

Abstract
Metal uptake by apo-manganese superoxide dismutase in vitro is a complex process exhibiting multiphase “gated” reaction kinetics and a striking sigmoidal temperature profile that has led to a model of conformationally gated metal binding, requiring conversion between “closed” and “open” forms. The present work systematically explores the structural determinants of metal binding in both WT apoprotein and mutational variants as a test of mechanistic models. The pH dependence of metallation under physiological conditions (37°C) shows it is linked to ionization of a single proton with a pKa of 7.7. Size exclusion chromatography demonstrates that the apoprotein is dimeric even when it is fully converted to the open form. The role of molecular motions in metal binding has been probed by using disulfide engineering to introduce covalent constraints into the protein. While restricting motion at domain interfaces has no effect, constraining the subunit interface significantly perturbs metal uptake, but does not prevent the process. Mutagenesis of residues in the active site environment results in a dramatic shift in the transition temperature by as much as 20°C or loss of pH-sensitivity. Based on these results, a mechanism for metal uptake by manganese superoxide dismutase is proposed involving reorientation of active site residues to form a metal entry channel.
Keywords: metal uptake, alternative conformation, gating, kinetics, mutagenesis, disulfide engineering, metallation, metalloprotein
Insertion of the catalytic metal is an essential step in the maturation of every metalloenzyme, yet in spite of its importance, relatively little is known about this aspect of metalloprotein function. A combination of in vivo and in vitro studies suggest that a variety of metallation mechanisms have evolved in biology (1), ranging from chaperoned delivery of toxic metal ions (Cu, Ni) (1-3) or complex metalloclusters (Fe-S) (4,5), to simple reversible binding of metal cofactors (e.g., Ca, Mg) (6). These studies are just beginning, and the mechanism of metal binding is completely unknown for the majority of metalloenzymes.
Manganese superoxide dismutase (MnSOD) is an antioxidant metalloenzyme that serves as a front-line defense against oxidative stress (7,8). It is a member of the highly conserved and ubiquitous (Mn,Fe)-SOD superfamily of enzymes, present in virtually all aerobic organisms and differing essentially only in their specific metal cofactor requirement (Mn or Fe) (7-9). Metal binding by SOD is particularly important because the presence of the metal cofactor is required for catalysis, but binding the wrong metal ion may have toxic effects (10). The proteins are symmetric dimers (11) (or tetramers (12)) formed from identical subunits that are in turn based on a two-domain architecture (including an N-terminal α-hairpin domain, residues 1-99; and a C-terminal α/β domain, residues 100-206, EC MnSOD numbering), with a metal binding site buried in the interior of the protein, surrounded by a second shell of residues on all sides (Fig. 1). In the holoprotein, the metal ion is ligated by amino acid side chains (His, Asp) arising from both N- and C-terminal regions, effectively cross-linking the two domains. Metal binding by E. coli MnSOD has been shown to be moderately strong, but kinetically irreversible as a result of large protein activation barriers (14). The binding of distinct metal ions (e.g., Mn2+, Co2+) is mutually exclusive and kinetically competitive, indicating that there is a single high affinity binding site in each subunit. Curiously, the apoprotein (prepared by chelation of the bound metal under denaturing conditions) is unable to rebind metal under ambient conditions (15). This observation initially led to the conclusion that the binding site of the renatured protein was occupied by an adventitious metal ion (16). We have recently shown that apo-MnSOD, while unable to bind metal at room temperature, undergoes a cooperative, thermally induced transition that allows metals to bind from solution (17-19). Conversion of apo-MnSOD to a form capable of binding metal ions occurs over a narrow temperature range for both mesophilic and thermophilic SOD apoproteins, but the midpoint temperature for the transition varies, typically lying near the optimal growth temperature of the source organism.
Figure 1.

