

1. Introduction
Human apolipoprotein E (apoE) is critical for neuronal maintenance and repair [1, 2]. The three common isoforms of apoE, apoE2, apoE3, apoE4, differ in their ability to perform these functions. ApoE4 is the major known genetic risk factor for late-onset familial and sporadic Alzheimer’s disease (AD), which accounts for more than 95% of AD cases [3]. In AD subjects and transgenic mice, apoE4 displays a gene dose effect in reducing presynaptic synaptophysin immunoreactivity and dendritic spine density [4, 5]. ApoE4 also impairs synaptic functions [6]. Human apoE4 knock-in mice have reduced long-term potentiation in the dentate gyrus than apoE3 mice, without evidence of advanced neurodegeneration [7]. ApoE4 mice also show reduced excitatory synaptic transmission and dendritic arborization in the amygdala [8].
Although several mechanisms have been proposed, (Mahley et al [1], Huang et al [9]), the molecular basis of the apoE isoform-specific effects remains unclear. However, the detrimental effects of apoE4 must reflect its unique structural and biophysical properties [9–11]. ApoE4 has three major structural characteristics that distinguish it from apoE2 and apoE3: domain interaction (an interaction between the amino- and carboxyl-terminal domains mediated by a salt bridge between Arg-61 and Glu-255) [12], a lower resistance to protein unfolding (resulting in a propensity to form a reactive folding intermediate or a molten globule state) [10, 13], and the lack of cysteine [14]. Molten globule formation and aggregation in apoE4 are neurotoxic to cultured neuronal cells, and the single cysteine in apoE3 results in hetero- and homo-disulfide-linked dimers that modulate lipid binding and low density receptor binding activity [10, 14]. Therefore, to understand mechanisms and to design potential therapeutic approaches, it is necessary to distinguish the relative contribution of these structural and biophysical differences to apoE4-associated neurodegeneration. This cannot be accomplished with the human apoE4 knock-in mouse model, which displays all apoE4-specific structural properties. Previously, domain interaction was proposed to underlie the association of apoE4 with AD and other neurodegenerative diseases [12]. However, this hypothesis has not been formally tested in vivo due to the lack of a suitable animal model specific for domain interaction.
To test this hypothesis directly, we took advantage of the fact that wildtype (WT) mouse apoE does not exhibit either domain interaction or molten globule formation. Regarding domain interaction, mouse apoE contains the equivalents of Arg-112 and Glu-255 but lacks the equivalent of the critical Arg-61 (instead, it contains threonine), making it functionally similar to apoE3 with a preference of high density lipoprotein [11]. To introduce domain interaction into mouse apoE, the threonine codon was replaced with an arginine codon in mouse Apoe by gene targeting, resulting in an “apoE4-like” functional molecule [15] with similar stability to WT mouse apoE and human apoE3 [16]. Thus, the Arg-61 apoE mouse provides a specific model of domain interaction, and any phenotype observed in this model, compared to WT, implicates domain interaction. Other features of the model include that expression of Arg-61 apoE is under the control of the natural mouse control elements and that the single mutation in WT mouse apoE minimizes any species effects. Secretion of Arg-61 apoE by astrocytes is decreased in these mice, leading to lower brain levels of apoE than in WT mice, suggesting that Arg-61 apoE may be selectively degraded by astroctyes [17].
In this study, the Arg-61 apoE model was examined to determine if domain interaction contributes to the synaptic deficits associated with apoE4 independent of an added Aβ stress. Our results demonstrate that domain interaction is associated with both morphologic and functional synaptic deficits.
2. Materials and Methods
2.1 Mice
Arg-61 apoE mice were generated as described [15] and backcrossed with WT C57BL/6J mice for eight generations [17]. In most experiments, 12-month-old male Arg-61 mice and WT controls were used. Male and female Arg-61 apoE mice express similarly lower levels of apoE than WT mice as reported [17]. Mice were housed and handled in accordance with the National Institutes of Health’s Giude for the Care and Use of Laboratory Animals.
2.2 Novel environment
Mice were singly housed for 3 days before the experiment. Mice (7 mice for each group) assigned to the novel environment exploration were then transferred to a larger uncovered cage (45 × 25 × 20 cm) that contained different bedding and five novel objects and was located in an adjacent room that differed markedly in size, shape, lighting, and furnishings. The mice were allowed to explore the new environment for 2 hours; control mice (6 WT and 7 Arg-61 apoE mice) remained undisturbed in their home cages. At the end of the experiment, mice were removed from either the novel environment or home cage and immediately sacrificed.
2.3 Morris Water Maze
Mice were trained to find a square platform (14 cm × 14 cm) submerged 1.5 cm below the surface in a 122-cm pool containing of water (18 ± 2°C) made opaque by the addition of nontoxic tempera powder. In a series of 4 trials, mice were pretrained to remain on the platform in which they were placed in a restricted (14 cm × 122 cm) alley within the pool and allowed to swim until they encountered the platform. The trial ended when the mouse remained on the platform for 5 sec without jumping off. If the mouse did not accomplish this task within 90 sec of testing, it was led to the platform by hand, placed on it, and allowed to sit for 5 sec.
After 1 day of pre-training, mice were trained to find the visibly cued platform with a 10-cm-high, black-and-white-striped marker at its center. Mice were trained in 2 daily sessions of two trials each over 3 consecutive days. The platform and the visual cue were moved for each trial. The mice were then trained to find a hidden platform in 2 daily sessions of 3 trials each over 5 consecutive days. The platform location was kept constant, but the starting point where each mouse was placed into the water was changed between trials. For both hidden and cued training, the intertrial interval was 10 minutes, and the time between sessions was 3 hours. Maximum trial length was 60 sec. Time to reach the platform (latency), path length, and swim speed were recorded with an EthoVision video tracking system (Noldus, Wageningen, The Netherlands).
Probe trials (60 sec) were performed 18 hours after the 30th trials of hidden platform training. For each trial, the platform was removed from the pool, and the amount of time spent searching in the target quadrant was recorded.
Learning curves for the hidden platform water maze training were generated by calculating the mean distance traveled to the platform for each mouse in each session. A repeated-measure ANOVA of distance traveled with genotype and session as independent factors was used to compare learning between the two genotypes. For the probe trial, time spent in the target quadrant was compared by ANOVA with genotype as the independent factor. Within each genotype, a paired t-test compared time spent in the target quadrant with time spent in each of the other three quadrants. One mouse was excluded from analysis because of slow swimming speeds (< 15 cm/sec) during both hidden training and probe trial.
2.4 Tissue preparation
All mice were anesthetized and perfused transcardially with cold normal saline (0.9% NaCl). Brains were removed and divided sagittally. One hemibrain was fixed in 4% phosphate-buffered paraformaldehyde, pH 7.4, at 4°C for 48 hours for vibratome sectioning. The other was snap-frozen and stored at −80°C for western blot analysis; some hemibrains were dissected into subregions (cerebral cortex and whole hippocampus) before freezing. All experiments were approved by the Committee on Animal Research of the University of California, San Francisco.
2.5 Immunohistochemistry and antibodies
All the antibodies used in the experiments are commercially available except the antibodies against mouse apoE [17], neuroligin 1 (gift from Dr. Nils Brose), and Arc (a gift from Dr. Steven Finkbeiner). The rabbit anti-mouse apoE was demonstrated to be specific against mouse apoE [17], whereas the sensitivity and specificity for antibodies against neurolgin−1 and Arc were reported by the providers [18,19].
Postfixed tissues were cut into 40-µm-thick sections with a vibratome (VT1000s, Leica Microsystems, Bannockburn, IL). Before further processing, specimens were assigned code numbers to ensure objective assessment. Three sagittal sections (320 µm apart between 1.00 and 2.40 mm from the sagittal suture) were washed in PBS (pH 7.4), blocked with 10% normal horse and/or goat serum (Vector Laboratories, Burlingame, CA) for 1 hour, and incubated overnight at 4°C with primary antibody. After PBS washes, the sections were incubated with secondary antibodies conjugated with fluorescein isothiocyanate (FITC) or Texas Red (TR). After three washes in PBS, immunofluorescently labeled sections were mounted in VectaShield (Vector Laboratories). All sections were stained simultaneously for each marker under identical conditions.
ApoE expression was assessed by double immunostaining with a polyclonal rabbit anti-mouse antibody against recombinant, full-length WT mouse apoE (1:500) [17] and mouse monocolonal (mAb) against either the neuron-specific marker NeuN (1:1,000, Chemicon, Temecula, CA) or the astrocyte-specific marker glial fibrillary acidic protein (GFAP; 1:500, Chemicon). Primary antibodies were detected with FITC-conjugated anti-rabbit IgG (1:100, Vector Laboratories) for apoE and TR-conjugated anti-mouse IgG (1:250) for NeuN or GFAP.
To assess the integrity of neuronal structures, the sections were labeled with mouse mAb against microtubule-associated protein 2 (MAP2; 1:500, Chemicon) and TR-conjugated anti-mouse IgG (1:250, Vector Laboratories) to identify dendrites. The presynaptic proteins synaptophysin and Bassoon were identified with mouse mAb against synaptophysin (1:50, Chemicon) or Bassoon (1:250, Chemicon). To identify postsynaptic sites, mouse mAbs against neuroligin-1 (NLG1; 1:500) [19] and biotinylated anti-mouse IgG (1:250, Vector Laboratories) were used. NLG1 was visualized with the Tyramide Signal Amplification fluorescence system (PerkinElmer, Boston, MA).
