

Abstract
In vivo imaging has revolutionized our understanding of biological processes in brain physiology and pathology. However, breathing-induced movement artifacts have impeded the application of this powerful tool in studies of the living spinal cord. Here we describe in detail a method to image stably and repetitively, using two-photon microscopy, the living spinal tissue in mice with dense fluorescent cells or axons, without the need for animal intubation or image post-processing. This simplified technique can greatly expand the application of in vivo imaging to study spinal cord injury, regeneration, physiology and disease.
Keywords: Spinal cord, Microglia, Axons, In vivo imaging, Two-photon microscopy
1. Introduction
In recent years, the advent of two-photon microscopy (Denk et al., 1990) and the generation of transgenic animals which express fluorescent proteins driven by tissue-specific promoters (Feng et al., 2000; Tsien, 1998) have allowed the direct observation of cells and their behaviors under both physiological and pathological conditions in vivo (Germain et al., 2006; Helmchen and Denk, 2005; Misgeld and Kerschensteiner, 2006; Svoboda and Yasuda, 2006). Imaging the living brain has revealed unexpected details of the dynamics and the functional properties of neuronal, astrocytic and microglial structures under physiological conditions as well as following stimulation or injury (Davalos et al., 2005; Grutzendler et al., 2002; Nimmerjahn et al., 2005; Svoboda et al., 1997; Trachtenberg et al., 2002; Wang et al., 2006). Moreover, the application of this technology in the brain has reshaped our understanding of the pathology of neurodegenerative diseases, such as Alzheimer's disease (Christie et al., 2001; Grutzendler and Gan, 2006; Takano et al., 2007; Tsai et al., 2004).
Conversely, limited work has been performed on imaging the living spinal cord. The close proximity of the animal's heart and lungs to the spinal column results in artifacts generated by the heartbeat and breathing movements, which significantly impede the acquisition of steady images from the spinal cord of anesthetized mice. As a result, imaging studies have been predominantly performed on ex vivo spinal cord slices (Kawakami et al., 2005). Recently a method to image single axons in the mouse spinal cord was described (Misgeld et al., 2007), which had previously been used to demonstrate single axon degeneration and regeneration for the first time in vivo (Kerschensteiner et al., 2005). However, this method requires a series of surgically invasive techniques, such as multiple serial laminectomies combined with a temperature-controlled perfusion system and animal intubation, as well as extensive image post-processing to remove out-of-focus frames and reconstruct aligned low-noise images (Misgeld et al., 2007). Moreover, this method requires the interruption of the animal's respiration during image acquisition and was performed on a transgenic line with sparsely labeled axons that made the imaging and reconstruction of a single axon per field of view possible in the living mouse (Kerschensteiner et al., 2005; Misgeld et al., 2007).
Here we describe a novel in vivo method to perform two-photon imaging of multiple axons, microglia and blood vessels in the mouse spinal cord with outstanding stability that generates images and/or timelapse movies that do not require image alignment. We expose a very small area of the spinal cord of an anesthetized mouse and subsequently immobilize its spinal column under the two-photon microscope. By combining a customized spinal stabilization device with a method of deep anesthesia, we significantly minimized respiratory-induced movements and performed in vivo imaging of fine cellular structures without animal intubation or respiratory control. By significantly reducing injury and bleeding we can maintain a stable physiological window over extended periods of time, and/or revisit and reimage the same area at later timepoints. Our technique allows the acquisition of high-resolution images that can be immediately visualized and interpreted, at a level of detail that permits the study of densely populated cellular processes and cell–cell interactions in the living mouse spinal cord.
2. Materials and methods
2.1. Animal surgery
The Cx3cr1GFP/+ transgenic line (Jung et al., 2000) (kindly provided by Dan Littman and Michael Dustin) and the YFP-H line (Feng et al., 2000) (kindly provided by Anthony Wynshaw-Boris) were used for the imaging of microglia and axons, respectively. Both transgenic lines are currently commercially available by the Jackson Laboratories. For imaging blood vessels mice were injected intravenously with a 3% rhodamine dextran solution (70 kDa, Invitrogen, Carlsbad, CA). Mice were housed at the University of California, San Diego animal facility and all experiments were approved by the University of California, San Diego Institutional Animal Care and Use Committee.
