waldmann H. ChemBioChem. 2004;5:1575. doi: 10.1002/cbic.200400135. [DOI] [PubMed] [Google Scholar]; (c) Manger M, Scheck M, Prinz H, von Kries JP, Langer T, Saxena K, Schwalbe H, Furstner A, Rademann J, Waldmann H. ChemBioChem. 2005;6:1749. doi: 10.1002/cbic.200500171. [DOI] [PubMed] [Google Scholar]
- 4.For a review, see Fürstner A. Angew Chem Int Ed. 2003;42:3582. doi: 10.1002/anie.200300582.. Syntheses of Racemic 1: Fürstner A, Weintritt H. J Am Chem Soc. 1998;120:2817.Fürstner A, Gastner T, Weintritt H. J Org Chem. 1999;64:2361.Kim SH, Figueroa I, Fuchs PL. Tetrahedron Lett. 1997;38:2601.Mochizuki T, Itoh E, Shibata N, Nakatani S, Katoh T, Terashima S. Tetrahedron Lett. 1998;39:6911.Harrington P, Tius M. Org Lett. 1999;1:649. doi: 10.1021/ol990124k.Robertson J, Hatley RJD, Watkin DJ. J Chem Soc Perkin Trans 1. 2000:3389.. Asymmetric Syntheses: Bamford SJ, Luker T, Speckamp WN, Hiemstra H. Org Lett. 2000;2:1157. doi: 10.1021/ol005750s.Trost BM, Doherty GA. J Am Chem Soc. 2000;122:3801.Harrington PE, Tius MA. J Am Chem Soc. 2001;123:8509. doi: 10.1021/ja011242h.Occhiato EG, Prandi C, Ferrali A, Guarna A. J Org Chem. 2005;70:4542. doi: 10.1021/jo0504058.. Approaches to the Roseophilin Core: Salamone SG, Dudley GB. Org Lett. 2005;7:4443. doi: 10.1021/ol051730k.Song C, Knight DW, Whatton MA. Org Lett. 2006;8:163. doi: 10.1021/ol052683z.
- 5.For applications of different Nazarov cyclization strategies toward roseophilin, see references 4f, 4j, 4k and 4m.
- 6.(a) He W, Sun X, Frontier AJ. J Am Chem Soc. 2003;125:14728. doi: 10.1021/ja037910b. [DOI] [PubMed] [Google Scholar]; (b) Malona JA, Colbourne JM, Frontier AJ. Org Lett. 2006;8:5661. doi: 10.1021/ol062403v. [DOI] [PubMed] [Google Scholar]; (c) He W, Herrick IR, Atesin TA, Caruana PA, Kellenberger CA, Frontier AJ. J Am Chem Soc. 2008;130:1003. doi: 10.1021/ja077162g. [DOI] [PubMed] [Google Scholar]
- 7.(a) Padwa A, Kline DN, Perumattam J. Tetrahedron Lett. 1987;23:913. [Google Scholar]; (b) Padwa A, Chiacchio U, Kline DN, Perumattam J. J Org Chem. 1988;53:2238. [Google Scholar]
- 8.(a) Canterbury DP, Frontier AJ, Um JM, Cheong PHY, Goldfeld DA, Huhn RA, Houk KN. Org Lett. 2008;10:4597. doi: 10.1021/ol8019154. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Canterbury DP, Herrick IR, Um JM, Houk KN, Frontier AJ. Tetrahedron. doi: 10.1016/j.tet.2008.10.003. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Claus RE, Schreiber SL. Org Synth. 1986 [Google Scholar]; Coll. 7:168. [Google Scholar]
- 10.Song C, Knight DW, Whatton MA. Tetrahedron Lett. 2004;45:9573. [Google Scholar]
- 11.Lau CK, Dufresne C, Bélanger PC, Piétré S, Scheigetz J. J Org Chem. 1986;51:3038. [Google Scholar]
- 12.Masanori S, Mori K. Eur J Org Chem. 2001:503. [Google Scholar]
- 13.Xu RX, Anderson HJ, Gogan NJ, Loader CE, McDonald R. Tetrahedron Lett. 1981;22:4899. [Google Scholar]
- 14.Tietze LF, Kettschau G, Heitmann K. Synthesis. 1996:851. [Google Scholar]
- 15.For similar difficulties with the N-tosyl group, see Liu J, Yang Q, Mak TCW, Wong HNC. J Org Chem. 2000;65:3587. doi: 10.1021/jo9915224.
- 16.(a) Casadei MA, Galli C, Mandolini L. J Am Chem Soc. 1984;106:1051. [Google Scholar]; (b) Brillon D, Deslongchamps P. Can J Chem. 1987;65:56. [Google Scholar]
- 17.For a review, see Trost BM. Angew Chem Int Ed. 1989;28:1173.
- 18.Kitagawa Y, Itoh A, Hashimoto S, Yamamoto H, Nozaki H. J Am Chem Soc. 1977;99:3864. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures, characterization data, X-ray crystal structure coordinates and files for compound 21 in CIF fromat. This material is available free of charge via the Internet at http://pubs.acs.org.