Expression of Arc and Fos, induced by a novel environment exposure, was detected in dentate gyrus granule neurons by staining with the avidin-biotin/peroxidase method. Briefly, after quenching endogenous peroxidase activity and blocking nonspecific binding, sections were incubated with rabbit anti-Arc (1:5000) or rabbit anti-Fos (1:10,000, Ab-5, Oncogene), detected with biotinylated goat anti-rabbit (1:200; Vector Laboratories, Burlingame, CA) antibody, and visualized with the Tyramide Signal Amplification fluorescence system (PerkinElmer).
Glutamate transporter 1 (GLT1) levels in astrocytes were determined by immunolabeling vibratome sections with rabbit polyclonal antibody against GLT1 (1:250, Abcam, Cambridge, MA). The sections were simultaneously co-stained with mouse mAb anti-GFAP. GLT1 was visualized with FITC-conjugated anti-rabbit IgG (1:100, Vector Laboratories) and GFAP with TR-conjugated anti-mouse IgG (1:250 Vector Laboratories).
2.6 Acquisition of immunofluorescencence images
Digital images of MAP2, synaptophysin, Bassoon, and NLG1 immunofluorescence were taken with a Bio-Rad Radiance 2000 laser-scanning confocal system (Bio-Rad, Hercules, CA) mounted on a BX60 microscope (Olympus, Melville, NY) with a 60× oil objective. The aperture (laser power, iris, and pin hole), contrast, and gain level were initially adjusted manually to obtain images with a pixel intensity within a linear range. For each marker, the linear range of the fluorescence intensity was determined in control sections from WT mice. This setting was used to collect all images analyzed in the same experiment. To obtain each final image, four scans were averaged (Kalman filter) with LaserSharp 2000 software (Bio-Rad). Images of ApoE-, NeuN-, GFAP-, Arc-, Fos-, and GLT1-immunolabeled sections were obtained with a digital camera (AxioCam HR, version 5.05.10, Carl Zeiss, Thornwood, NY), a BX-60 microscope (4× or 10× objective), and AxioVision software (version 3.1.2.1, Carl Zeiss). Immunofluorescence was examined in the L2–5 layer of the neocortex and in the stratum lucidum (CA3) and stratum radiatum (CA1) of the hippocampus. Randomly chosen areas (3–4 per subregion with an area of 2×2 µm) were evaluated with Image-Pro Plus (Version 5.05, Media Cybernetics, Silver Spring, MD). The data from each selected region was averaged as the data for each slide/section, and the data from three sagittal sections (320 µm apart between 1.00 and 2.40 mm from the sagittal suture) were averaged as the final results for each tested mouse.
2.7 Quantitation of immunoreactive structures
The area of synaptophysin-immunoreactive (SYN-IR) presynaptic terminals of defined signal intensity was quantitated and expressed as a percentage of the total image area, as described [20]. This method has been validated with quantitative immunoblots and enzyme-linked immunosorbent assays [4, 20, 21]. Similar procedures were used to quantify Bassoon-positive presynaptic boutons, the area of the neuropil occupied by MAP-2-immunolabeled dendrites, and NLG1-positive postsynaptic sites. NeuN-positive neurons were counted in selected CA1 areas of hippocampus (2×2 µm in the high magnification pictures). Integrated optical densities of apoE, GFAP, or GLT1 in defined brain region were determined.
The relative numbers of Arc-immunoreactive (Arc-IR) or Fos-IR granule cells were determined by counting Arc-IR- or Fos-IR-positive cells in dentate gyrus in every 8th serial saggittal section (total six sections containing hippocampus region). Results are presented as the total number of positive cells per hemibrain [22].
2.8 Western blotting and quantitation of protein levels
Levels of apoE and synaptophysin were quantitated by immunoblotting. Brain tissues were homogenized in ice-cold lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.1% sodium dodecyl sulfate, 0.5% Nonidet P−40, 0.5% sodium deoxycholate, and a mixture of protease and phosphatase inhibitors) and centrifuged at 30,000 rpm for 30 min at 4°C in a TLA 100.3 rotor in an Optima TL Ultracentrifuge (Beckman Instruments, Fullerton, CA). Supernatants were collected and analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (150 µg of protein was loaded to detect apoE and 30 µg to detect synaptophysin) and western blotting. Mouse WT apoE and Arg-61 apoE were detected with an antibody against recombinant, full-length WT mouse apoE (1:10,000) [17]. Synaptophysin was detected with anti-synaptophysin mAb (1:50,000). Protein bands were detected by chemiluminescence with horseradish peroxidase–coupled anti-rabbit or anti-mouse IgG secondary antibody. The blots were scanned, and protein levels were quantitated by densitometry (ImageJ, Bethesda, MD).
2.9 Quantitation of total cholesterol secreted from primary astrocyte cultures
Primary astrocytes from 3-day-old pups were grown to 80% confluence in medium containing Dulbecco’s modified Eagle medium and 20% fetal bovine serum in 15-cm tissue culture dishes. The cells were washed twice with PBS and once with serum-free medium containing phenol-red free Dulbecco’s modified Eagle medium (Sigma, St. Louis, MO), 1% glutamine, and B-27 supplement (Invitrogen, Carlsbad, CA). The cells were then incubated in the serum-free medium for 72 hours. The conditioned medium was collected, and centrifuged at 1000 rpm for 5 minutes. The supernatant was lyophilized, and the lipids were extracted three times with chloroform:methanol (2:1). The extract was evaporated under nitrogen, and the lipids were dissolved in 2-propanol and analyzed for total cholesterol with the cholesterol/cholesteryl ester quantitation kit (Calbiochem, San Diego, CA) according to the manufacturer’s instructions. For total cellular protein determination, the cells on ice were washed gently three times with PBS and lysed for 5 minutes with 0.1 N NaOH. The cell lysate was centrifuged at 1000 rpm for 5 minutes and analyzed for total protein. The cholesterol data were normalized to cellular protein levels and expressed as the amount of secreted cholesterol per mg cell protein.
2.10 Statistical analysis
Quantitative data are expressed as mean ± SEM. Differences between means were assessed with the unpaired, two-tailed t test. Differences among means were evaluated by ANOVA followed by Dunnett's or Tukey-Kramer post hoc test.
3. Results
3.1 Arg-61 apoE is expressed at a lower level in the brain than WT apoE
The apoE expression pattern in the neocortex (not shown) and hippocampus (Figure 1A, B) was similar in 12-month-old WT and Arg-61 apoE mice. As shown by double immunofluorescence staining, in both groups, apoE was expressed predominantly in GFAP-positive protoplasmic and fibrous astrocytes (Figure 1A). ApoE was not detected in NeuN-positive neurons (Figure 1B). Strikingly, apoE staining revealed the full cell volume of astrocytes with very fine resolution of cellular processes; whereas anti-GFAP staining highlighted only large processes (Figure 1A), similar to results in the EGFPapoE mice [23]. In low magnification pictures, the apoE expressions showed a diffused staining pattern in hippocampus, which was due to the staining of fine distal astrocytic processes (Figure 1C). However, Arg-61 apoE mice had lower levels of apoE in the hippocampus than WT mice, as shown immunohistochemically (Figure 1C and quantified as apoE immuno-reactive intensity in Figure 1D) and, as in our previous study [17], by western blot analysis (Figure 1E, F).
Figure 1.
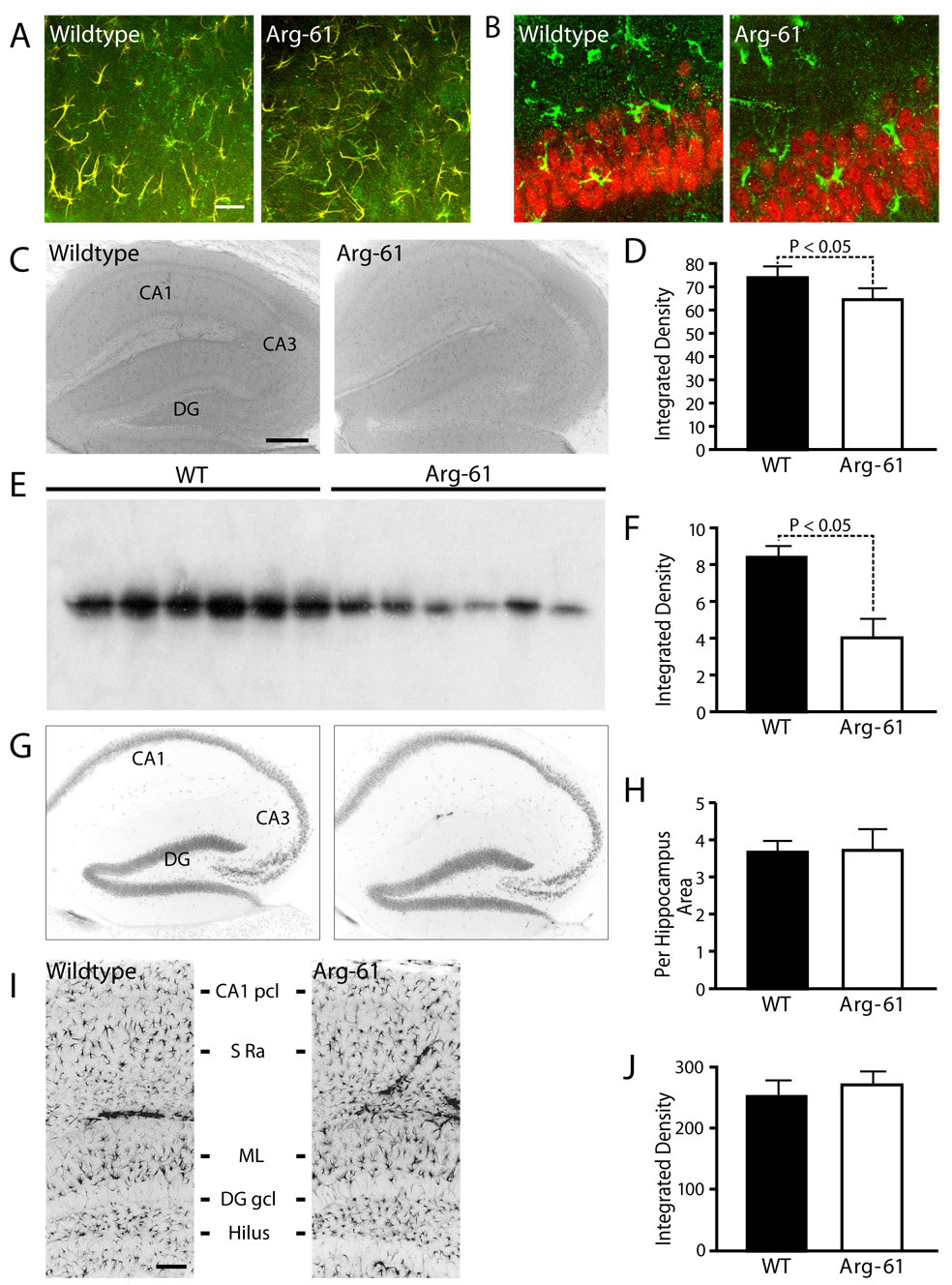
(A) Double immunostaining for apoE (green) and GFAP (red) showed co-localization (yellow) of apoE in astrocytes somas and larger cellular processes in hippocampus. (B) Double immunostaining for apoE (green) and NeuN (red) showed no co-localization. (C–F) ApoE expression and protein levels in the brains of 12-month-old WT and Arg-61 apoE mice. ApoE immunoreactivities in the hippocampus were assessed immunohistochemically (C), quantitated as immunochemical integrated density (D, n = 9 per group) and by western blot of mouse brain homogenates (E, F, n = 6 mice per group). (G) Distribution of NeuN-positive hippocampal neurons in the brain of WT and Arg-61 apoE mice, and (H) the relative numbers of neurons quantitated in selected CA1 areas of hippocampus (n = 9 mice for each group). (I) Distribution of GFAP-positive astrocytes in different layers of hippocampus (CA1 pcl – CA1 pyramidal cell layer, S ra – strata radiatum, ML – molecular layer, DG gcl – dentate gyrus granule cell layer) in the brain of WT and Arg-61 apoE mice, and (J) the GFAP immuno-reactivities were quantitated in the hippocampus (n = 10 for each group). Scale Bars: (A) 10 µm, (C) 250 µm, (I) 50 µm.