Adult transgenic mice were anesthetized intraperitoneally with 100 mg ketamine, 15 mg xylazine and 2.5 mg acepromazine per kg in 0.9% NaCl solution and were kept anesthetized for the duration of the imaging experiments with hourly injections of half this dose. For the experiments performed under urethane anesthesia animals were injected intraperitoneally at a dose of 1.5 g/kg of body weight. The back of the animal was shaved and swabbed with Betadine. A midline incision of the skin (∼1.5 cm in length) exposed the back musculature over the thoracic vertebrae. The paravertebral muscles at the desired level (T11) were carefully separated from the vertebral column and a laminectomy of a single vertebra exposed the spinal cord underneath as described (Zheng et al., 2003). Small pieces of Gelfoam absorbable gelatin sponges (Pharmacia, Pfizer Inc.) were used during the operation to control bleeding.
2.2. Stabilization of the spinal column
We made a spinal column stabilization device, by mounting the Narishige STS-A Compact Spinal Cord Clamps and the Narishige MA-6N head holding adaptor on a steel base plate that was cut to fit on the miscroscope's stage (Fig. 1A). The two Narishige parts were aligned on the plate so that the animal's head would be supported while its spinal column and its tail were clamped (Fig. 1B). After performing the laminectomy and exposing a small segment of the spinal cord, we placed two of the spinal clamps of the STS-A device along the anterior–posterior axis of the animal by making two very small incisions of the muscles on both sides of the spinal column (Fig. 1B). The two clamps were placed at an angle of ∼45° to allow enough space for lowering a water immersion lens over the exposed spinal cord (Fig. 1B). We placed the third clamp of the STS-A device at the base of the tail so the animal's entire body was suspended in the air for the duration of the imaging experiments (Fig. 1B). An animal suspension method has been previously used for immobilizing animals, such as cats (Frank and Fuortes, 1955) and rats (Beaumont and Gardiner, 2002) for recording from the spinal cord with microelectrodes. A small well of Gelseal (Amersham Biosciences Corp.) was built around the exposed spinal cord to facilitate the maintenance of the tissue in a drop of artificial cerebrospinal fluid (ACSF) and the immersion of the microscope lens in this solution for in vivo imaging.
Fig. 1.
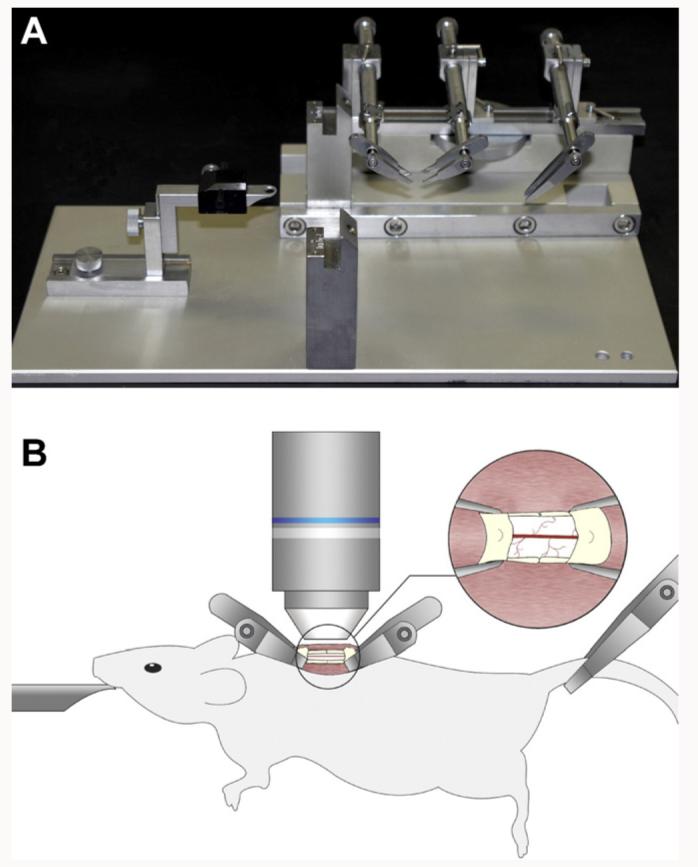
In vivo two-photon imaging of the stabilized mouse spinal cord. (A) A spinal stabilization device that could fit on a lowered microscope stage was built as shown here, using a steel base plate to support and align the STS-A Narishige compact spinal cord clamps and the MA-6N Narishige head holding adaptor. (B) Adult transgenic mice anesthetized with a KXA mix were positioned on the spinal stabilization device as shown here. A small retraction of the paravertebral muscles allowed the insertion of the fine tips of the clamping device and a laminectomy exposed the spinal cord. The entire device was placed in a temperature controlled chamber under the two-photon microscope and a heated water immersion lens dipped in a drop of ACSF was used for in vivo imaging of the fluorescently labeled spinal cord.
