


Abstract
A simple isothermal nucleic-acid amplification reaction, primer generation–rolling circle amplification (PG–RCA), was developed to detect specific nucleic-acid sequences of sample DNA. This amplification method is achievable at a constant temperature (e.g. 60°C) simply by mixing circular single-stranded DNA probe, DNA polymerase and nicking enzyme. Unlike conventional nucleic-acid amplification reactions such as polymerase chain reaction (PCR), this reaction does not require exogenous primers, which often cause primer dimerization or non-specific amplification. Instead, ‘primers’ are generated and accumulated during the reaction. The circular probe carries only two sequences: (i) a hybridization sequence to the sample DNA and (ii) a recognition sequence of the nicking enzyme. In PG–RCA, the circular probe first hybridizes with the sample DNA, and then a cascade reaction of linear rolling circle amplification and nicking reactions takes place. In contrast with conventional linear rolling circle amplification, the signal amplification is in an exponential mode since many copies of ‘primers’ are successively produced by multiple nicking reactions. Under the optimized condition, we obtained a remarkable sensitivity of 84.5 ymol (50.7 molecules) of synthetic sample DNA and 0.163 pg (∼60 molecules) of genomic DNA from Listeria monocytogenes, indicating strong applicability of PG–RCA to various molecular diagnostic assays.
INTRODUCTION
The invention of polymerase chain reaction (PCR) notably changed biological research and diagnostics, since specific DNA sequence in genome can be amplified and detected with enormous sensitivity and specificity (1). Its applications to DNA cloning, genetic fingerprints, in vitro diagnostics and others have been already well documented (2). However, PCR requires precise control of temperature cycling for successful DNA amplification, and the resultant instrumental restraint has been hampering its wider and more versatile applications (e.g. point-of-care use in hospitals and miniaturized systems for high-throughput analysis).
Accordingly, several isothermal amplification methods have been developed, which are free from these problems. Rolling circle amplification (RCA) utilizes circular single-stranded DNA probes as everlasting templates for DNA polymerization. Among several modes of RCA proposed (3–7), the following two are especially well known. In linear rolling circle amplification (LRCA), circular probes are combined with sense primers, and long concatenated sequence copies of the circular probes are produced by DNA polymerase (3,4). No temperature cycling is required, however the signal amplification of LRCA is inherently linear and is not necessarily satisfactory. On the other hand, hyper-branched RCA (also known as ramification or cascade RCA) shows exponential signal amplification under isothermal conditions (5,6). Here, circular probes (detection targets) are combined with a pair of sense and antisense primers and enormous amplification can be achieved. However, in either modes of RCA, in order to detect internal nucleic-acid sequences, circular probes must be obtained through circularization of padlock probes in separate reactions and it is difficult to conduct both reactions simultaneously (8). Also, there has been a controversy that padlock probes topologically linked on long linear single-stranded DNA might inhibit RCA (9,10).
In this article, we developed a novel mode of RCA, dubbed primer generation–RCA (PG–RCA), in which nucleic-acid sequences of sample DNA are detected with high sensitivity and wide dynamic range. One of the distinctive advantages over conventional nucleic-acid amplification technologies is being free from troublesome design and usage of exogenous primers since ‘primers’ are generated successively during the reaction. By simple design of circular probe and addition of nicking enzyme, conventional linear RCA was successfully converted to an exponential amplification mode without complicated topological factors. A remarkably high sensitivity and wide dynamic range, which are comparable to those of PCR, were obtained under isothermal conditions. Detection capability of low copy number of nucleic-acid sequences allows diverse applications of PG–RCA in several various molecular diagnostic assays.
MATERIALS AND METHODS
Reagents
Oligonucleotides were purchased from Integrated DNA Technologies or Sigma Genosys. Vent (exo-) DNA polymerase, Nb.BsmI, BsmI, BbvCI and exonuclease III were purchased from New England Biolabs. Circligase ssDNA ligase and exonuclease I were from Epicentre Biotechnologies.
Preparation of circular probes
Circular probe I, II or LM was prepared by self ligation of 20 µM circular probe precursor I, II or LM, respectively (Table 1). The self ligation reaction was conducted at 60°C for 3 h in a 20 µl reaction containing 50 mM MOPS buffer (pH 7.5), 10 mM KCl, 5 mM MgCl2, 2.5 mM MnCl2, 50 µM ATP, 1 mM DTT and 200 U Circligase ssDNA ligase. Circularized probes were treated with 10 U exonuclease I and 100 U exonuclease III at 37°C overnight and purified twice with 15% or 20% polyacrylamide/7 M urea gel electrophoresis.
Table 1.