3.2 No evidence of neuronal loss or gliosis
Arg-61 apoE mice did not display gross structural abnormalities in any brain region or significant loss of neurons or astrocytes (Figure 1G, I). Specifically, the relative numbers of NeuN-positive neurons (Figure 1H) and the densities GFAP-positive astrocytes (Figure 1J) were similar in the hippocampus in WT and Arg-61 apoE mice. Astrocytes were similarly distributed in the hippocampal CA1 pyramidal cell layer, strata radiatum, and molecular layer of the dentate gyrus in both mice (Figure 1I). Finally, there was no significant evidence of astrogliosis in the Arg-61 apoE mice (Figure 1J).
3.3 Presynaptic synaptophysin levels in Arg-61 mice decline with age
ApoE is important for neuronal remodeling and maintenance of synapto-dendritic connections. To assess synaptic content, we measured synaptophysin levels in 12-month-old Arg-61 apoE and WT mice (Figure 2). The density of synaptophysin-immunoreactive (SYN-IR) presynaptic terminals was 10–25% lower in Arg-61 apoE mice in hippocampal subregions (CA1, 25.2 ± 0.5% vs. 28.4 ± 0.6%, P < 0.05; CA3, 19.8 ± 0.6% vs. 26.6 ± 1.4%, P < 0.05) and in neocortex (25.2 ± 0.4% vs. 28.3 ± 0.8%, P < 0.05) (Figure 2A,B). Western blot analysis showed that synaptophysin levels in the Arg-61 apoE mice were 11.3 ± 2.0% lower in the cortex and 21.1 ± 3.6% lower in the hippocampus (Figure 2C,D).
Figure 2.
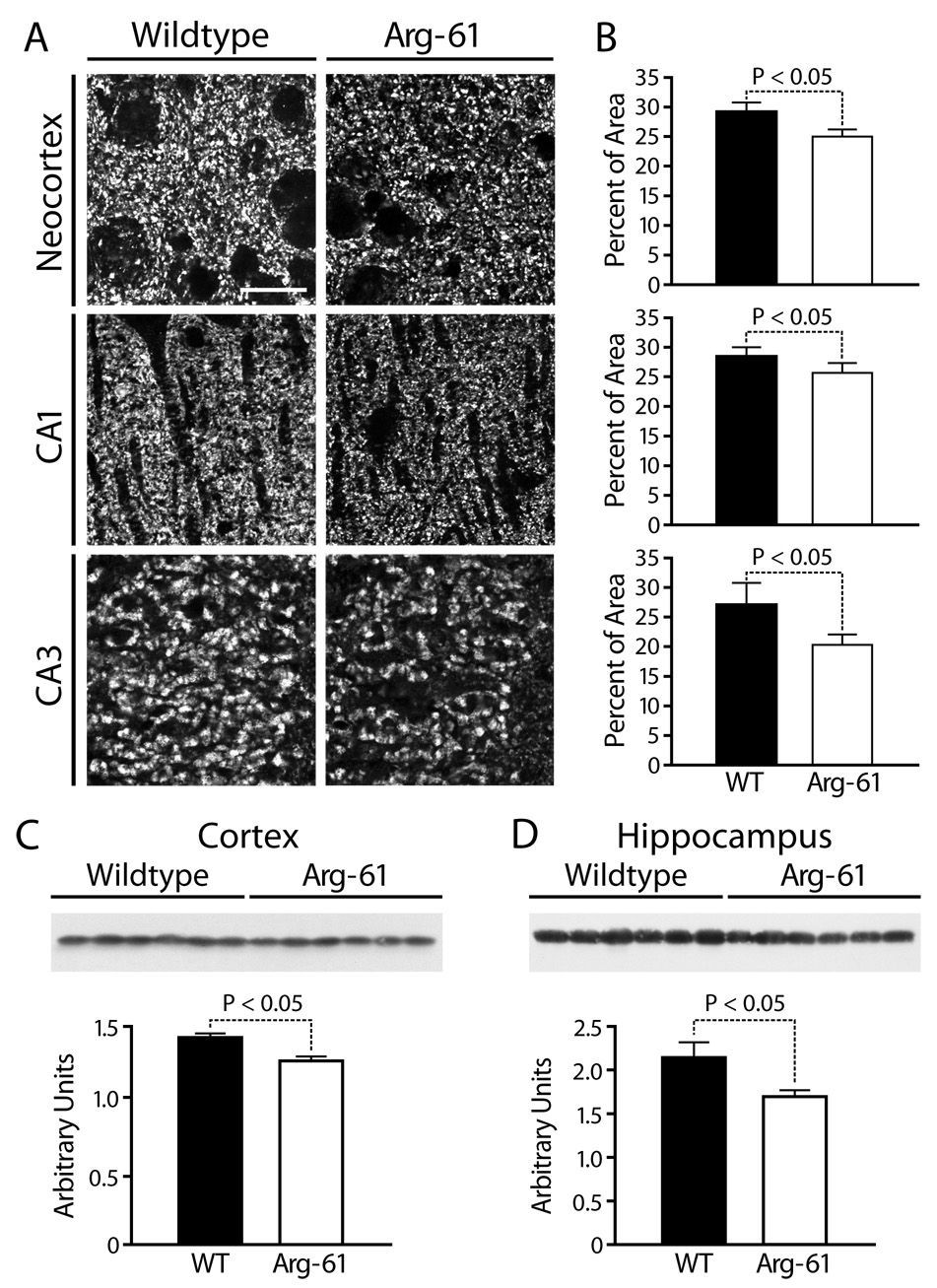
Synaptophysin expression in the brains of WT and Arg-61 apoE mice. (A, B) Immunostaining for synaptophysin and quantitation in the neocortex and the CA1 and CA3 areas of the hippocampus (n = 9 mice per group). (C, D) The protein levels of synaptophysin in the cortex and hippocampus were quantitated by western blot analysis (n = 6 mice per group). Scale Bar: 5 µm.
To assess age-dependent loss of SYN-IR presynaptic terminals, we examined mice at 6, 12, and 21–24 months of age (Figure 3A). WT mice had no significant age-dependent loss of SYN-IR. In Arg-61 apoE mice, SYN-IR was not reduced at 6 months but was significantly lower than in WT mice in all brain subregions at 12 months, with a further decline at 21–24 months (neocortex 24.1 ± 1.8% vs. 28.0 ± 1.5%, CA1 24.9 ± 1.5% vs. 29.0 ± 1.3%, CA3 18.9 ± 1.6% vs. 26.0 ± 1.3%) (Figure 3B).
Figure 3.