The buried metal binding site in manganese superoxide dismutase. A stereoview cross-section through a single subunit of the homodimeric Escherichia coli MnSOD is shown, with a surface clipping plane bisecting the Mn coordination polyhedron. The solvent-excluded molecular surface of the protein is displayed (probe radius, 1.4 Å). The bound Mn ion (red sphere) and metal-ligating amino acid side chains (white: His26, His81, Asp167, His171) as well as substrate gateway residues (yellow: His30, Tyr34) are rendered as ball-and-stick structures using Chimera (Ref. 13). Based on PDB ID 1vew.
Recently, a direct continous fluorimetic assay has been developed for measuring metal binding by apo-MnSOD based on quenching of intrinsic tryptophan luminescence when Co2+ ion binds in the active site (19). Analysis of the metal binding mechanism of E. coli apo-MnSOD by this method reveals kinetic behavior consistent with conformationally gated metal binding with the protein converting between “open” and “closed” states, ion accessibility being defined empirically in terms of the metal uptake assay (Scheme 1). This gating process must reflect a conformational change associated with one of the protein interfaces, making the buried binding site accessible to metal ions in solution. The structure of the “open” state of apo-MnSOD is presently unknown. The X-ray crystal structure of a hyperthermophilic SOD apoprotein (from Pyrobaculum aerophilum), lacking bound metal ion, is virtually identical to the metallated form, but the structure presumably represents the “closed” state of the apoprotein (20,21). Two models previously proposed for the open state (subunit dissociation and domain separation) (19) both involve large-scale reorganization of the protein structure, a requirement that has never been tested experimentally. The present study uses protein mutagenesis to test both open state models and to probe the structural basis of the gating transition required for metal uptake by apomanganese superoxide dismutase.
Scheme 1.

Conformationally gated metal uptake by MnSOD.
MATERIALS AND METHODS
Biochemical materials
All reagents were from commercial sources and used without purification.
Culture media
Terrific Broth (TB) (12 g/L tryptone, 24 g/L yeast extract, 2.31 g/L potassium phosphate monobasic, 12.54 g/L potassium phosphate dibasic), Luria-Bertani medium (LB) (5 g/L NaCl, 5 g/L yeast extract, 10 g/L tryptone), yeast-peptone-dextrose (YPD) (10 g/L yeast extract, 20 g/L peptone, 20 g/L glucose), supplemented with antibiotics as required for selection (carbenicillin, 100 mg/L; chloramphenicol, 25 mg/L). Super Optimal Broth (SOB) (0.5 g/L NaCl, 2.5 mL 1 M g/L KCl, 1.2 g/L MgSO4, 5 g/L yeast extract, 20 g/L tryptone,; SOC (SOB supplemented with 3 g/L glucose).
Biological materials
Ultracompetent Escherichia coli XL2-Blue cells were from Stratagene (La Jolla, CA). Electrocompetent E. coli cells of other strains were routinely prepared by standard procedures (22) and electrotransformed using an Eppendorf 2510 electoporator with 1 mm cuvette (18 kV/cm).
Construction of the E. coli ΔsodA expression host
An E. coli ΔsodA deletion strain was constructed for MnSOD protein expression, using phage λ Red recombineering methods (23,24). Escherichia coli BW25113 and pKD3, pKD43 and pCP20 plasmids were obtained from Dr. F. Wayne Outten, University of South Carolina. DNA for sodA gene deletion was prepared using oligos REDSOD1-4 (Table 1) with pKD3 and pQGBsodA (18,25) as templates. After reaction, the PCR mixtures were treated with DpnI restriction enzyme to digest the templates, and subsequently heated to 65°C to inactivate the restriction enzyme. Separate PCR products for the sodA 5′ and 3′ genomic flanking regions and the CAT (CmR) insert were joined by overlap extension and the resulting 1320 bp product was used to transform electrocompetent E. coli BW25113 | pKD43 grown in SOB overnight with L-arabinose and carbenicillin. Transformants were recovered in SOC medium and spread on LB/Cm plates for selection. Positive clones were subsequently screened by colony PCR using primers specific for the sodA coding region. One of these clones was made electrocompetent, transformed with pCP20 and selected on LB/C agar at 30°C. Single colonies were streaked onto YPD non-selective media and grown at the non-permissive temperature (43°C) to cure the pCP20 plasmid. Single colonies were subsequently cleaned by restreaking and cultivation at 37°C, and the ΔsodA genotype was verified by PCR using genomic DNA as template.