Oligonucleotide sequences used in PG–RCA
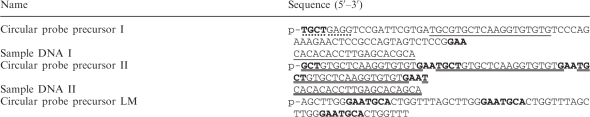 |
‘p-’ indicates a 5′ phosphate modification. Solid underline and double solid underline indicate hybridization sequences between circular probes and their corresponding sample DNAs. Bold characters indicate recognition sequences of Nb.BsmI and BsmI, and perforated underline indicates that of BbvCI.
PG–RCA
Unless specified, PG–RCA was conducted at 60°C in 10 µl reaction containing 20 mM Tris–HCl buffer (pH 8.8), 10 mM (NH4)2SO4, 10 mM KCl, 6 mM MgSO4, 400 µM each dNTP, 0.1% Triton X-100, 1/100 000 diluted SYBR Green I (Invitrogen), 7.5 nM circular probe, 0.4 U Vent (exo-) DNA polymerase, 1 U Nb.BsmI and a different dose of corresponding sample DNA with overlay of 10 µl mineral oil. PG–RCA product was analyzed by 1.0% agarose gel electrophoresis with SYBR Gold (Invitrogen) staining. For real-time detection, fluorescent intensity of each reaction was monitored in iCycler real-time PCR instrument (Bio-rad) with excitation at 490 nm and emission at 530 nm. Threshold time (TT) was estimated from the reaction time when the fluorescent intensity of the reaction exceeds an arbitrary threshold, which was set right above the background fluorescent intensity.
Restriction enzyme digestion analysis of PG–RCA product
PG–RCA products using circular probe I were digested with either BsmI or BbvCI to confirm that the products are the concatenated sequence copies of the circular probe. Enzymatic digestion was conducted by 2 U BsmI at 65°C or 0.4 U BbvCI at 37°C in 20 µl reaction containing 10 mM Tris–HCl buffer (pH 8.8), 50 mM NaCl, 10 mM MgCl2, 1 mM dithiothreitol (DTT) and 500 nM digestion adaptor (5′-TCCGGAATGCTGAGGTCCG-3′). The digested products were analyzed by 1.5% agarose electrophoresis with SYBR Gold staining.
Genomic DNA samples
Listeria monocytogenes (ATCC no. 19115), Listeria innocua (ATCC no. BAA-680), Escherichia coli (ATCC no. 11775) and Salmonella enterica (ATCC no. 14028) were cultured in 5 ml of brain heart infusion broth at 37°C overnight. Genomic DNA from each culture was purified using ZR Fungal/Bacterial DNA Kit (Zymo Research) following the manufacture's protocol and quantified by measuring the optical density at 260 nm with DU7400 spectrophotometer (Beckman Coulter).
PG–RCA detection of L. monocytogenes genomic DNA
Prior to PG–RCA reaction, genomic DNA samples were heat denatured at 95°C for 2 min in 5 µl reaction containing 15 nM circular probe LM, 20 mM Tris–HCl buffer (pH 8.8), 10 mM (NH4)2SO4, 10 mM KCl, 6 mM MgSO4, 0.1% Triton X-100 and 1/10 000 diluted SYBR Green I (Invitrogen). PG–RCA reaction was conducted at 60°C by adding 5 µl of enzyme mix containing 20 mM Tris–HCl buffer (pH 8.8), 10 mM (NH4)2SO4, 10 mM KCl, 6 mM MgSO4, 0.1% Triton X-100, 1/10 000 diluted SYBR Green I (Invitrogen), 8 µM each dNTP, 0.05 U Vent (exo-) DNA polymerase and 1 U Nb.BsmI to the denatured genomic DNA samples. The fluorescent intensity of each reaction was monitored in iQ5 real-time PCR instrument (Bio-rad) with a SYBR Green filter set. TT was estimated from the reaction time when the fluorescent intensity of the reaction exceeds an arbitrary threshold, which was set right above the background fluorescent intensity.
RESULTS
PG–RCA
This method allows sensitive detection of specific 3′-end sequences of sample DNA, using circular single-stranded DNA probes as signal amplification tools. The circular probe is required to carry only two sequences for signal amplification: a hybridization sequence to the specific 3′-end sequence of sample DNA and a complementary sequence of a nicking site.
PG–RCA initiates from hybridization of a circular probe to a sample DNA, and is followed by a cascade reaction of LRCA and nicking reaction (Figure 1). Under the same reaction condition, LRCA reaction is designed to produce long concatenated sequence copy of the circular probe and nicking reaction to generate multiple ‘primers’ for the circular probe from the LRCA product, therefore those reactions continuously initiate each other until some of the reaction components, most likely dNTP substrates, are depleted. Also, as multiple reaction cycles can be initiated from a single cycle, PG–RCA reaction accumulates LRCA products and ‘primers’ in an exponential manner over a time.
Figure 1.