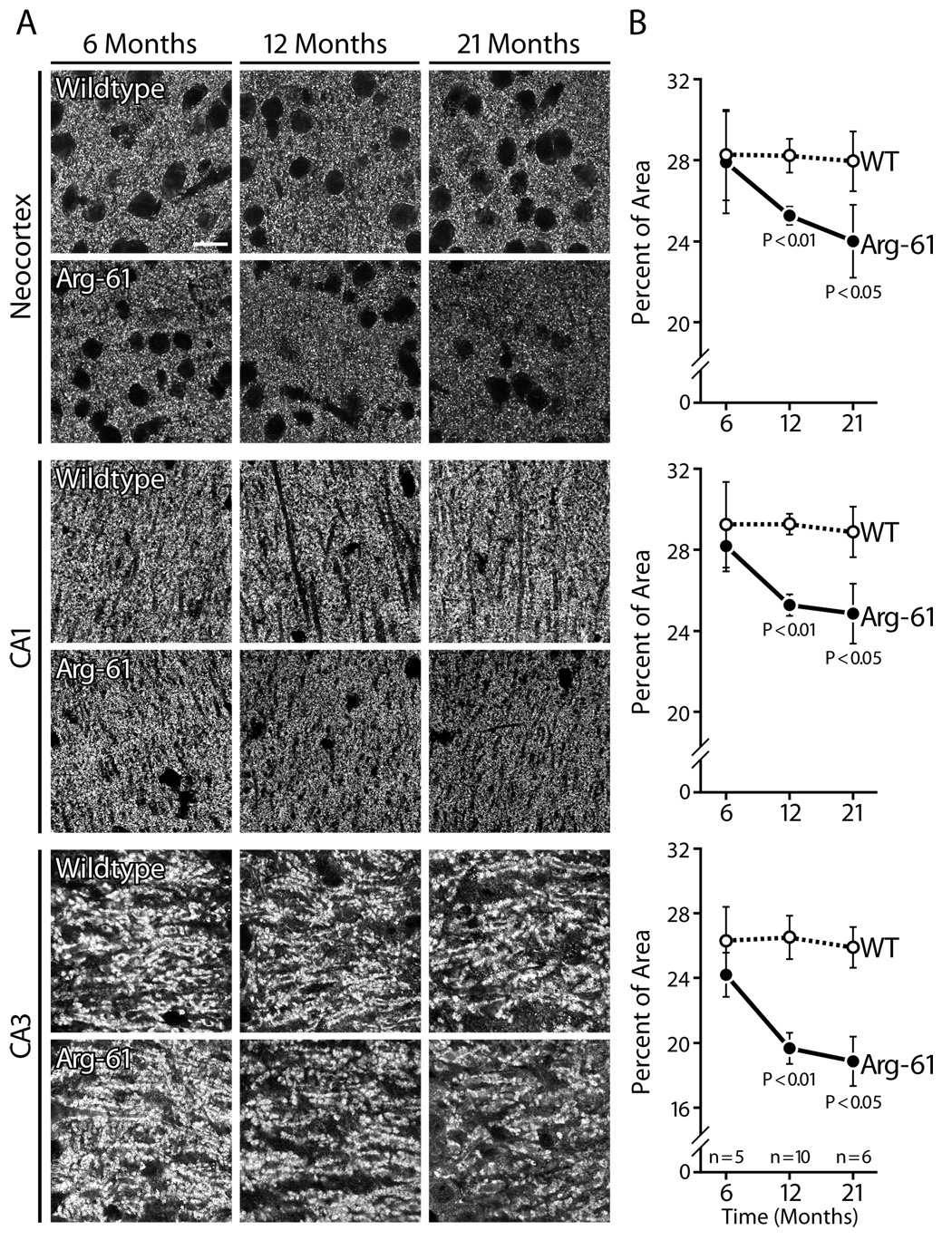
Age-dependent change of synaptophysin immunoreactivity in Arg-61 apoE mice compared to WT. Scale Bar: 5 µm.
The active zone is an important component of presynaptic terminals and is crucial for efficient synaptic transmitter release. To examine the effect of Arg-61 apoE on the active zone, immunostaining was performed for Bassoon, an active zone component involved in structural organization and neurotransmitter release [24]. Bassoon-immunoreactive presynaptic boutons were less numerous in the hippocampus of Arg-61 apoE mice than WT mice (CA3, 26.3 ± 0.9% vs. 28.7 ± 0.7%, P < 0.05; CA1, 27.1 ± 0.8% vs. 29.7 ± 0.8%, P = .05); no significant difference was observed in the neocortex (27.6 ± 1.5% vs. WT 29.5 ± 2.2%) (Figure 4A, B).
Figure 4.
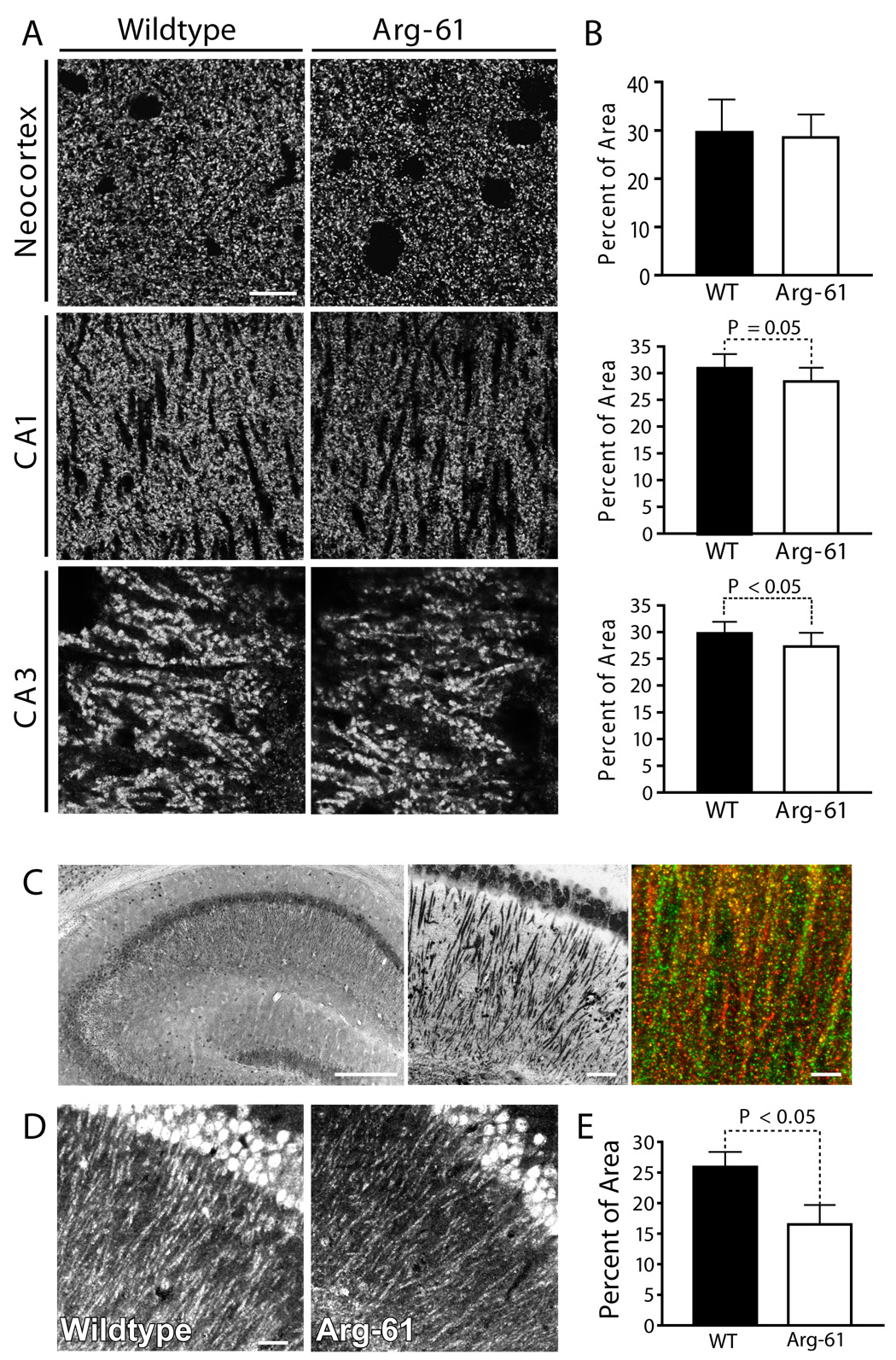
Presynaptic integrity accessed by Bassoon levels and determination of NLG1 positive dendrites and postsynaptic terminals in the brains of WT and Arg-61 apoE mice. (A) Immunostaining for Bassoon in neocortex and hippocampus and (B) quantitated by percent of immuno-reactive area (n = 9 per group). (C) Sample picture of immunostaining of hippocampal CA1 pyramidal neurons (left panel) and dendrites (central panel) by NLG1 in WT mouse brain. (C, right panel) Sample picture of WT mouse hippocampal CA1 region, double labeling postsynaptic terminals with GluR2/3 (green) and NLG1 (red), colocalization of GluR2/3 with NLG1 are shown in yellow and orange. (D) Immunostaining of positive postsynaptic terminals by NLG1 and (E) quantitation by percent of area of NLG1 immunoreactivity (n = 6 per group). Scale Bars: (A) 5 µm, (C) left panel 250 µm, central 50µm, right 5 µm, (D) 25 µm.
3.4 Neuroligin1-immunoreactive dendrites are less abundant in Arg-61 apoE mice
To determine if Arg-61 apoE is also associated with postsynaptic changes, we analyzed the total dendritic area and postsynaptic density by immunostaining. No differences in total dendritic area, detected by MAP2 staining, were observed between Arg-61 apoE and WT mice (hippocampal CA1 region, 26.8 ± 0.6% vs. 27.2 ± 0.3%, P > 0.05). However, the Arg-61 mice had significantly fewer NLG1-immunoreactive postsynaptic densities in the hippocampus than WT mice (Figure 4C). NLG1 is a postsynaptic cell-adhesion molecule specifically localized to postsynaptic densities of excitatory synapses [19]. NLG1 immunostaining was prominent in hippocampal CA1 pyramidal neuron somata and apical dendrites and, to a lesser extent, in basal dendrites (Figure 4C). In most dendrites, NLG1 colocalized with excitatory synaptic GluR2/3 glutamate receptors (Figure 4C). In WT mice, the level of NLG1 (defined as percent area of NLG1 immunoreactive postsynaptic terminals) in the hippocampus (26%) was comparable to the levels of synaptophysin (28%) and Bassoon (29%). However, in Arg-61 apoE mice, the density of NLG1-immunoreactive postsynaptic terminals was ~40% lower (16.5 ± 3.2% vs. 25.9 ± 2.5%, P < .05) (Figure 4D, E), demonstrating post-synaptic deficit in this model.