Table 1.
Oligonucleotides used in this work.
| Name | Sequence |
|---|---|
| RED-SOD-1 | 5′-GGGCATTTTCCTGCAAAAC-3′ |
| RED-SOD-2 | 5′-ACCACATCAATTGAAACGCTG-3′ |
| RED-SOD-3 | 5′-GAAGCAGCTCCAGCCTACACCCAGTATTGTCGGGCGG-3′ |
| RED-SOD-4 | 5′-GGACCATGGCTGATTCCCATGCATTTGCCGCCTGCTG-3′ |
| RED-SOD-5 | 5′-CCGCCCGACAATACTGGGTGTAGGCTGGAGCTGCTTC-3′ |
| RED-SOD-6 | 5′-CAGCAGGCGGCAAATGCATGGGAATTAGCCATGGTCC-3′ |
| BAD-QC-1 | 5′-P-GGTATTTCACACCGCATAAGTGCACTCTCAGTACAATCTG-3′ |
| BAD-QC-2 | 5′-P-GATCTGCAGCTGGTACCAAATGGGAATTCGAAGCTTTC-3′ |
| BAD-QC-3 | 5′-P-GGGCTAACAGGAGGAATTACATATGAAAAAACTGCTGTTCGC-3′ |
| S126C | 5′-P-CGCTTTGGTTGCGGCTGGGCATGG-3′ |
| Y174C | 5′-P-GTGGGAACATGCTTACTGCTTGAAATTCCAGAACCGC-3′ |
| E170C | 5′-P-GCCTGGATGTGTGGTGCCATGCTTACTACTTGAAATTC-3′ |
| R72C | 5′-P-GAAAACCGTACTGTGCAACAACGCTGGC-3′ |
| D147C | 5′-P-CTACTGCTAACCAGTGTTCTCCGCTGATGGG-3′ |
| H17C | 5′-P-ACGATGCCCTGGAACCGTGTTTCGATAAGCAGACCA-3′ |
| N190C | 5′-P-AAAGAGTTCTGGTGCGTGGTTAACTGGGACGAAGCAG-3′ |
| H171A | 5′-P-GCCTGGATGTGTGGGAAGCTGCTTACTACCTGAA-3′ |
Construction of the pBAD2sodA expression vector
The commercially available expression vector pBAD/gIII-A (Invitrogen, Carlsbad, CA) was modified for MnSOD expression, eliminating two NdeI restriction sites (one flanking the pBR322 ori region of the vector and a second NdeI restriction site within the multilinker), and introducing a new NdeI restriction site at the translational initiation site under the PBAD promoter by triple mutagenesis using the mutagenic oligos BAD-QC-1, -2 and -3 (Table 1) and the Quik-Change Multi mutagenesis kit (Stratagene, La Jolla, CA). The pBAD2 product was verified by restriction mapping and/or nucleotide sequence analysis. The sodA coding sequence obtained by NdeI/HindIII digestion of the pQGBsodA plasmid (18,25) was ligated to a similarly digested pBAD2 vector to form pBAD2sodA.
Site-directed mutagenesis
Mutational variants of EC MnSOD were prepared by site-directed mutagenesis of the pBAD2sodA expression plasmid template, using the Quik-Change Multi mutagenesis kit (Stratagene, La Jolla, CA) and the appropriate oligonucleotide primers (Table 1). The sequences of the products were verified by direct sequence analysis (Molecular Biology Core Sequencing Facility, Oregon Health and Science University). Plasmids were transformed into electrocompetent E. coli BW25113 ΔsodA cells for expression.