2.3. In vivo imaging of the mouse spinal cord
The mouse positioned on the stabilizing device was inserted in a heat-controlled chamber underneath a customized FV300 Olympus microscope and imaging was performed using an Olympus 40× 0.8NA water-immersion lens and a Spectra Physics Mai-Tai IR laser tuned at 920 nm for two-photon excitation of either GFP or YFP. To accommodate the extra space required for the stabilization device to fit under the microscope lens as shown in Fig. 1B, we removed the mounting of the FV300 Olympus microscope's stage and used metallic rods directly mounted on the air-table to support the stage at a lowered position, allowing approximately 10 cm of working distance between the stage and the objective lens. Moreover, in order to maintain the exposed tissue under physiological conditions, we kept the ACSF solution applied on the spinal cord at 37 °C, by using an objective lens heater (Bioptechs).
2.4. Repetitive imaging and post-operative care
For repetitive imaging we removed the mice from the stabilizing device and removed the Gelseal from the area around the laminectomy. We subsequently restored the back muscles and sutured them over the laminectomy. We applied neomycin in the form of powder (Neo-Predef, Pfizer Inc.) over the sutured muscles. The area was swabbed with Betadine and antibiotic powder was applied externally over the sutured skin. Mice received 1.5 ml Lactated Ringers solution (Baxter Healthcare) as a nutrient and hydration supplement, as well as analgesic treatment sub-cutaneously (0.1 mg/kg Buprenorphine, Buprenex, Reckit Benckiser Pharmaceuticals Inc.). Mice were placed on a heating pad until full recovery from anesthesia and were then individually housed. Additional antiseptic treatment was administered intra-peritoneally daily for the first 3–5 days after surgery (0.2 mg/kg Ampicillin, Abraxin Bioscience Inc., in Bacterio-static NaCl 0.9%, Abbott/Ross) and mice were monitored daily to ensure recovery. For re-imaging through the same laminectomy the sutured skin and muscles were reopened and mice were treated as described in Sections 2.1–2.3. Re-imaging was performed using the blood vasculature as a map as previously described (Grutzendler et al., 2002).
2.5. Image processing and quantification
Image acquisition was performed using the Olympus FluoView software. Subsequently, all z-stacks of images were projected along the z-axis to recreate two-dimensional representations of the three-dimensional structures within the imaged tissue. Supplementary movies 1 and 3 were generated by z-projections of stacks of images taken sequentially over time. To ensure full representation of the cells, in all z-stacks we set the upper and lower limits so that all processes of the cell bodies shown were included in the raw imaging data and in their subsequent z-projections. Supplementary movies 2 and 4 show a single z-plane of the imaged spinal cord scanned continuously for approximately 15 s at a rate of 2.8 frames per second. According to standard laser-scanning microscopy procedures and as we have previously shown (Davalos et al., 2005), during image acquisition laser power and detector gain and offset levels were set as to minimize background noise and partly saturate cell bodies so that cell processes can be visible and sharp. Post-acquisition, images and movies were only adjusted for brightness, contrast and background noise by using ImageJ, the software developed and freely distributed by the US National Institutes of Health. For brightness and contrast adjustments the depth of pixel intensities that spanned the entire 8-bit range (0–255) was readjusted for display optimization. The range, mean value and standard deviation of the pixel intensity distribution are mentioned in Figure and Supplementary Movie Legends.