Reaction mechanism of PG–RCA. PG–RCA initiates from hybridization between a sample DNA and a circular probe (Step 1). Once a sample DNA and circular probe form a complex, DNA polymerase synthesizes a long concatenated sequence copy of the circular probe through linear rolling circle amplification (LRCA) (Step 2). Then, multiple circular probes hybridize to multiple sites of the LRCA product, and nicking enzyme recognition sequences of the LRCA product are activated by double strand formation (Step 3). Nicking enzyme recognizes and cleaves the recognition sequences and produces multiple complexes of a ‘primer’ and circular probe (Step 4). Each complex can initiate the next round of PG–RCA (go back to Step 2).
PG–RCA was demonstrated using Vent (exo-) DNA polymerase and thermostable strand specific nicking enzyme, Nb.BsmI. Circular probe I used in the reaction is a 72-base single-strand sequence and contains a complementary sequence to a 19-base specific 3′-end sequence (sample DNA I) and a nicking site for Nb.BsmI (Table 1). The reaction was conducted at 60°C for up to 120 min and the product was analyzed by agarose gel electrophoresis (Figure 2A). With 500 amol sample DNA, smear and ladder DNA products were observed together over time, which could be LRCA products and its nicked fragments (‘primers’), respectively. On the other hand, we did not observe such products without sample DNA, however smear and ladder products were observed as background amplification in a prolonged incubation, which will be discussed later.
Figure 2.

Endpoint product analysis of PG–RCA. (A) Endpoint products in the presence or absence of 500 amol sample DNA were analyzed on 1.0% agarose gel electrophoresis. The reaction was conducted at 60°C for 30, 45, 60, 75, 90, 105 and 120 min and each reaction was analyzed separately. Lane M was loaded with a 100 bp DNA marker (100, 200, 300, 400, 500/517, 600, 700, 800, 900, 1000, 1200 and 1517 bp from the bottom). (B) Endpoint products of 90-min reactions with all or partial reaction components were analyzed on 1.0% agarose gel electrophoresis. DNA, POL and NICK indicate sample DNA, Vent (exo-) DNA polymerase and Nb.BsmI, respectively. Lane M is a 100 bp DNA marker. (C) Endpoint product of a 120-min PG–RCA reaction was digested with either BsmI or BbvCI and analyzed on 1.5% agarose gel electrophoresis. Lane ‘-’ was loaded with the 120 min reaction product before restriction enzyme digestion. Lane M is a 100 bp DNA marker.
In order to confirm whether both DNA polymerase and nicking enzyme are required for PG–RCA, the reaction was conducted without either of the enzymes (Figure 2B). Agarose gel analysis of the reaction shows that PG–RCA products were observed only when both enzymes exist with sample DNA as expected from the reaction mechanism (Figure 1). When the reaction mixture contains only DNA polymerase, the reaction is expected to work as LRCA (linear amplification) and very little amount of product was observed here. Its amplification efficiency is not as great as PG–RCA and its product is not abundant enough for SYBR gold staining used in the agarose gel analysis. This data suggests that simple addition of nicking enzyme to LRCA can convert linear amplification in LRCA to exponential amplification in PG–RCA and enhance the amplification efficiency to a great extent.
Furthermore, the PG–RCA product was digested by either of two restriction enzymes, BsmI and BbvCI, in order to confirm that the product was the expected concatenated sequence copy of circular probe I (Figure 3C). BsmI recognizes the same 7-base sequence as Nb.BsmI, which is used in PG–RCA, while BbvCI recognizes a different 7-base sequence that is only found in the circular probe I (Table 1). As both enzymes do not recognize single-strand DNA, excess amount of a 19-base oligonucleotide complementary to those nicking sites was supplemented during the digestion reaction. PG–RCA product was successfully digested into a single small fragment whether BsmI or BbvCI was used. The small fragment was further analyzed by 15% denaturing polyacrylamide gel analysis and found to be the same size as a linear 72-base circular probe precursor I (data not shown). These data indicate that PG–RCA product is a concatenated sequence copy of the circular probe and we concluded that PG–RCA is working as shown in Figure 1.
Figure 3.

Real-time product analysis of PG–RCA. (A) Fluorescent intensity of PG–RCA reaction was monitored in real time. PG–RCA was conducted at 60°C with 400 µM each dNTP, 0.4 U Vent (exo-) DNA polymerase, 1 U Nb.BsmI and 7.5 nM circular probe II as described in Materials and methods section. Sample DNA concentration in each reaction was prepared by 10-fold serial dilution from 500 amol to 0.5 zmol, and their signal amplification curves were indicated by colored lines (blue, light blue, purple, dark green, green, brown and orange, respectively) (n=3). Negative controls are indicated by black lines (n=3). (B) Threshold time TT (the reaction time when fluorescent intensity of each reaction exceeds a threshold, indicated by a perforated line in Figure 3A) was plotted against the sample DNA concentration (S) of the reaction. Solid line indicates linear least squares fitting between 500 and 0.5 amol sample DNA and its formulation is TT=−8.65 log10(S)+28.5 (R2=0.997). Perforated line indicates average TT value of the negative controls. Limit of detection is 58.4 zmol or 3.50 × 104 molecules of sample DNA by calculation from the intersection of both lines.