3.5. Induced expression of Arc and Fos is reduced in Arg-61 apoE mice
Activity-dependent plastic changes in the strength of synaptic connections are considered to underlie learning and memory. To assess the functional consequence of the losses of synaptic proteins in the Arg-61 apoE mice, we tested two markers: Arc and Fos, products of the immediate-early gene response. Increased hippocampal expression of these proteins in response to stimulation is thought to be obligatory for spatial memory formation [25]. Arc, an effector immediate-early gene expressed predominantly in cortical and hippocampal glutamatergic neurons, is required for maintenance of long-term potentiation and for memory consolidation [25]. Exposure to a novel environment rapidly increases Arc expression in neurons, allowing the identification of neuronal network activity [25].
At baseline, the number of Arc-IR granule neurons in the dentate gyrus of the hippocampus did not differ significantly in WT and Arg-61 apoE mice (Arg-61 87 ± 11 vs. WT 92 ± 10, P > .05) (Figure 5A, B). Environmental exploration markedly increased Arc IR in hippocampal areas in WT mice, numbers of Arc IR granule cells increased by 3.5-fold; whereas, there was only a moderate increase (by 2.3-fold) of Arc-positive granule cells in Arg-61 apoE mice (Figure 5B, Arg-61 200 ± 14 vs. WT 320 ± 34, P < 05). These differences do not reflect reduced exploration of the novel environment by Arg-61 apoE mice, since no differences in exploratory behaviors or locomotion were seen in the elevated plus maze or open field test (see supplement data). We also noted that, in hippocampal CA1 strata radiatum, Arc-immunoreactivities in the dendrites of CA1 pyramidal neurons were higher in both non-stimulated and novel environment-stimulated WT mice than those of Arg-61 apoE mice (Figure 5A), however as discussed by Palop et al. [22], the quantification of Arc-mRNA expression more accurately reflects the novel environment-induced Arc expression in dendrites.
Figure 5.
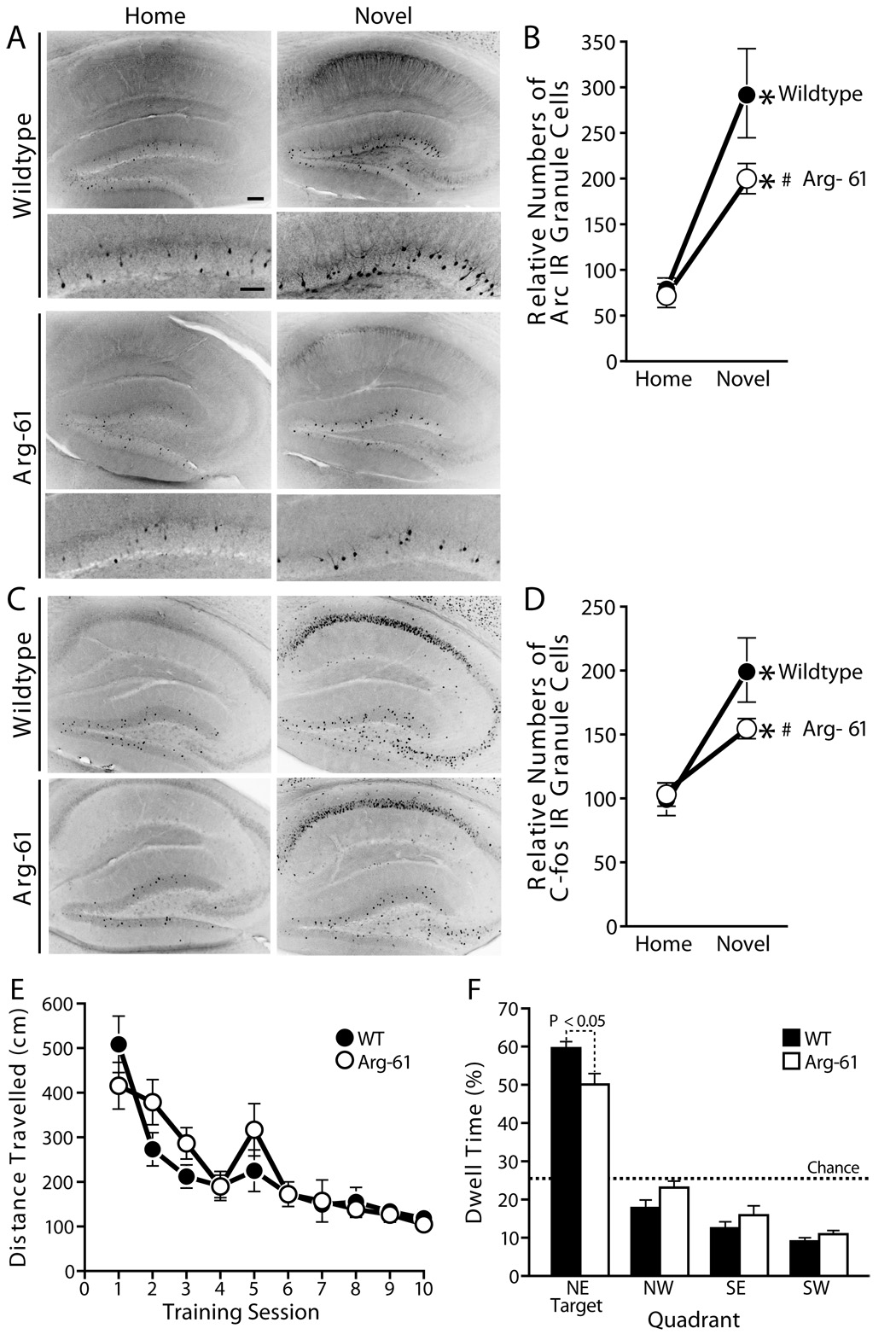
Novel environment induced Arc and Fos expression in hippocampal dentate gyrus granule neurons and mild memory deficit in Arg-61 apoE mice in water maze test. (A) Arc induction in Arg-61 apoE and WT mouse hippocampus, and (B) quantitation of relative numbers of Arc-immunoreactive dentate gyrus granule neurons. (C) Fos induction in Arg-61 apoE and WT mouse hippocampus, and (D) quantitation of relative numbers of Fos-immunoreactive dentate gyrus granule neurons. *P < .05 vs. mice in home cage, #P < .05 vs. WT mice. In water maze test, (E) There were no genotype differences in distance traveled to hidden platform during training (P > .47; n = 13 per group). (F) In probe trial when hidden platform was removed, Arg-61 mice spent significantly less time in the correct quadrant than WT controls. Scale Bars: (A) upper panel 100 µm, lower panel 50 µm.
To determine if induction of other immediate-early genes in granule cells was also affected in Arg-61 apoE mice, we examined Fos expression. At baseline, WT and Arg-61 apoE mice had similar numbers of Fos-positive granule neurons in the dentate gyrus (Arg-61 140 ± 8 vs. WT 127 ± 11, P > .05) (Fig 5C). After the 2-h novel environment stimulation, the number of Fos-positive granule neurons increased by 1.7 fold in WT mice but by only 1.2 fold in Arg-61 apoE mice (Figure 5D, Arg-61 apoE, 161 ± 8 vs. WT, 217 ± 15, P < .05). The levels of Fos and Arc expression in Arg-61 apoE mice were tightly correlated both at baseline (R2 = 0.81; P = .02) and after environmental exploration (R2 = 0.68; P = .04), suggesting that Arg-61 apoE affects a regulatory mechanism common to different immediate-early genes.
3.6. Memory is impaired in Arg-61 apoE mice
To determine if the changes in synaptic plasticity–related proteins have behavioral consequences in Arg-61 apoE mice, we tested spatial learning in the Morris water maze. Arg-61 mice learned to find a hidden platform submerged below the surface of a pool of water as quickly as WT mice (Figure 5E, repeated measures ANOVA genotype effect P > .05). However, when the platform was removed from the pool for a probe trial 18 hours after the completion of training, Arg-61 apoE mice spent less time than WT littermates in target quadrant (Figure 5D, ANOVA genotype effect F(1,24) = 7.1, P < .05), indicating that Arg-61 apoE mice showed an impairment of hippocampus-related reference memory. Although they performed less well than WT mice, Arg-61 apoE mice still displayed a preference for the target quadrant compared to any other quadrant of the pool (paired t tests, P < .0001 for all quadrants). This is consistent with the mild but significant synaptic pathology observed in these mice.
3.7. GLT1 immunoreactivity is lower in Arg-61 apoE mice
We speculated that the decreased levels of synaptophysin, Bassoon, and NLG1 might reflect a chronic excitotoxic effect due to inefficient glutamate clearance by astrocytes. To test this possibility, we examined GLT1 levels (Figure 6). Double immunohistochemical labeling showed GLT1 immunoreactivity in GFAP-positive astrocytes but not in NeuN-positive neurons (data not shown). Laser scanning confocal microscopy showed GLT1 expression in the soma and proximal processes of astrocytes (data not shown), as well as in the distal astrocytic processes and surrounding synapses (Figure 6B). The expression pattern of GLT1 was similar in the hippocampus of Arg-61 apoE and WT mice (Figure 6A), but the level of expression was lower in Arg-61 apoE mice, as shown by the integrated intensity of GLT1 fluorescence (CA1, 75.8 ± 3.4% vs. WT, 92.4 ± 2.9%, P < .05; CA3, 55.0 ± 2.7% vs. WT, 66.8 ± 1.8%, P < .05) (Figure 6C).