To quantify image stability, we acquired timelapse images of microglial cell bodies in a single z-plane scanned at different frequencies ranging from 1 to 3 frames per second and wrote custom scripts in Matlab (The Mathworks, Inc.) to compute their x–y displacement over time. Briefly, small regions of interest (ROI) were drawn such that an individual cell soma was within the ROI throughout an entire time series. For each individual image in the time series, a threshold was set based on the mean pixel intensity plus two times the standard deviation of all pixels in the ROI. This thresholding method was used to ensure that a consistent segment of the soma was captured, independent of changes in pixel intensity as a function of time. Once the threshold was set, the center of mass (or ‘tcentroid’) of the thresholded pixels was computed on each time point, and the Euclidean distance between centroids of consecutive time points was tabulated. This distance represents the displacement in (x,y) of cell bodies in the image between consecutive frames acquired from a single z-plane. We calculated the average displacement for the entire time series for each cell and then plotted the mean displacement of all cells that were imaged per anesthetic group.
3. Results
3.1. Stable in vivo imaging of densely populated fluorescent microglia and blood vessels in the spinal cord without the need for image alignment using a custom stabilization device
In vivo imaging of microglia has only been performed in the brain using the Cx3cr1GFP/+ transgenic line, which expresses a high density of fluorescently labeled microglia and allows the study of their rapid process dynamics (Davalos et al., 2005; Nimmerjahn et al., 2005). The dense population of microglia and their rapid process movements would require an imaging technique of considerable stability to allow their study in the spinal cord in vivo. We reasoned that to stably image the living spinal cord, we would need to develop a technique that would minimize the breathing and heartbeat movements of the mouse and/or prevent them from being relayed to the animal's spinal column. As shown in Fig. 1, we developed a stabilization device that is compatible with the two-photon microscope and allows adequate space underneath the animal's body to accommodate breathing and heartbeat movements, which are thus not transferred to the imaged tissue. In addition, we used a ketamine–xylazine–acepromazine (KXA) anesthetic cocktail that minimizes the breathing movements of the animal and achieves a very calm yet steady breathing rhythm. Using this technique, we imaged the spinal cord of the Cx3cr1GFP/+ mouse and detailed the highly motile processes of GFP-labeled cells and their close interactions with the vasculature (Fig. 2A). We were able to project entire z-stacks of serial images acquired over minutes to hours and construct timelapse movies of the imaged tissue without the need for any image alignment within either the same or consecutive image stacks (Supplementary movie 1). Individual processes can be clearly seen changing shape, volume and length within minutes. While some microglial cell bodies and processes appear constantly attached to the vasculature, other processes extend and retract towards blood vessel walls as well as towards the spinal cord parenchyma (Supplementary movie 1). High magnification image shows a single microglial cell body attached to the wall of a blood vessel and its processes extended both around the vessel and towards the spinal cord parenchyma (Fig. 2B). Microglia appear to be in close proximity with both arteries and veins ranging from small capillaries to large vessels.
Fig. 2.

In vivo imaging of high-density microglial cells and blood vessels in the spinal cord of anesthetized mice. Projected but non-aligned z-stacks of (A) highly dense GFP-positive microglia (green) in the spinal cord of a CX3CR1GFP/+ mouse in close proximity with blood vessels (red, labeled with i.v. injection of rhodamine dextran). Pixel intensities for the green channel range from 0 to 255 with a mean value of 37.3 and standard deviation 65.5 and for the red channel from 0 to 255 with a mean value of 13.8 and standard deviation 25.5. (B) High magnification image labeled as in (A) of a single microglial cell attached to the wall of a blood vessel with processes extended around the vessel and towards the spinal cord parenchyma. Pixel intensities for the green channel range from 0 to 255 with a mean value of 35.6 and standard deviation 64.7 and for the red channel from 0 to 255 with a mean value of 34.7 and standard deviation 63.5. Scale bars, 10 μm.
Since the normal breathing rhythm of the mouse exceeds 100 breaths per minute (∼1.5–2 breaths per second), we additionally performed a fast scan of a single z-plane in the spinal cord of Cx3cr1GFP/+ mice at a rate of 2.8 frames per second. Supplementary movie 2 shows minimal to no movement between consecutive frames of the same z-plane using the stabilization device described in Fig. 1 and KXA anesthesia. Notably, the resolution and detail of imaging was similar to that achieved in our previous studies in the brain (Davalos et al., 2005).