One of the ways to improve the amplification efficiency of PG–RCA further is to increase the efficiency of LRCA and nicking reaction in each reaction cycle. To do that, we designed circular probe II and increased the number of sample DNA hybridization sequences and nicking enzyme recognition sequences to three per circular probe (Table 1). The PG–RCA product with circular probe II contained mainly a smear DNA product and very little ladder product on 1.0% agarose gel analysis (data not shown). This is probably because the LRCA product from circular probe II has nicking sites every 22 bases and even the ladder products could look like smear. Instead, it was confirmed that the product contained a ladder product ranging from ∼22 bases, which is the expected smallest ‘primer’, on 15% denaturing polyacrylamide gel analysis (data not shown).
Real-time analysis of PG–RCA
PG–RCA reaction was monitored in real time by measuring the fluorescent intensity of the reaction reagent using circular probe II (Figure 3A). The fluorescent intensity in each reaction remained rather constant in early stage of the reaction but abruptly increased at a certain reaction time due to the accumulation of the PG–RCA product. These fluorescent intensity–reaction time curves synchronized very well among triplicate experiments containing the same amount of sample DNA. Furthermore, the abrupt signal increases appeared earlier when the reaction contained larger amount of sample DNA. In order to quantitatively analyze PG–RCA reactions, threshold time (TT: the time when the fluorescent intensity exceeds an arbitrary threshold), was defined in analogy to threshold cycle (CT) in real-time PCR analysis (11), and plotted against the amount of sample DNA in log scale (Figure 3B). We obtained a good linearity between 500 and 0.5 amol sample DNA or four orders of magnitude in linear least squares fitting. PG–RCA accumulated its products exponentially, and the slope of the fitting indicated that PG–RCA products increased by 10-fold every 8.7 min. The detection limit of PG–RCA was 58.4 zmol or 3.50 × 104 molecules of sample DNA by calculating from the intersection of the fitting and average TT value of the negative controls, which showed background signal amplifications without sample DNA.
Suppression of background signal amplification and improvement of detection limit
The negative control reactions containing only circular DNA probe, enzymes and dNTP substrates provided similar products to those of the positive control reactions (Figure 3). Apparently, the cascade reaction cycle of PG–RCA was initiated even without sample DNA or any 3′-end sequences. This false reaction initiation was as efficient as the reaction initiation by 58.3 zmol sample DNA (the detection limit described above).
In order to improve the limit of detection further, it is necessary to suppress the background signal amplification of PG–RCA. Several concentrations of dNTP substrates, Vent (exo-) DNA polymerase and nicking enzyme, Nb.BsmI, were compared in the presence or absence of 500 zmol sample DNA (Figure 4). It was found that decreasing the concentration of dNTP or Vent (exo-) DNA polymerase could increase the TT values of the negative controls more significantly than those with 500 zmol sample DNA and improve the limit of detection (Figures 4A and 4B). At 400 µM dNTP (Figure 4A) or 0.4 U Vent (exo-) DNA polymerase (Figure 4B), the TT values of the positive and negative controls were almost identical. On the other hand, at reduced concentrations of dNTP substrates (0.4 µM in Figure 4A) or Vent (exo-) DNA polymerase (0.02 U in Figure 4B), the TT value of the positive control was clearly smaller than that of the negative control, suggesting that the false reaction initiation in the negative control may be related with dNTP substrates and DNA polymerase. However, changing concentration of Nb.BsmI did not change the TT values of the positive and negative controls at all (Figure 4C). The reaction conditions were further optimized to improve the detection limit of PG–RCA (Figure 4D). At 0.4 µM dNTP, 1000-fold lower concentration than the original condition, Vent (exo-) DNA polymerase concentration was also reduced to differentiate the positive and negative controls even better, and we obtained the current optimized reaction condition of PG–RCA: 0.4 µM dNTP and 0.05 U Vent (exo-) DNA polymerase.
Figure 4.

Optimization of reaction conditions for PG–RCA. Threshold time in the presence (open circle) or absence (cross) of 500 zmol sample DNA was compared at various concentrations of each reaction component (n=2). (A) At constant concentrations of Vent (exo-) DNA polymerase (0.4 U), Nb.BsmI (1 U) and circular probe II (7.5 nM), dNTP concentrations were compared among 400 (original), 40, 4 and 0.4 µM. (B) At constant concentrations of dNTP (400 µM), Nb.BsmI (1 U) and circular probe II (7.5 nM), Vent (exo-) polymerase concentrations were compared among 0.4 (original), 0.08, 0.04 and 0.02 U. (C) At constant concentrations of dNTP (400 µM), Vent (exo-) DNA polymerase (0.4 U) and circular probe II (7.5 nM), Nb.BsmI concentrations were compared among 1 (original), 0.5, 0.25 and 0.125 U. (D) At constant concentrations of dNTP (0.4 µM, 1000-fold dilution of the original concentration), Nb.BsmI (1 U) and circular probe II (7.5 nM), Vent (exo-) polymerase concentrations were compared among 0.4, 0.2, 0.1 and 0.05 U.