Recombinant protein expression
E. coli BW25113 | pBAD2sodA expression strains were inoculated into 1 L of Terrific Broth (TB) supplemented with 2 g/L glucose and 100 mg/L carbenicillin and grown to saturation at 37°C with shaking. The cells were collected and resuspended in 1 L TB (minus phosphate) supplemented with 5 mM MnCl2, 2 g/L glycerol and 0.5 g/L L-arabinose. Cells were harvested after incubation at 37°C for 24 h with shaking. EC MnSOD H30A, Y34A, Y34F, E170A, and Y174F variants were expressed as previously described (26-29).
Protein purification
EC MnSOD was purified by a modification of previously described methods (26). The cell-free extract from 15 g of cells was loaded onto an ion exchange column (DE-52, 5×45 cm) and the flow-through SOD fractions were further purified by CM-52 chromatography or chromatofocussing (PBE-94 Polybuffer Exchanger developed with Polybuffer-74 ampholyte).
Formation of apoprotein
Metal-containing MnSOD was denatured in 3.5 M Guanidinium HCl (GnHCl) (pH 3.5) containing 10 mM EDTA and renatured by dialysis against 20 mM Tris/EDTA (pH 7.6) (15,30). EDTA was removed by gel filtration (BioGel P-6). For thiol-containing variants, 2-mercaptoethanol (2-ME) was included when required to prevent disulfide formation. The apoprotein product was stored at -80°C.
Metal reconstitution
Apo-MnSOD (approximately 0.2 mM) in 40 mM MOPS pH 7.8 containing 8 mM MnCl2 was incubated at 37°C for 0.5-2 h. After cooling on ice, the solution was made 8 mM in EDTA and desalted by gel filtration (BioGel P-30) in 20 mM potassium phosphate buffer (pH 7). These reconstituted samples were used for Mn analysis and superoxide dismutase activity assays. Some samples were reconstituted by denaturation/renaturation in GnHCl as described above for preparation of apoprotein, but without EDTA and including 1 mM MnCl2.
Fluorimetric metal uptake assay
Metal uptake measurements were performed using a Cary Eclipse Spectrofluorimeter (Varian, Inc., Walnut Creek, CA) equipped with a Cary temperature controller and a Peltier 4-position multicell holder. Protein samples (50 μg/mL in 20 mM MOPS or TAPS buffer) were excited at 280 nm and emission intensity was monitored at 333 nm using a dynode voltage of 650 V. Metal binding timecourses were initiated by adding an aliquot of 6 mM CoCl2 stock solution to a thermally equilibrated, stirred solution of apo-MnSOD to give a final Co2+ concentration of 24 μM (10 equivalents). Both the protein sample and the metal stock solution were equilibrated at the target temperature for 7 min before starting the reaction. Fluorescence emission intensity was recorded at 1 s-1 sampling frequency for 30 min after addition of metal salts. The kinetic timecourses were imported into a data analysis program (Scientist, Micromath Research, St. Louis, MO) and fit to a multi-exponential relaxation process including two exponential relaxation phases, a linear time-dependent term, and a constant offset (19).
Protein characterization
The concentration of purified SOD was determined by optical absorption measurements, using the published molar extinction coefficient (ε280 nm =8.66×104 M-1cm-1) (31), or by the method of Lowry et al. (32). Superoxide dismutase activity was measured with the xanthine oxidase/cytochrome c inhibition assay (33). Metal analyses were performed using a Varian Instruments SpectrAA Model 20B atomic absorption spectrometer equipped with a GTA 96 graphite furnace.
Protein homogeneity was routinely evaluated using SDS-PAGE. Non-reducing discontinuous SDS-PAGE electrophoresis was performed using 12% Tris-HCl Ready-Gel (Bio-Rad Laboratories, Hercules, CA) to detect formation of covalent cross-links. For reducing SDS-PAGE, 5% 2-ME was included in the sample buffer.