3.2. Evaluation of the anesthetic mix in the imaging stability of the spinal cord
To evaluate the contribution of reduced breathing movement to image stability, we performed a direct comparison between KXA and urethane, which preserves vigorous breathing. Using urethane anesthesia under the same image acquisition parameters as in Supplementary movie 1, the majority of microglial processes appear blurry and the detail of their fine morphology very poor (Supplementary movie 3), when compared to KXA (Supplementary movie 1). A fast scan of a single z-plane at a rate of 2.8 frames per second using urethane anesthesia shows image instability between consecutive frames of the same z-plane (Supplementary movie 4). We also acquired 30 μm-deep z-stacks from the spinal cord of animals under KXA and urethane and subsequently projected them along the z-axis. At the level of a single z-stack, microglial processes appear blurred and distorted under urethane anesthesia (Fig. 3B), when compared to the fine detail of even high-order processes under KXA (Fig. 3A), using the same image acquisition parameters (scanning frequency, resolution, z-step, z-stack depth). To quantify the difference in stability between the two anesthetic regimens, we performed single z-plane scanning of 10–20 microglial cells from three animals per group at different scanning frequencies. Use of urethane resulted in a 2.4-fold increase of microglial cell displacement on the x–y plane scanned at one frame per second, when compared to KXA anesthesia (Fig. 3C, p < 0.0001). Similar results were observed at faster scanning frequencies of 2 or 3 frames per second (data not shown). Overall, these results suggest that KXA provides increased stability for in vivo imaging of the spinal cord using a custom-made stabilization device for two-photon microscopy.
Fig. 3.

Effect of anesthesia on image stability during in vivo spinal cord imaging. Z-stack projection of individual microglial cells in the spinal cord of mice anesthetized with KXA (A) or urethane (B). Both z-stacks were of the same depth of z-planes. Quantification shows a 2.4-fold increase in microglia cell body displacement along the x–y plane when mice are anesthetized with urethane, when compared to KXA (p < 0.0001 one-way ANOVA). Scale bar, 10 μm.
3.3. Repetitive imaging of the same axonal segments or microglia in the living spinal cord
To illustrate the ability to image axons repetitively, we used a mouse expressing YFP in a neuronal subpopulation driven by the Thy1 promoter (YFP H-Line) (Feng et al., 2000) and imaged its spinal cord with multiple labeled axons per field of view (Fig. 4A). Using the blood vasculature as a map, we relocated and reimaged the same axons in the spinal cord 5 days later (Fig. 4B). Even though the spinal cord of mice in the YFP-H line has a dense population of labeled ascending and descending axons, the stability of the acquired data on both days allowed us to identify the same axonal segments without the need to digitally reconstruct the raw images. Similarly, we were able to reimage microglia in the spinal cord of Cx3cr1GFP/+ mice. Some microglia could be identified at a similar position in the spinal cord, while others showed changes in their location and process extension (Fig. 4C and D). Overall, these results show that both axons and microglia can be reimaged in the spinal cord in vivo.
Fig. 4.
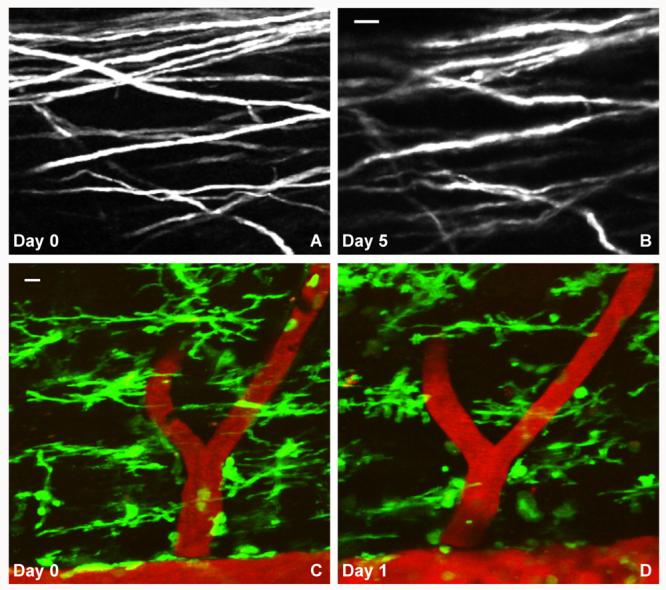
Repetitive in vivo imaging of high-density axons and microglia in the spinal cord of anesthetized mice. YFP-labeled – most likely dorsal column sensory – axons on days 0 (A) and 5 (B). Pixel intensities range from 0 to 255 with a mean value of 62.5 and standard deviation 78.4 (A), and a mean value of 46.9 and standard deviation 64.4 (B). Repetitive in vivo imaging of microglia and the vasculature (labeled as in Fig. 2) in the spinal cord of CX3CR1GFP/+ mice on days 0 (C) and 1 (D). Pixel intensities for the green channel range from 11 to 255 with a mean value of 56.7 and standard deviation 66.9 and for the red channel 18–255 with a mean value of 49.5 and standard deviation 46.4 (C). Pixel intensities for the green channel range from 10 to 253 with a mean value of 42.6 and standard deviation 54.1 and for the red channel 17–253 with a mean value of 59.3 and standard deviation 57.8 (D). Scale bars, 10 μm.