Under the optimized reaction condition, the fluorescent intensities of PG–RCA reactions with different concentrations of sample DNA were monitored in real time and the TT value of each reaction was plotted against the amount of the sample DNA in log scale (Figure 5). The background signal amplifications were suppressed under this reaction condition in comparison to the original condition, therefore the signal amplification curves with low concentrations of sample DNA were clearly separated from the background amplifications. A fair linearity was obtained in seven orders of magnitude of sample DNA concentration (from 5 fmol to 0.5 zmol). It is noteworthy that the detection limit was now as low as 84.5 ymol (50.7 molecules) of sample DNA (see the intersection of the two straight lines in Figure 5B). Although the amplification efficiency decreased because of lower dNTP and DNA polymerase concentrations, PG–RCA can still accumulate its products by 10-fold every 19.2 min and the whole reaction can be completed within 150 min.
Figure 5.

PG–RCA under optimized reaction condition. (A) Fluorescent intensity of PG–RCA reaction at the optimized reaction condition (0.4 µM dNTP, 0.05 U Vent (exo-) DNA polymerase, 1 U Nb.BsmI and 7.5 nM circular probe II) was monitored in real time. Sample DNA concentration in each reaction was prepared by 10-fold serial dilution from 5 fmol to 0.5 ymol and their signal amplification curves were indicated by colored lines (dark blue, blue, light blue, purple, dark green, green, brown, orange, yellow, red and gray, respectively) (n=2). Negative controls are indicated by black lines (n=2). (B) Threshold time (TT) was plotted against the sample DNA concentration (S) of the reaction. Solid line indicates linear least squares fitting between 5 fmol and 0.5 zmol sample DNA and its formulation is TT=−19.2 log10(S)+75.6 (R2=0.998). Perforated line indicates average TT value of the negative controls. Limit of detection is 84.5 ymol or 50.7 molecules of sample DNA by calculation from the intersection of both lines.
Initiation mechanism of background signal amplification
The false reaction initiation in the negative control could be related with dNTP substrates and DNA polymerase, and not with the nicking enzyme (Figures 4A–4C). In order to investigate how the background signal amplification initiates further, negative control PG–RCA reactions were conducted with dNTP substrates pre-incubated in the presence or absence of circular probe together with DNA polymerase, nicking enzyme or no enzyme, and it was investigated which combination affects the background signal amplification by comparing the TT value of each PG–RCA reaction (Supplementary Figure S1).
In the absence of circular probe, the dNTP substrates pre-incubated with DNA polymerase increased the background signal amplification significantly, however those pre-incubated with nicking enzyme or by itself did not change the background level at all. This data clearly confirmed that dNTP and DNA polymerase caused the false initiation of PG–RCA. On the other hand, in the presence of circular probe, the dNTP substrates pre-incubated with DNA polymerase did not much affect the background level as well as the other samples, suggesting that the circular probe is not involved with the false initiation of PG–RCA.
Detection and quantification of bacterial genomic DNA
PG–RCA was applied to detect genomic DNA from foodborne pathogen, L. monocytogenes. Circular probe LM was designed to detect a complementary strand of its virulence gene, hly (GeneBank GeneID 2797098), encoding a cholesterol-dependent cytolysin, listeriolysin O (LLO) (Figure 6A) (12). Circular probe LM contains three 26-base sequences complementary to the gene including a nicking site for Nb.BsmI. When this circular probe hybridizes to the gene, Nb.BsmI can nick the genomic DNA and generate a 3′-end sequence to initiate the first cycle of PG–RCA. BLAST (Basic Local Alignment Search Tool, http://blast.ncbi.nlm.nih.gov) search of the 26-base target sequence against L. innocua, E. coli and S. enterica genome showed no exact match and 13-base partial match (including a nicking site for Nb.BsmI) in E. coli genome at maximum.
Figure 6.