Protein quaternary structure was determined by size exclusion chromatography (SEC) over BioGel P-100 or Sephacryl 100-HR (1.5 × 100 cm) in a thermostatted column with temperature controlled by a circulating water bath. For measurement of SOD apoprotein, the elution buffer contained 1 mM EDTA and the metal-free character of the sample was verified by fluorimetric metal uptake after gel filtration.
Quantitation of free thiols was performed by reaction of protein samples with Ellman's reagent (5,5′-dithio-bis-(2-nitrobenzoic acid), DTNB) with or without 4 M GnHCl, monitoring product formation at 412 nm according to the published procedure (34). The analysis was calibrated using L-cysteine standard.
Thiol oxidation
Protein thiols were oxidized by treating the protein (0.2 mM) in 100 mM potassium phosphate buffer (pH 7.8) containing 1 mM EDTA with 1.2 equivalents (relative to free SH) of 2-iodosobenzoic acid (IBA) (35) for 24 h at room temperature followed by desalting (Bio-Gel P-6). The extent of the reaction was determined by nonreducing SDS-PAGE analysis and the free thiol content of the product was determined by reaction with DTNB.
Bis-bromobimane covalent bridge modification
Apo-MnSOD (R72C/D147C) (0.5 mM) in 50 mM HEPES pH 7.5 containing 4 mM EDTA was reacted with bis-bromobimane (bBm) (36) (1.5 equivalents) at 37°C for 1 h. The mixture was centrifuged and desalted (BioGel P-6) in the dark. The product of the reaction was analyzed by reaction with DTNB, and a sample prepared by extensive dialysis against ammonium acetate buffer and water for mass spectrometry.
Mass analysis
Mass analysis was performed by the Proteomics Shared Resource (Oregon Health and Science University). Prior to mass analysis samples were mixed 1:1 with a solution of 49.9% acetonitrile 49.9% deionized water, and 0.2% formic acid. Samples were then infused into a Applied Biosystems QSTAR XL mass spectrometer with an electrospray ionization source using a syringe pump at a flow rate of 10ul per minute. The resulting mass spectrum was deconvoluted using the Bayesian Protein Reconstruct utility of the Bioanalyst extensions module for Analyst QS 1.1.
RESULTS AND DISCUSSION
1. Biochemical characterization of the gating transition
The biphasic kinetics that have been previously reported for metal uptake by apo-MnSOD (19) imply that metal binding is a conformationally gated, sequential process in which the rate-limiting step is a structural change and not the intrinsic metal binding reaction. In this process, the protein interconverts between the reactive (open gate) and nonreactive (closed gate) conformations (Scheme 1) in a two-state dynamic equilibrium, as demonstrated in earlier temperature-dependence studies. The fraction of open form present in the equilibrium mixture is reflected in the ratio of the fast phase amplitude to the total amplitude (sum of fast and slow phase amplitudes), (A0/ATOT) (Fig. 2, Inset).
Figure 2.

pH dependence of metal binding by Apo-MnSOD. Co2+ uptake was monitored fluorimetrically as described in the Methods. MnSOD (50 ug/mL) in 2.5 mL sample buffer was equilibrated at 37°C for 10 min, and the reaction initiated by addition of 24 μM CoCl2 (approximately 10 stoichiometric equivalents). The pH of each sample was measured directly in the reaction cuvette. (Top) pH dependence of the fast phase gating fraction (A0/ATOT). A fit of the data to a modified Henderson-Hasselbalch equation as described in the text is shown (pKa = 7.7). (Bottom) pH dependence of the slow phase rate constant, kslow. A linear fit to the data is shown (-m = +1.1).