Figure 6.
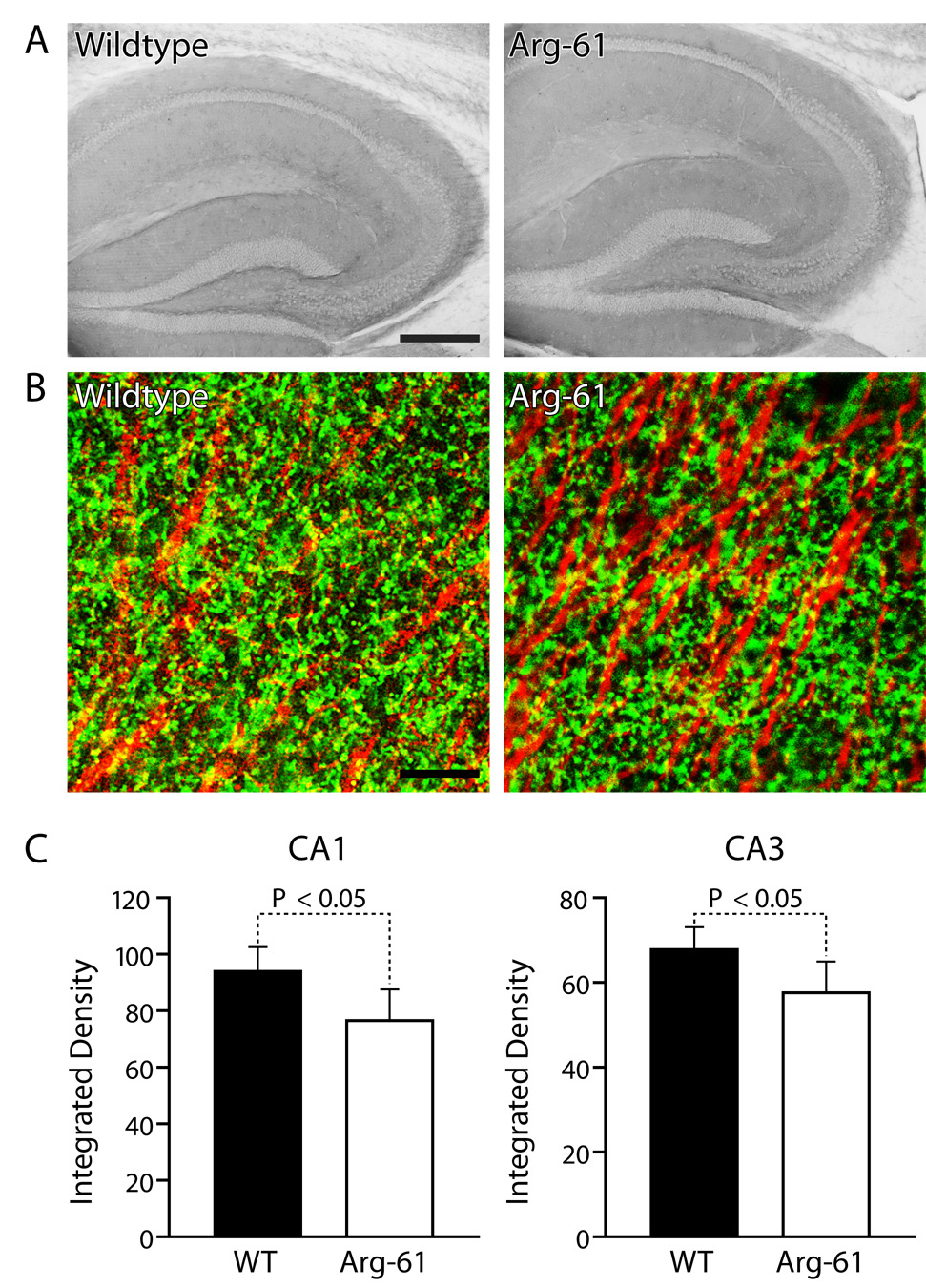
Localization and quantitation of astrocyte glutamate transporter, GLT1, levels in the brains of WT and Arg-61 apoE mice. (A) Immunostaining of GLT1 in mouse brain hippocampus. (B) Double immunostaining of GLT1 (green) and MAP2 (red) in hippocampal CA1 region. (C) Quantitation of GLT1 levels in CA3 and CA1 by integrated optical density (n = 9 per group). Scale Bars: (A) 250 µm, (B) 10 µm.
3.8. Primary Arg-61 apoE mouse astrocyte cultures secrete less cholesterol than WT
Since cholesterol is required for synaptic maintenance, and astrocytes in Arg-61 apoE mice secrete reduced amounts of Arg-61 apoE [17], we assessed cholesterol secretion in primary astrocyte cultures. Astrocytes from Arg-61 apoE mice secreted 34% less cholesterol than those from WT mice (2.4 ± 0.16 µg/mg vs. 3.7 ± 0.25 µg/mg protein).
4. Discussion
Defining the impact of domain interaction, one of the distinguishing structural features of apoE4, is essential for understanding the isoform-specific effects of apoE in neurodegeneration and for identifying potential therapeutic strategies. In this study, we found that Arg-61 apoE mice, a specific model of apoE4 domain interaction, displayed early neurodegenerative changes in the brain in the absence of neuronal loss or gliosis. The synaptic organization in the hippocampus and neocortex was disrupted, as demonstrated by an age-dependent decline in SYN-IR presynaptic terminals, a reduction in Bassoon, which participates in neurotransmitter release, and decreased levels of NLG1, which is a critical cell-adhesion molecule determining synapse formation and function [26]. As a functional consequence of the losses of synaptic proteins, Arg-61 apoE mice showed an impaired induction of Arc and Fos expression in response to novel environment stimulation and a mild hippocampus-dependent memory deficit in the water maze test. These alterations mirror the pathological changes that characterize the early stages of AD.
In AD patients and non-demented subjects, apoE4 is associated with lower levels of synaptic proteins, including a substantial decrease in synaptophysin and a detectable decrease in the presynaptic membrane protein syntaxin [27]. AD begins with subtle changes in synaptic efficacy well before neuronal degeneration is apparent, and loss of synapses likely disrupts hippocampal and cortical circuits, with functional consequences contributing to progressive cognitive impairment [28–30]. Even in non-demented middle-aged and elderly human subjects, apoE4 is associated with greater cognitive decline, and the decline shows a gene dose-response [31–33]. A significant association was found between apoE4 and transition from normal to mild cognitive impairment [34], which represents early-stage AD and with high positive predictive value for eventual conversion to AD [35].
Early hippocampal pathology, as seen in altered expression of synaptic proteins, occurs in the pre-symptomatic stage of AD and other dementias [30]. These changes might reflect a disrupted connection between the hippocampus and both limbic and neocortical areas, which is necessary for the integration of cognitive function. Our results demonstrating morphologic and functional synaptic deficits in Arg-61 apoE mouse hippocampus reflect the pathological changes and cognitive decline observed in the early stages of mild cognitive impairment or AD. Moreover, the results are also comparable with those observed in both human subjects and apoE4 transgenic and knock-in mice, indicating that domain interaction is a major contributor to the observed synaptic deficits.
Since astrocytes are the major producers of apoE in the central nervous system [23], astrocyte-generated apoE4 might be important in the initial stage of neurodegeneration. The age-dependent synapse degeneration in Arg-61 apoE mice occurred without any additional stress and with apoE expression under the control of the natural promoters and regulatory elements. Our present observation and previous report demonstrate that Arg-61 apoE is expressed at a lower level, 40%~50% lower, in the brain than WT apoE, as a result of domain interaction causing an increased intracellular degradation [36]. Similar reduced apoE levels also occur in the human–apoE4 knock-in mouse brains [17]. However, the morphological and functional synaptic deficits that we demonstrate are not simply due to the reduced apoE levels. Heterozygous mice with one null-apoE allele, with similarly reduced levels of the apoE (60% lower), do not display reduced synaptophysin-immunoreactivity and other neurodegenerative features compared to WT mice [21]. The functional consequences resulting from the single mutation, i.e., domain interaction, in Arg-61 apoE mice is the basis for the synaptic deficits. Previously, we suggested that domain interaction might cause apoE4 or Arg-61 apoE to be processed abnormally in astrocytes, activating a stress response pathway in the endoplasmic reticulum [17]. We recently reported that intracellular Arg-61 apoE levels in primary astrocytes were ~40% lower that WT, suggesting an increased degradation of Arg-61 apoE in the astrocytes. Our results also indicate that apoE4 domain interaction results in a chronic ER stress response that activates all three unfolded protein response pathways in primary astrocytes [36]. Potentially this stress/response is likely to result in a global astrocyte dysfunction in the Arg-61 apoE mouse brain, suggested by, but not necessarily limited to reduced GLT1 level in Arg-61 apoE mice. Global cellular effects in protein misfolding diseases, particularly in astrocytes, is an emerging concept [37, 38].
We speculate two potential mechanisms that might explain the association of apoE4 domain interaction with the synaptic deficits observed in this model: chronic glutamate excitoxicity and decreased cholesterol secretion, both of which involve the interaction of astrocytes with synapses and are not necessarily mutually exclusive. The effects of these mechanisms could represent early stages in the neurodegenerative process preceding the neuronal expression of apoE in response to stress, that leads to the generation of neurotoxic apoE4 carboxyl-terminal truncated fragments [9]. If astrocyte function is compromised by domain interaction, as we suggest, dysfunctional astrocytes may not effectively support stressed neurons and dysfunctional astrocytes leading to neuronal degeneration.