Real-time quantification of hly gene in L. monocytogenes genomic DNA by PG–RCA. (A) Circular probe LM for detection of pathogenic L. monocytogenes genomic DNA. The probe targets the complementary strand of virulence gene, hly (GeneBank GeneID 2797098), encoding a cholesterol-dependent cytolysin, listeriolysin O (LLO). Circular probe LM contains three repeats of a 26-base sequence complementary to the gene including a nicking site for Nb.BsmI. Since these repeat sequences have 5-base overlaps each other, the circular probe comprises three repeats of a 21-base sequence (red, blue and green). (B) Genomic DNA from L. monocytogenes (0.1–100 pg) was analyzed by real-time PG–RCA with circular probe LM. Threshold time (TT) was plotted against the L. monocytogenes genomic DNA concentration (S) of the reaction. Solid line indicates linear least squares fitting between 0.1 and 100 pg L. monocytogenes genomic DNA and its formulation is TT = −19.1 log10(S) + 233 (R2=0.964). Perforated line indicates average TT value of the negative controls (n=2). Limit of detection is 0.163 pg (∼60 molecules) of L. monocytogenes genomic DNA by calculation from the intersection of both lines. (C) Genomic DNA (100 pg) from L. monocytogenes, L. innocua, E. coli and S. enterica were analyzed by real-time PG–RCA with circular probe LM and their threshold times were compared with the values for L. monocytogenes (100 pg). ‘No DNA’ indicates the negative controls.
A quantity of 0.1–100 pg of L. monocytogenes genomic DNA was analyzed by real-time PG–RCA at the optimized condition determined above, except using circular probe LM and the initial incubation of the genomic DNA with this circular probe at 95°C for 2 min. Threshold times were determined as described in Figures 3 and 5, and a clear linearity between the threshold time and genomic DNA concentration was observed (Figure 6B). From the intersection of the two straight lines in Figure 6B, the detection limit of this assay is 0.163 pg (∼60 molecules) of L. monocytogenes genomic DNA. For the purpose of comparison, 100 pg of genomic DNA samples from L. innocua, E. coli and S. enterica were analyzed by PG–RCA using the circular probe LM. Their threshold times were much larger than that for 100 pg of L. monocytogenes genomic DNA and close to the negative controls or background amplifications of PG–RCA (Figure 6C), indicating that circular probe LM specifically recognizes the hly gene sequence of L. monocytogenes genome to initiate PG–RCA.
DISCUSSION
By simple design of circular probe and addition of nicking enzyme, conventional linear RCA (LRCA) was successfully converted to an exponential amplification. PG–RCA can detect specific 3′-end sequences just like LRCA, however PG–RCA works in an exponential reaction mechanism and is much more sensitive than LRCA. After optimizing the reaction condition, we obtained a great sensitivity with detection limit of 84.5 ymol or 50.7 molecules of synthetic sample DNA and a large dynamic range of seven orders of magnitude, which is comparable to PCR and hyper-branched RCA. Furthermore, PG–RCA is applicable to detection of internal DNA sequences containing nicking sites without compromising its sensitivity, and it was demonstrated that PG–RCA can detect as low as 0.163 pg (∼60 molecules) of genomic DNA from L. monocytogenes and distinguish clearly L. monocytogenes genomic DNA from other bacterial DNA.
The background signal amplification of PG–RCA was successfully suppressed by decreasing dNTP and DNA polymerase concentrations and the reaction sensitivity was improved by almost 700-fold from the original reaction condition, which is similar concentrations of dNTP and DNA polymerase to other nucleic-acid amplification reactions such as PCR and hyper-branched RCA (Figures 3 and 5). We also confirmed that magnesium concentration affect the intensity of the background signal amplification (the optimum concentration: 4–6 mM MgSO4), presumably affecting the polymerase activity although Nb.BsmI also requires magnesium ions as cofactors. Different reaction temperatures, DNA polymerase inhibitors such as acyclonucleotide, triphophosphatesnucleotide triphosphates (NTPs), bicalin, genistin, N-ethylmaleimide and reaction supplements such as single-strand binding protein (SSB), betaine and dimethyl sulfoxide (DMSO) were also tested to suppress the background signal amplification, however any of them did not selectively suppress the background signals of PG–RCA.
The false initiation of PG–RCA could be related with primer/template-independent polymerization, which has been reported for several thermophilic DNA polymerases including Vent (exo-) DNA polymerase (13,14). In one of the proposed reaction mechanisms, it is considered that DNA polymerase synthesizes short oligonucleotides from dNTP substrates without primer or template, and elongates palindrome sequences in the early oligonucleotide products to large DNA products by repeating self priming and strand displacement extension (15). Also, it was reported that the reaction could be further accelerated by thermophilic DNA restriction enzymes (16). We observed that primer/template-independent polymerization or the dNTP substrates incubated with DNA polymerase increased the false initiation of PG–RCA (Supplementary Figure S1). However, we ruled out the possibility that primer/template-independent polymerization is the background signal amplification itself because we did not observe any signal amplification at the optimized reaction condition, i.e. 0.4 µM dNTP and 0.05 U Vent (exo-) DNA polymerase, even after 10 h reaction without circular probes. Therefore, we propose the following mechanism for the false initiation of PG–RCA: first, random short oligonucleotides are generated from dNTP substrates by primer/template-independent polymerization. Then, 3′-ends of the oligonucleotides hybridize to circular probes non-specifically. DNA polymerase extends the 3′-ends on the circular probes and initiates the reaction cycle of PG–RCA without sample DNA (Supplementary Figure S2).