The earlier studies have been extended to investigate the pH sensitivity of the apo-MnSOD metallation process by performing the fluorimetric metal uptake assay in buffers spanning a range of pH values (6.5 - 8.5) at constant temperature (37°C). The open fraction (A0/ATOT) increases from < 0.2 to > 0.8 over this pH range, reflecting extensive conversion to the “open” form of the protein at higher pH (Fig. 2). The data were fit to a modified Henderson-Hasselbalch equation for a pH-dependent equilibrium (Eqn 1), including a Hill coefficient (n) to model titration of interacting sites (37):
| (1) |
This analysis yields an apparent pKa of 7.7 for the pH-dependent process, and a Hill coefficient n = 0.7 (Fig. 2, Top). The magnitude of the Hill coefficient suggests that interaction with a second site titrating over this pH range gives rise to a modest anti-cooperativity for the deprotonation event governing the metal uptake kinetics.
As expected, the rate constant associated with the slow phase (kslow) is also pH dependent. The pH sensitivity of the kinetics arises from a hydrogen ion ([H+]) term in the underlying empirical rate law, leading to expressions that allow proton ionization steps to be detected through the pH dependence of the rate constant (38) (Eqns 2&3):
| (2) |
| (3) |
Eqn 3 describes a log-linear pH-rate relation, with a slope (-m) whose sign (plus or minus) indicates whether the transition state involves proton loss or gain, respectively, and whose magnitude may be interpreted as the number of protons actively involved in the transition state for the rate process. In the present case, the data exhibits a clear a log-linear rate profile with a slope (-m = +1.1) reflecting ionization of a single proton in the closed→open transition (Fig. 2, Bottom).
Repeating the pH profile measurement over a range of temperatures (25 - 49°C) reveals a coupling between protonation and the temperature-dependence of gating equilibria in the protein (Fig. 3). Titration isotherms recorded at different temperatures are dramatically shifted, yielding very different values of the apparent pKa for metal uptake (9 vs. 6) at the low- and high-temperature limits of the range. These results imply that metal uptake is facilitated by deprotonation of a site in the protein, while raising the temperature permits metal uptake to occur at lower pH by increasing the thermal excitation.
Figure 3.

Temperature dependence of the apo-MnSOD gating equilibrium. (Left) pH titration isotherms measured for Co2+ uptake by apo-MnSOD at temperatures T = 25 - 49°C at 2°C intervals (1-13). Data were fit to a modified Henderson-Hasselbalch equation as described in the text and the fitted lines are shown. (Right) Apparent pKa values for metal uptake determined for temperatures ranging from 25°C to 49°C are shown with a polynomial fit to guide the eye. Inset: van'T Hoff analysis of the temperature-dependent apparent pKa for the gating transition. Van'T Hoff parameters: (1) high temperature limit, ΔHvH = 160 kJ/mol, ΔSvH = 450 J/mol·K. (2) low temperature limit, ΔHvH = 70 kJ/mol, ΔSvH = 164 J/mol·K.
The combination of the pH dependence of the open fraction in the fast phase and the log-linear pH-rate profile provide evidence for an essential single proton ionization step in the conformational gating transition, associated with a group having an apparent pKa of 7.7 at 37°C. Based on published values for intrinsic pKas of protein constituents (39), the apparent pKa of 6 observed at temperatures approaching the global unfolding temperature of apo-MnSOD (Tm = 52°C)(14) is in the range typical of solvent-exposed His side chains in proteins. At lower temperatures, the apparent pKa increases to values consistent with His in a salt bridge (e.g., carboxylate complex) (40,41). The temperature dependence of the apparent pKa suggests a decrease in the strength of the salt bridge at elevated temperatures. While these results appear to implicate a His residue in the gating transition, it does not specify which of the eight His residues in EC MnSOD is involved, or the nature of the conformational gating process.
A van't Hoff analysis of the data (Fig. 3, Inset) allows the temperature-dependence of the apparent pKa to be interpreted in terms of thermodynamic parameters. The limiting slope at low temperature (25°C) yields estimates ΔHvH = 70 kJ/mol, ΔSvH = 164 J/mol·K; and at high temperature (45°C), ΔHvH = 160 kJ/mol, ΔSvH = 450 J/mol·K. The latter results are similar to those previously obtained from analysis of the temperature dependence of the gating equilibrium at constant pH (7.0).