4.1. Chronic glutamate excitotoxicity
Astrocytes are responsible for removing neurotransmitters released by active synapses, including glutamate, a prominent excitatory neurotransmitter and a potent neurotoxin [39, 40]. Astrocytic uptake of glutamate by the astrocyte-specific transporter GLT1 is important in controlling synaptic transmission and efficacy [39]. In humans, decreased glutamate transporter activity is associated with increased excitotoxicity and with neurodegeneration initially reflected in synapse loss and reduced synaptophysin levels [41]. Also in a transgenic mouse model of AD, reduced GLT1 protein levels correlate with significantly reduced glutamate transport activity [41]. Thus, reduced GLT1 levels in Arg-61 apoE mice may reflect inefficient clearance of glutamate by astrocytes that generates a chronic excitotoxicity, resulting in the reduced levels of the synaptic proteins synaptophysin, Bassoon, and NLG1. Ultimately, these synaptic deficits could result in the functional deficits, demonstrated by the decreased induction of neuronal activity–regulated Arc and Fos expression in Arg-61 apoE mice, and mild memory deficit.
4.2. Reduced cholesterolsSecretion
Previously, we demonstrated in human apoE knock-in and Arg-61 apoE mice that domain interaction results in primary astrocytes secreting less apoE4 than apoE3, and less Arg-61 apoE than WT mouse apoE [17]. In the present study, we found that Arg-61 apoE astrocytes also secrete less cholesterol. Since cholesterol secreted by astrocytes and transported by apoE-containing lipoproteins mediates synapse-promoting effects [42–44], any reduction in cholesterol delivery might lead to degeneration of synapses. Astrocyte-delivered cholesterol also contributes to the biogenesis and cycling of synaptic vesicles and synaptophysin is a cholesterol-binding protein [45, 46]. Thus, synaptophysin may function with cholesterol in renewing synaptic-vesicle membranes [46], suggesting that cholesterol availability is critical for synaptic vesicle docking and fusion [45]. Although there is regional variation in the glial influence on synapse development and neurite outgrowth [47], data from brain slice and primary neuron cultures also suggest a critical role for astrocyte-derived signals, including astrocyte-secreted cholesterol, as endogenous modulators of neurotransmission in both cortex and hippocampus [47, 48]. The local interaction between the perisynaptic astroglial processes and the synapses are critical for synaptogenesis and fine manipulation of synaptic plasticity in mature brain [49]. Therefore, decreased secretion of cholesterol by Arg-61 apoE astrocytes may not effectively support synaptic structure and function and neuronal maintenance. The reduced astrocyte glutamate transporter and lower secreted cholesterol suggest an astrocyte dysfunction that could contribute to the age-dependent reduction in synaptophysin and the decrease in Bassoon and NLG1 levels in Arg-61 apoE mice. It should also be noted that astrocytes affect numerous pathways supporting and maintaining normal neuronal and brain functions [50].
In summary, our findings clearly demonstrate that apoE4 domain interaction is a contributor to apoE4-associated synaptic degeneration and functional synaptic deficits, possibly through adverse effects on astrocyte function. The linking of apoE4 domain interaction and astrocyte dysfunction represents a new paradigm for the association of apoE4 with neurodegenerative diseases and indicates that apoE4 domain interaction represents a viable therapeutic target. It was recently reported that disrupting domain interaction with small molecules abolishes the apoE4 enhanced Aβ production in cultured cells [51]. The relative contributions of molten globule formation and the lack of cysteine in apoE4 will await the generation of mice specific for these features, which are currently in progress.
Supplementary Material
Acknowledgements
This work was supported in part by: (1) American Health Assistance Foundation Pilot Grant for Alzheimer’s Disease Research, A2006-231 (NZ) “ApoE4 structural properties and synaptic deficits.” (2) National Institutes of Health grants: (a) PO1 AG022074 (Dr. Lennart Mucke, KW) “Project 2: Protein structure in apoE4-associated neurodegeneration.” (b) RO1 AG20235 (KW) “Role of apoE4 domain interaction in neurodegeneration.” and (c) AG028793 (KW) “Role of apoE structure and metabolism in neurodegeneration.” (3) This publication is made possible by grant CO6 RR0118928 from the NIH National Center for Research Resources. We thank Dr. Eliezer Masliah (University of California, San Diego) for training in immunochemical image analysis, Belinda Bituin and James McGuire for excellent technical assistance, Hilary Gerstein and Tiffany Wu in Gladstone Behavioral Core Laboratory for excellent technical assistance in the mouse behavioral and novel environmental experiments, John Carroll and Chris Goodfellow for graphics, Stephen Ordway and Gary Howard for editorial assistance, and Linda Turney for manuscript preparation.
Abbreviations
- Apo
apolipoprotein
- AD
Alzheimer’s Disease
- Arc
activity-regulated cytoskeleton-associated protein
- FITC
fluorescein isothiocyanate
- GFAP
glial fibrillary acidic protein
- GLT1
glutamate transporter 1
- mAb
mouse monocolonal
- MAP2
microtubule-associated protein 2
- NLG1
neuroligin-1
- SYN-IR
synaptophysin-immunoreactive
- TR
Texas Red
- WT
wildtype
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: All the authors verify that there are no conflicts of interest including financial, personal or other relationships with other people or organizations regarding this work.
References
- 1.Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: A causative factor and therapeutic target in neuropathology, including Alzheimer's disease. Proc. Natl. Acad. Sci. USA. 2006;103:5644–5651. doi: 10.1073/pnas.0600549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weisgraber KH, Mahley RW. Human apolipoprotein E: The Alzheimer’s disease connection. FASEB J. 1996;10:1485–1494. doi: 10.1096/fasebj.10.13.8940294. [DOI] [PubMed] [Google Scholar]
- 3.Roses AD. Apolipoprotein E alleles as risk factors in Alzheimer’s disease. Annu. Rev. Med. 1996;47:387–400. doi: 10.1146/annurev.med.47.1.387. [DOI] [PubMed] [Google Scholar]
- 4.Buttini M, Orth M, Bellosta S, Akeefe H, Pitas RE, Wyss-Coray T, Mucke L, Mahley RW. Expression of human apolipoprotein E3 or E4 in the brains of Apoe−/− mice: Isoform-specific effects on neurodegeneration. J. Neurosci. 1999;19:4867–4880. doi: 10.1523/JNEUROSCI.19-12-04867.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ji Y, Gong Y, Gan W, Beach T, Holtzman DM, Wisniewski T. Apolipoprotein E isoform-specific regulation of dendritic spine morphology in apolipoprotein E transgenic mice and Alzheimer's disease patients. Neuroscience. 2003;122:305–315. doi: 10.1016/j.neuroscience.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 6.Levi O, Jongen-Relo AL, Feldon J, Roses AD, Michaelson DM. ApoE4 impairs hippocampal plasticity isoform-specifically and blocks the environmental stimulation of synaptogenesis and memory. Neurobiol. Dis. 2003;13:273–282. doi: 10.1016/s0969-9961(03)00045-7. [DOI] [PubMed] [Google Scholar]
- 7.Trommer BL, Shah C, Yun SH, Gamkrelidze G, Pasternak ES, Ye GL, Sotak M, Sullivan PM, Pasternak JF, LaDu MJ. ApoE isoform affects LTP in human targeted replacement mice. Neuroreport. 2004;15:2655–2658. doi: 10.1097/00001756-200412030-00020. [DOI] [PubMed] [Google Scholar]
- 8.Wang C, Wilson WA, Moore SD, Mace BE, Maeda N, Schmechel DE, Sullivan PM. Human apoE4-targeted replacement mice display synaptic deficits in the absence of neuropathology. Neurobiol. Dis. 2005;18:390–398. doi: 10.1016/j.nbd.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 9.Huang Y, Weisgraber KH, Mucke L, Mahley RW. Apolipoprotein E. Diversity of cellular origins, structural and biophysical properties, and effects in Alzheimer’s disease. J. Mol. Neurosci. 2004;23:189–204. doi: 10.1385/JMN:23:3:189. [DOI] [PubMed] [Google Scholar]
- 10.Hatters DM, Peters-Libeu CA, Weisgraber KH. Apolipoprotein E structure: insights into function. Trends Biochem Sci. 2006;31(8):445–454. doi: 10.1016/j.tibs.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 11.Weisgraber KH. Apolipoprotein E: Structure-function relationships. Adv. Protein Chem. 1994;45:249–302. doi: 10.1016/s0065-3233(08)60642-7. [DOI] [PubMed] [Google Scholar]
- 12.Dong L-M, Weisgraber KH. Human apolipoprotein E4 domain interaction. Arginine 61 and glutamic acid 255 interact to direct the preference for very low density lipoproteins. J. Biol. Chem. 1996;271:19053–19057. doi: 10.1074/jbc.271.32.19053. [DOI] [PubMed] [Google Scholar]
- 13.Morrow JA, Hatters DM, Lu B, Höchtl P, Oberg KA, Rupp B, Weisgraber KH. Apolipoprotein E4 forms a molten globule: A potential basis for its association with disease. J. Biol. Chem. 2002;277:50380–50385. doi: 10.1074/jbc.M204898200. [DOI] [PubMed] [Google Scholar]