It is difficult to specify which reaction step dominates the overall PG–RCA kinetics because of the complexity of the reaction mechanism, however it is possible to deduce which reaction factor is involved more significantly than the others by comparing the reaction kinetics at different reaction parameters. Reduction of dNTP and DNA polymerase concentrations significantly increased the TT values of the positive control reactions or decreased those reaction speeds (Figure 4A and 4B), while that of nicking enzyme did not affect the reaction speeds at all (Figure 4C). Therefore, at the reaction conditions we tested, the linear RCA reaction may control the reaction speed of PG–RCA more significantly than the nicking reaction. We also speculate that circular probe concentration, which controls hybridization speed of circular probes to LRCA products, affects the reaction kinetics significantly as the reaction speed was maximum around 7.5–15 nM circular probe and the reaction speed decreased out of this range (data not shown).
L. monocytogenes genomic DNA detection using circular probe LM indicated that a short 26-base sequence with nicking enzyme recognition is specific enough to distinguish its hly gene sequence from other bacterial genomic DNA. Thus, PG–RCA can target relatively short nucleotide regions as long as those include nicking sites, and should be advantageous especially when only limited nucleotide regions are available for detection such as heavily damaged genomic DNA samples.
PG–RCA with circular probe LM required longer incubation than that with circular probe II, however amplification efficiency was similar by judging from the slopes of the least square fittings (−19.1 and −19.2, respectively) (Figures 5B and 6B). This may be because circular probe LM may have lower background amplification than circular probe II, while genomic DNA was less efficient to initiate PG–RCA than synthetic sample DNA.
CONCLUSION
PG–RCA is useful to detect specific nucleic-acid sequences sensitively under isothermal conditions. Compared with PCR, no requirement of precise control of thermal cycling (thus no need of sophisticated thermal cyclers) should be a great advantage, especially in point-of-care molecular diagnostics and high-throughput systems. Comparing with other exponential nucleic-acid amplification reactions, PG–RCA generates ‘primers’ during the reaction, thereby being free from troublesome design and usage of exogenous primers. The reaction design of PG–RCA is much simpler than the other isothermal amplification methods since the circular probe is required to have only primer hybridization and nicking sequences.
PG–RCA is applicable to various molecular diagnostic assays by simply designing nucleic-acid probes to initiate the cascade reaction of PG–RCA on target molecule recognition. For example, specific gene sequences of genomic DNA can be detected sensitively and specifically by PG–RCA as long as the sequences include nicking sites. The same assay scheme may be applicable to specific RNA sequence detection since nicking reaction of DNA strands in DNA/RNA hybrids was recently reported (17). Alternatively, PG–RCA can be combined with nucleic-acid probe technology such as cycling probe and invader probe to detect specific DNA or RNA sequences and generate specific 3′-end sequence for PG–RCA detection (18–20). When it is combined with aptamer or 3′-end sequence-labeled antibody, PG–RCA may be applicable to specific protein detection like immuno-RCA and proximity ligation assay (21,22). Furthermore, as PG–RCA can distinguish relatively short nucleotide sequences specifically, highly multiplexed reaction may be possible by designing circular probes with different sequences around nicking sites to avoid cross reaction among those probes. Multiplex capacity may depend on the cross reactivity among circular probes when only a single nicking enzyme is used, however use of multiple nicking enzymes could increase the multiplex capacity further.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: Ministry of Education, Culture, Sports, Science and Technology of Japan.