The sensitivity of the gating transition to both temperature and pH has made it possible to identify experimental conditions (pH 7.5, 45°) under which the apoprotein is fully converted to the open state. Analysis of the protein under these conditions should allow the structural basis for the conformational gating to be defined. Specifically, it suggests a critical test of a requirement for dissociation of the subunits in the gating transition.
Size exclusion chromatography (SEC) apo-MnSOD in 20 mM MOPS buffer (pH 7.5) containing 1 mM EDTA at 45°C (or 20 mM TAPS buffer pH 9 at room temperature) indicates that under these conditions the protein remains dimeric, with no significant dissociation of the subunits, although elution of the apoprotein is delayed slightly compared to the Mn2-MnSOD holoprotein (Fig. 4). Under the same conditions, the MnSOD E170A variant, which has been previously shown to extensively dissociate in solution (26), is found to be purely monomeric by SEC (Fig. 4). These results appear to exclude subunit dissociation as a requirement for conversion to the open state. However, the subunit dissociation model can be tested even more stringently by introducing covalent constraints into the protein structure (see below).
Figure 4.

Size exclusion chromatography of apo-MnSOD. A thermostatted column of BioGel P-100 (1.5×100 cm) equilibrated with 20 mM MOPS (pH 7.5) containing 1 mM EDTA at 45°C was loaded with apo-MnSOD (trace A), Fe2-MnSOD (E170A) (trace B); or a MW calibration mixture (trace C: (1) bovine serum albumin; (2) Mn2-MnSOD; (3) carbonic anhydrase).
2. Engineering disulfide constraints
Two models previously proposed for the conformational gating transition of apo-MnSOD (subunit dissociation and domain separation), both require the folded protein to open up at specific molecular interfaces. One way to experimentally test whether displacement at an interface is involved in metal uptake is to constrain the interface with a covalent cross-link and measure the effect on the process. The absence of cysteine residues in WT EC MnSOD suggests the possibility of performing Cys substitution mutagenesis to constrain the structure with disulfides, an approach that has proven effective in probing molecular motions in other proteins (42-44).
In order to implement this strategy it is first necessary to identify residues that should readily form unstrained disulfides in the protein when replaced with Cys. Structural studies suggest that Cβ carbons in a protein disulfide should optimally be separated by 3.5 - 5 Å (45,46), so residue pairs in EC MnSOD having Cβ carbons within this separation range were identified by importing the Cβ atomic coordinates for metallated EC Mn2-MnSOD (PDB ID 1vew) into a spreadsheet analysis program (Microsoft EXCEL) and using the geometric constraint as a search criterion. The targets identified by this method include three residues on the symmetric dimer interface of the protein (Ser126, Glu170 and Tyr174)(Fig. 5) where Cys substitution is predicted to allow disulfides for form with the corresponding residue in the opposing subunit, as well as a pair of residues (His17, Asn190) whose replacement by Cys could serve to constrain the first helix of the N-terminal domain, which covers the metal binding site (Fig. 5). Based on the selection criterion, no suitable residue pairs were found that could be used to introduce a disulfide constraint at the opposite end of the domain interface. However, an Arg-Asp salt-bridge “clasp” feature (Arg72, Asp147) that is highly conserved over MnSOD structures was selected as a starting point for constraining the domain motions through Cys engineering.
Figure 5.