4. Discussion
We have developed a novel in vivo technique for two-photon microscopy that allows the direct, stable and repetitive imaging of densely populated cells and axons in the living spinal cord of transgenic mice. By using commercially available spinal clamps and a head mounting piece we custom-built a spinal stabilization device that allows breathing space underneath the mouse body. Using this stabilization scheme in combination with an anesthetic mix that reduces respiratory movements, we developed a technique that significantly improves the spinal column's stability under the two-photon microscope. Stability is achieved without the need for respiratory control and allows direct data acquisition from the raw images without image post-processing. This technique allows the detailed study of multiple axonal and glial processes, as well as the interactions of glial cells with the vasculature by in vivo imaging and can thereby provide functional information of cell–cell interactions in the living spinal cord.
Our method exposes a small segment of the living spinal cord, by performing a single laminectomy without perturbing the dura matter. This ensures the integrity of the imaged tissue underneath the meninges and minimizes the overall injury that is incurred to the animal's spine. If, however, a larger imaging window is required, our technique can accommodate the removal of 2–3 serial laminae and the acquisition of stable images from a broader area of the spinal cord (not shown). By building a wall of GelSeal around the spinal window the exposed spinal segment can be easily maintained in a small well of warm ACSF for in vivo imaging. Moreover, building a physical barrier between the exposed tissue and the paravertebral muscles keeps any residual bleeding from the retracted muscles away from the imaging solution and ensures the stability of the tissue throughout the imaging experiments. In the brain repetitive imaging of the Cx3cr1GFP/+ mouse-induced microglial activation as a response to the open skull surgery where the dura was covered with agarose and a coverglass (Xu et al., 2007). Our data show that our technique can be used to relocate and reimage microglia in the spinal cord 1 day later. Given that microglia are the prime responders to changes in their extracellular environment, repetitive imaging studies at later time points should take into consideration potential microglial responses to the laminectomy and/or the repetitive opening and closing of the wound. Overall, we have developed a technique that allows the application of two-photon microscopy, which can be used to image deeper in the living spinal cord, and allow the study of cellular structures and their changes over time by providing high-resolution images and/or timelapse movies.
In vivo imaging inherently presents with slight movement between sequential timepoints in timelapse movies, mainly due to the animal heartbeat that is present even under deep anesthesia. Subsequent alignment between shifted timepoints can easily produce a “perfectly steady” timelapse movie when necessary. The initial stability of the imaged tissue and the quality of the raw imaging data determines the extent of image post-processing required for data presentation and analysis. If the initial stability is low, timelapse image processing becomes far more complicated and time consuming, because alignment is required not only between shifted timepoints, but also within the individual stacks of each time point. The stability of our technique provides stacks of images for each time point that can be used without alignment to make timelapse movies. Indeed, to demonstrate the stability of the raw data presented here, we performed no image alignment at any step of making our figures and movies. The unavoidable residual shift between some frames of the timelapse movies 1 and 2 is comparable to that observed when imaging other living tissues, such as the brain through a thin skull or a craniotomy. With this very limited residual movement, our method generates raw in vivo data that can be studied and quantified without the need for respiratory control or image post-processing. We were able to use commercially available transgenic lines and image densely populated glial cells interacting with the vasculature over minutes to days, as well as multiple axonal segments in the spinal cord that we were able to relocate and reimage over a period of days. It is worth noting that like most in vivo methods, the results that can be obtained by using our in vivo imaging method may greatly depend on the use of proper anesthesia. A wide range of anesthetic mixes have been used in the literature for in vivo imaging studies, many of them using various ratios of ketamine, xylazine and acepromazine, including this particular KXA mix (Lindquist et al., 2004; Schwickert et al., 2007; Shakhar et al., 2005; Tadokoro et al., 2006). This KXA mix was selected solely based on its effect on breathing movements, and was indeed superior to the use of urethane for stable imaging of the spinal cord. Other anesthetic approaches may produce comparable results to KXA used in this study. The degree to which this particular KXA mix and/or the state of anesthesia can affect the physiological properties of the spinal tissue should be taken into account along with the specific requirements of a given study.