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Saiki RK, Scharf S, Faloona F, Mullis KB, Horn GT, Erlich HA, Arnheim N. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science. 1985;230:1350–1354. doi: 10.1126/science.2999980. [DOI] [PubMed] [Google Scholar]
- 2.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 3.Fire A, Xu SQ. Rolling replication of short DNA circles. Proc. Natl Acad. Sci. USA. 1995;92:4641–4645. doi: 10.1073/pnas.92.10.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu D, Daubendiek S, Zillman M, Ryan K, Kool E. Rolling circle DNA synthesis: small circular oligonucleotides as efficient templates for DNA polymerases. J. Am. Chem. Soc. 1996;118:1587–1594. doi: 10.1021/ja952786k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lizardi PM, Huang X, Zhu Z, Bray-Ward P, Thomas DC, Ward DC. Mutation detection and single-molecule counting using isothermal rolling-circle amplification. Nat. Genet. 1998;19:225–232. doi: 10.1038/898. [DOI] [PubMed] [Google Scholar]
- 6.Zhang DY, Brandwein M, Hsuih TCH, Li H. Amplification of target-specific, ligation-dependent circular probe. Gene. 1998;211:277–285. doi: 10.1016/s0378-1119(98)00113-9. [DOI] [PubMed] [Google Scholar]
- 7.Dahl F, Banér J, Gullberg M, Mendel-Hartvig M, Landegren U, Nilsson M. Circle-to-circle amplification for precise and sensitive DNA analysis. Proc. Natl Acad. Sci. USA. 2004;101:4548–4553. doi: 10.1073/pnas.0400834101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nilsson M, Malmgren H, Samiotaki M, Kwiatkowski M, Chowdhary BP, Landegren U. Padlock probes: circularizing oligonucleotides for localized DNA detection. Science. 1994;265:2085–2088. doi: 10.1126/science.7522346. [DOI] [PubMed] [Google Scholar]
- 9.Banér J, Nilsson M, Mendel-Hartvig M, Landegren U. Signal amplification of padlock probes by rolling circle replication. Nucleic Acids Res. 1998;26:5073–5078. doi: 10.1093/nar/26.22.5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuhn H, Demidov VV, Frank-Kamenetskii MD. Rolling-circle amplification under topological constraints. Nucleic Acids Res. 2002;30:574–580. doi: 10.1093/nar/30.2.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Higuchi R, Fockler C, Dollinger G, Watson R. Kinetic PCR analysis: real-time monitoring of DNA amplification reactions. Nat. Biotech. 1993;11:1026–1030. doi: 10.1038/nbt0993-1026. [DOI] [PubMed] [Google Scholar]
- 12.Nelson KE, Fouts DE, Mongodin EF, Ravel J, DeBoy RT, Kolonay JF, Rasko DA, Angiuoli SV, Gill SR, Paulsen IT, et al. Whole genome comparisons of serotype 4b and 1/2a strains of the food-borne pathogen Listeria monocytogenes reveal new insights into the core genome components of this species. Nucleic Acids Res. 2004;32:2386–2395. doi: 10.1093/nar/gkh562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanaki K, Odawara T, Muramatsu T, Kuchino Y, Masuda M, Yamamoto K, Nozaki C, Mizuno K, Yoshikura H. Primer/template-independent synthesis of poly d(A-T) by Taq polymerase. Biochem. Biophys. Res. Commun. 1997;238:113–118. doi: 10.1006/bbrc.1997.7197. [DOI] [PubMed] [Google Scholar]
- 14.Ogata N, Miura T. Genetic information ‘created’ by archaebacterial DNA polymerase. Biochem. J. 1997;324:667–671. doi: 10.1042/bj3240667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ogata N, Miura T. Creation of genetic information by DNA polymerase of the archaeon Thermococcus litoralis: influences of temperature and ionic strength. Nucleic Acids Res. 1998;26:4652–4656. doi: 10.1093/nar/26.20.4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liang X, Jensen K, Frank-Kamenetskii MD. Very efficient template/primer-independent DNA synthesis by thermophilic DNA polymerase in the presence of a thermophilic restriction endonuclease. Biochemistry. 2004;43:13459–13466. doi: 10.1021/bi0489614. [DOI] [PubMed] [Google Scholar]
- 17.Gao W, Li X, Zeng L, Peng T. Rapid isothermal detection assay: a probe amplification method for the detection of nucleic acids. Diagn. Microbiol. Infect. Dis. 2008;60:133–141. doi: 10.1016/j.diagmicrobio.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 18.Duck P, Alvarado-Urbina G, Burdick B, Collier B. Probe amplifier system based on chimeric cycling oligonucleotides. BioTechniques. 1990;9:142–148. [PubMed] [Google Scholar]
- 19.Lyamichev V, Mast AL, Hall JG, Prudent JR, Kaiser MW, Takova T, Kwiatkowski RW, Sander TJ, de Arruda M, Arco DA, et al. Polymorphism identification and quantitative detection of genomic DNA by invasive cleavage of oligonucleotide probes. Nat. Biotech. 1999;17:292–296. doi: 10.1038/7044. [DOI] [PubMed] [Google Scholar]
- 20.Eis PS, Olson MC, Takova T, Curtis ML, Olson SM, Vener TI, Ip HS, Vedvik KL, Bartholomay CT, Allawi HT, et al. An invasive cleavage assay for direct quantitation of specific RNAs. Nat. Biotech. 2001;19:673–676. doi: 10.1038/90290. [DOI] [PubMed] [Google Scholar]
- 21.Schweitzer B, Wiltshire S, Lambert J, O'Malley S, Kukanskis K, Zhu Z, Kingsmore SF, Lizardi PM, Ward DC. Immunoassays with rolling circle DNA amplification: A versatile platform for ultrasensitive antigen detection. Proc. Natl Acad. Sci. USA. 2000;97:10113–10119. doi: 10.1073/pnas.170237197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gullberg M, Gústafsdóttir SM, Schallmeiner E, Jarvius J, Bjarnegård M, Betsholtz C, Landegren U, Fredriksson S. Cytokine detection by antibody-based proximity ligation. Proc. Natl Acad. Sci. USA. 2004;101:8420–8424. doi: 10.1073/pnas.0400552101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.