- 14.Weisgraber KH, Rall SC, Jr, Mahley RW. Human E apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the apo-E isoforms. J. Biol. Chem. 1981;256:9077–9083. [PubMed] [Google Scholar]
- 15.Raffaï RL, Dong L-M, Farese RV, Jr, Weisgraber KH. Introduction of human apolipoprotein E4 “domain interaction” into mouse apolipoprotein E. Proc. Natl. Acad. Sci. USA. 2001;98:11587–11591. doi: 10.1073/pnas.201279298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hatters DM, Peters-Libeu CA, Weisgraber KH. Engineering conformational destabilization into mouse apolipoprotein E. A model for a unique property of human apolipoprotein E4. J. Biol. Chem. 2005;280:26477–26482. doi: 10.1074/jbc.M503910200. [DOI] [PubMed] [Google Scholar]
- 17.Ramaswamy G, Xu Q, Huang Y, Weisgraber KH. Effect of domain interaction on apolipoprotein E levels in mouse brain. J. Neurosci. 2005;25:10658–10663. doi: 10.1523/JNEUROSCI.1922-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rao VR, Pintchovski SA, Chin J, Peebles CL, Mitra S, Finkbeiner S. AMPA receptors regulate transcription of the plasticity-related immediate-early gene Arc. Nat Neurosci. 2006;9(7):887–895. doi: 10.1038/nn1708. [DOI] [PubMed] [Google Scholar]
- 19.Song JY, Ichtchenko K, Sudhof TC, Brose N. Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc Natl Acad Sci U S A. 1999;96(3):1100–1105. doi: 10.1073/pnas.96.3.1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Masliah E, Terry RD, Alford M, DeTeresa R. Quantitative immunohistochemistry of synaptophysin in human neocortex: An alternative method to estimate density of presynaptic terminals in paraffin sections. J Histochem Cytochem. 1990;38(6):837–844. doi: 10.1177/38.6.2110586. [DOI] [PubMed] [Google Scholar]
- 21.Masliah E, Mallory M, Ge N, Alford M, Veinbergs I, Roses AD. Neurodegeneration in the central nervous system of apoE-deficient mice. Exp. Neurol. 1995;136:107–122. doi: 10.1006/exnr.1995.1088. [DOI] [PubMed] [Google Scholar]
- 22.Palop JJ, Chin J, Bien-Ly N, Massaro C, Yeung BZ, Yu G-Q, Mucke L. Vulnerability of dentate granule cells to disruption of Arc expression in human amyloid precursor protein transgenic mice. J. Neurosci. 2005;25:9686–9693. doi: 10.1523/JNEUROSCI.2829-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y. Profile and regulation of apolipoprotein E (apoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the apoE locus. J. Neurosci. 2006;26:4985–4994. doi: 10.1523/JNEUROSCI.5476-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.tom Dieck S, Sanmarti-Vila L, Langnaese K, Richter K, Kindler S, Soyke A, Wex H, Smalla KH, Kampf U, Franzer JT, Stumm M, Garner CC, Gundelfinger ED. Bassoon, a novel zinc-finger CAG/glutamine-repeat protein selectively localized at the active zone of presynaptic nerve terminals. J Cell Biol. 1998;142(2):499–509. doi: 10.1083/jcb.142.2.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tzingounis AV, Nicoll RA. Arc/Arg3.1: linking gene expression to synaptic plasticity and memory. Neuron. 2006;52(3):403–407. doi: 10.1016/j.neuron.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 26.Varoqueaux F, Aramuni G, Rawson RL, Mohrmann R, Missler M, Gottmann K, Zhang W, Sudhof TC, Brose N. Neuroligins determine synapse maturation and function. Neuron. 2006;51(6):741–754. doi: 10.1016/j.neuron.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 27.Tannenberg RK, Scott HL, Tannenberg AE, Dodd PR. Selective loss of synaptic proteins in Alzheimer's disease: Evidence for an increased severity with APOE varepsilon4. Neurochem Int. 2006;49(7):631–639. doi: 10.1016/j.neuint.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 28.Honer WG. Pathology of presynaptic proteins in Alzheimer's disease: More than simple loss of terminals. Neurobiol Aging. 2003;24(8):1047–1062. doi: 10.1016/j.neurobiolaging.2003.04.005. [DOI] [PubMed] [Google Scholar]
- 29.Reddy PH, Mani G, Park BS, Jacques J, Murdoch G, Whetsell W, Jr, Kaye J, Manczak M. Differential loss of synaptic proteins in Alzheimer's disease: Implications for synaptic dysfunction. J Alzheimers Dis. 2005;7(2):103–117. doi: 10.3233/jad-2005-7203. discussion 173-80. [DOI] [PubMed] [Google Scholar]
- 30.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 31.Blair CK, Folsom AR, Knopman DS, Bray MS, Mosley TH, Boerwinkle E. APOE genotype and cognitive decline in a middle-aged cohort. Neurology. 2005;64(2):268–276. doi: 10.1212/01.WNL.0000149643.91367.8A. [DOI] [PubMed] [Google Scholar]
- 32.Flory JD, Manuck SB, Ferrell RE, Ryan CM, Muldoon MF. Memory performance and the apolipoprotein E polymorphism in a community sample of middle-aged adults. Am J Med Genet. 2000;96(6):707–711. doi: 10.1002/1096-8628(20001204)96:6<707::aid-ajmg1>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 33.Martins CA, Oulhaj A, de Jager CA, Williams JH. APOE alleles predict the rate of cognitive decline in Alzheimer disease: a nonlinear model. Neurology. 2005;65(12):1888–1893. doi: 10.1212/01.wnl.0000188871.74093.12. [DOI] [PubMed] [Google Scholar]
- 34.Casselli RJ, Reiman EM, Locke DonaEC, Hutton ML, Hentz JG, Hoffman-Snyder C, Woodruff BK, Alexander GE, Osborne D. Cognitive domain decline in healthy apolipoprotein E {varepsilon}4 homozygotes before the diagnosis of mild cognitive impairment. Arch Neurol. 2007;64(9):1306–1311. doi: 10.1001/archneur.64.9.1306. [DOI] [PubMed] [Google Scholar]
- 35.Kryscio RJ, Schmitt FA, Salazar JC, Mendiondo MS, Markesbery WR. Risk factors for transitions from normal to mild cognitive impairment and dementia. Neurology. 2006;66(6):828–832. doi: 10.1212/01.wnl.0000203264.71880.45. [DOI] [PubMed] [Google Scholar]
- 36.Ramaswamy G, Weisgraber K. Apolipoprotein E4 domain interaction results in an endoplasmic reticulum stress response in primary astrocytes - Program No. 154.1/R22 - Society for Neuroscience - Neuroscience Meeting Planner - online; 2007. [Google Scholar]
- 37.Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- 38.Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterie H, Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10(5):615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seifert G, Schilling K, Steinhauser C. Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci. 2006;7(3):194–206. doi: 10.1038/nrn1870. [DOI] [PubMed] [Google Scholar]
- 40.Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: The revolution continues. Nat. Rev. Neurosci. 2005;6:626–640. doi: 10.1038/nrn1722. [DOI] [PubMed] [Google Scholar]
- 41.Masliah E, Alford M, DeTeresa R, Mallory M, Hansen L. Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1996;40:759–766. doi: 10.1002/ana.410400512. [DOI] [PubMed] [Google Scholar]
- 42.Göritz C, Mauch DH, Pfrieger FW. Multiple mechanisms mediate cholesterol-induced synaptogenesis in a CNS neuron. Mol Cell Neurosci. 2005;29(2):190–201. doi: 10.1016/j.mcn.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 43.Mauch DH, Nägler K, Schumacher S, Göritz C, Müller E-C, Otto A, Pfrieger FW. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- 44.Slezak M, Pfrieger FW. New roles for astrocytes: Regulation of CNS synaptogenesis. Trends Neurosci. 2003;26(10):531–535. doi: 10.1016/j.tins.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 45.Mitter D, Reisinger C, Hinz B, Hollmann S, Yelamanchili SV, Treiber-Held S, Ohm TG, Herrmann A, Ahnert-Hilger G. The synaptophysin/synaptobrevin interaction critically depends on the cholesterol content. J Neurochem. 2003;84(1):35–42. doi: 10.1046/j.1471-4159.2003.01258.x. [DOI] [PubMed] [Google Scholar]
- 46.Thiele C, Hannah MJ, Fahrenholz F, Huttner WB. Cholesterol binds to synaptophysin and is required for biogenesis of synaptic vesicles. Nat. Cell Biol. 2000;2:42–49. doi: 10.1038/71366. [DOI] [PubMed] [Google Scholar]
- 47.Steinmetz CC, Buard I, Claudepierre T, Nagler K, Pfrieger FW. Regional variations in the glial influence on synapse development in the mouse CNS. J Physiol. 2006;577(Pt 1):249–261. doi: 10.1113/jphysiol.2006.117358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wasser CR, Ertunc M, Liu X, Kavalali ET. Cholesterol-dependent balance between evoked and spontaneous synaptic vesicle recycling. J Physiol. 2007;579(Pt 2):413–429. doi: 10.1113/jphysiol.2006.123133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Witcher MR, Kirov SA, Harris KM. Plasticity of perisynaptic astroglia during synaptogenesis in the mature rat hippocampus. Glia. 2007;55(1):13–23. doi: 10.1002/glia.20415. [DOI] [PubMed] [Google Scholar]
- 50.Suzumura A, Takeuchi H, Zhang G, Kuno R, Mizuno T. Roles of glia-derived cytokines on neuronal degeneration and regeneration. Ann NY Acad Sci. 2006;1088(1):219–229. doi: 10.1196/annals.1366.012. [DOI] [PubMed] [Google Scholar]
- 51.Ye S, Huang Y, Müllendorff K, Dong L, Giedt G, Meng EC, Cohen FE, Kuntz ID, Weisgraber KH, Mahley RW. Apolipoprotein (apo) E4 enhances amyloid β peptide production in cultured neuronal cells: ApoE structure as a potential therapeutic target. Proc. Natl. Acad. Sci. USA. 2005;102:18700–18705. doi: 10.1073/pnas.0508693102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.