Our technique overcomes a long-standing hurdle in the field of in vivo imaging in the spinal cord, namely the strong movement artifacts generated by the breathing and heartbeat of the anesthetized mouse. As such, this technique has the potential to broaden the studies of cellular and molecular functions in the spinal cord in vivo, since it can accommodate sophisticated experimental designs, such as local injection of substances or electrophysiological recordings in the spinal cord while imaging. Moreover, application of our technique in different animal models of spinal cord injury can elucidate mechanisms of axonal degeneration and regeneration. In particular, simplified, repetitive in vivo imaging allows to unequivocally identify regenerating axons in the injured spinal cord, a major technical challenge in spinal injury and regeneration research. This technique can also allow the imaging of inflammatory and neurodegenerative processes in animal models of disease such as multiple sclerosis and amyotrophic lateral sclerosis, where the disease pathogenesis is prominent in the spinal cord. The availability of a reproducible and steady technique that allows the imaging of complex cellular interactions can be invaluable for spinal cord research, since it could facilitate the widespread application of this powerful tool in studies of the spinal cord in vivo.
Supplementary Material
Acknowledgements
We are grateful to Drs. Dan Littman and Michael Dustin for providing us with the Cx3cr1GFP/GFP mouse, Dr. Anthony Wynshaw-Boris for the YFP H-line, and Drs. Guy Shakhar and James Feramisco for experimental advice. We thank Shoana Sikorski, Xiaolin Tan and Adrienne Andres for technical assistance and John Lewis for the artwork. This work was supported by the NIH RR04050 to the National Center for Microscopy and Imaging Research at San Diego awarded to MHE, NIH/NINDS P30 NS047101 grant to the UCSD Neuroscience Microscopy Shared Facility, grants from the Roman Reed Spinal Cord Injury Research Fund of California, the Christopher Reeve Paralysis Foundation and the International Spinal Research Trust to BZ and the NMSS Research Grant RG3782, Christopher Reeve Foundation and Sam Schmidt Paralysis Foundation Grant AA2-0601-2, the Dana Foundation Program in Brain and Immuno-imaging and the NIH/NINDS grants NS051470 and NS052189 to KA.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.jneumeth.2007.11.011.
References
- Beaumont E, Gardiner P. Effects of daily spontaneous running on the electrophysiological properties of hindlimb motoneurones in rats. J Physiol. 2002;540:129–38. doi: 10.1113/jphysiol.2001.013084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie RH, Bacskai BJ, Zipfel WR, Williams RM, Kajdasz ST, Webb WW, et al. Growth arrest of individual senile plaques in a model of Alzheimer's disease observed by in vivo multiphoton microscopy. J Neurosci. 2001;21:858–64. doi: 10.1523/JNEUROSCI.21-03-00858.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–8. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73–6. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Frank K, Fuortes MG. Potentials recorded from the spinal cord with microelectrodes. J Physiol. 1955;130:625–54. doi: 10.1113/jphysiol.1955.sp005432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain RN, Miller MJ, Dustin ML, Nussenzweig MC. Dynamic imaging of the immune system: progress, pitfalls and promise. Nat Rev Immunol. 2006;6:497–507. doi: 10.1038/nri1884. [DOI] [PubMed] [Google Scholar]
- Grutzendler J, Gan WB. Two-photon imaging of synaptic plasticity and pathology in the living mouse brain. NeuroRx. 2006;3:489–96. doi: 10.1016/j.nurx.2006.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grutzendler J, Kasthuri N, Gan WB. Long-term dendritic spine stability in the adult cortex. Nature. 2002;420:812–6. doi: 10.1038/nature01276. [DOI] [PubMed] [Google Scholar]
- Helmchen F, Denk W. Deep tissue two-photon microscopy. Nat Methods. 2005;2:932–40. doi: 10.1038/nmeth818. [DOI] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, et al. Analysis of fractalkine receptor CX3CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–14. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami N, Nagerl UV, Odoardi F, Bonhoeffer T, Wekerle H, Flugel A. Live imaging of effector cell trafficking and autoantigen recognition within the unfolding autoimmune encephalomyelitis lesion. J Exp Med. 2005;201:1805–14. doi: 10.1084/jem.20050011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschensteiner M, Schwab ME, Lichtman JW, Misgeld T. In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat Med. 2005;11:572–7. doi: 10.1038/nm1229. [DOI] [PubMed] [Google Scholar]
- Lindquist RL, Shakhar G, Dudziak D, Wardemann H, Eisenreich T, Dustin ML, et al. Visualizing dendritic cell networks in vivo. Nat Immunol. 2004;5:1243–50. doi: 10.1038/ni1139. [DOI] [PubMed] [Google Scholar]
- Misgeld T, Kerschensteiner M. In vivo imaging of the diseased nervous system. Nat Rev Neurosci. 2006;7:449–63. doi: 10.1038/nrn1905. [DOI] [PubMed] [Google Scholar]
- Misgeld T, Nikic I, Kerschensteiner M. In vivo imaging of single axons in the mouse spinal cord. Nat Protoc. 2007;2:263–8. doi: 10.1038/nprot.2007.24. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–8. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Schwickert TA, Lindquist RL, Shakhar G, Livshits G, Skokos D, Kosco-Vilbois MH, et al. In vivo imaging of germinal centres reveals a dynamic open structure. Nature. 2007;446:83–7. doi: 10.1038/nature05573. [DOI] [PubMed] [Google Scholar]
- Shakhar G, Lindquist RL, Skokos D, Dudziak D, Huang JH, Nussenzweig MC, et al. Stable T cell-dendritic cell interactions precede the development of both tolerance and immunity in vivo. Nat Immunol. 2005;6:707–14. doi: 10.1038/ni1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda K, Yasuda R. Principles of two-photon excitation microscopy and its applications to neuroscience. Neuron. 2006;50:823–39. doi: 10.1016/j.neuron.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Svoboda K, Denk W, Kleinfeld D, Tank DW. In vivo dendritic calcium dynamics in neocortical pyramidal neurons. Nature. 1997;385:161–5. doi: 10.1038/385161a0. [DOI] [PubMed] [Google Scholar]
- Tadokoro CE, Shakhar G, Shen S, Ding Y, Lino AC, Maraver A, et al. Regulatory T cells inhibit stable contacts between CD4+ T cells and dendritic cells in vivo. J Exp Med. 2006;203:505–11. doi: 10.1084/jem.20050783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Han X, Deane R, Zlokovic B, Nedergaard M. Two-photon imaging of astrocytic Ca2+ signaling and the microvasculature in experimental mice models of Alzheimer's disease. Ann N Y Acad Sci. 2007;1097:40–50. doi: 10.1196/annals.1379.004. [DOI] [PubMed] [Google Scholar]
- Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, et al. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 2002;420:788–94. doi: 10.1038/nature01273. [DOI] [PubMed] [Google Scholar]
- Tsai J, Grutzendler J, Duff K, Gan WB. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat Neurosci. 2004;7:1181–3. doi: 10.1038/nn1335. [DOI] [PubMed] [Google Scholar]
- Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–44. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- Wang X, Lou N, Xu Q, Tian GF, Peng WG, Han X, et al. Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nat Neurosci. 2006;9:816–23. doi: 10.1038/nn1703. [DOI] [PubMed] [Google Scholar]
- Xu HT, Pan F, Yang G, Gan WB. Choice of cranial window type for in vivo imaging affects dendritic spine turnover in the cortex. Nat Neurosci. 2007;10:549–51. doi: 10.1038/nn1883. [DOI] [PubMed] [Google Scholar]
- Zheng B, Ho C, Li S, Keirstead H, Steward O, Tessier-Lavigne M. Lack of enhanced spinal regeneration in Nogo-deficient mice. Neuron. 2003;38:213–24. doi: 10.1016/s0896-6273(03)00225-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.