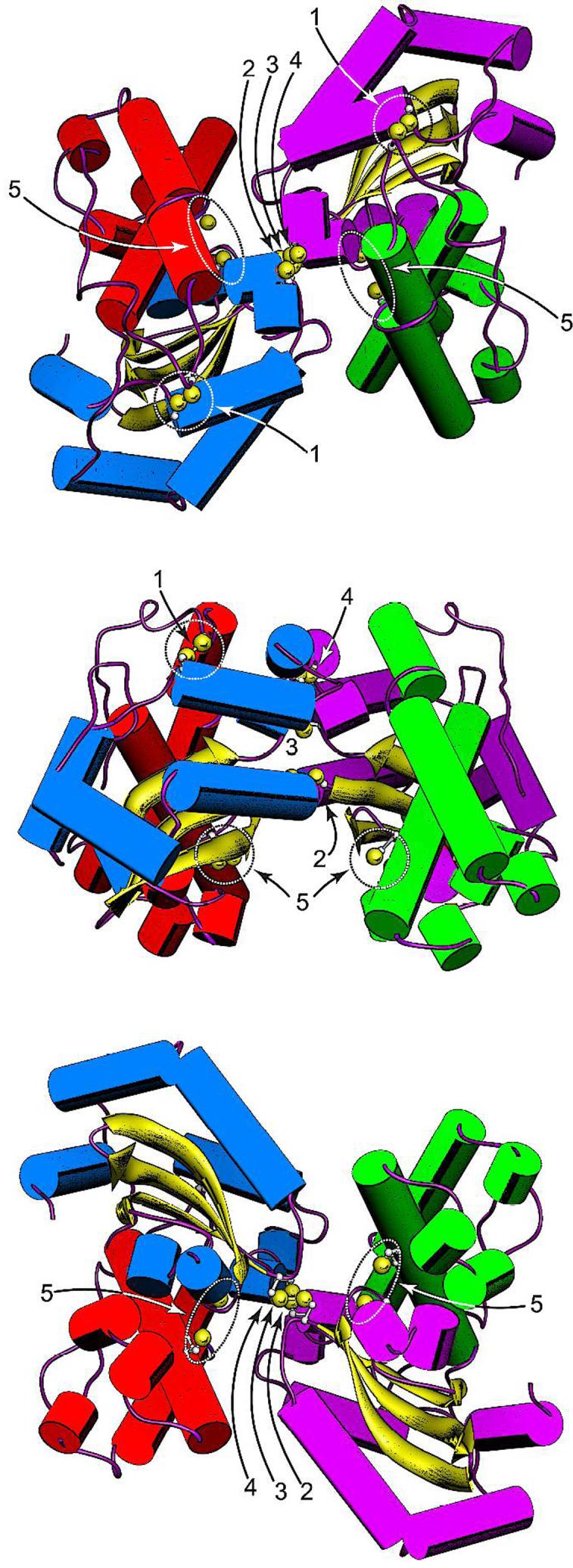
Disulfide constraints introduced into the EC MnSOD structure by Cys substitution mutagenesis. (Top) Top view of the MnSOD dimer, parallel to the molecular two-fold axis on the N-terminal domain side. (Middle) Side view of the MnSOD dimer, perpendicular to the molecular twofold axis. (Bottom) Bottom view of the MnSOD dimer, parallel to the molecular two-fold axis on the C-terminal domain side. The cysteines shown are hypothetical structures based on replacement of the corresponding residues in WT MnSOD (PDB ID 1vew) with Cys and manually adjusting the side chain dihedrals. Cysteines are rendered with ball-and-stick side chains, with Cys SG sulfur shown as a van der Waals sphere. In each view, the individual domains and subunits are identified by helix color. Subunit A: N-domain, green; C-domain, purple. Subunit B: N-domain, red; C-domain, blue. (1) H17CN190C disulfide; (2) S126CA-S126CB disulfide; (3) E170CA-E1706CB disulfide; (4) Y174CA-Y174CB disulfide; (5) R72C, D147C pair. Rendered using Chimera (Ref. 13).
Each of the mutational variants was constructed by site-directed mutagenesis of the pBAD2sodA expression vector, which places the EC MnSOD gene under control of the tight, strong arabinose promoter (47), and the protein products were purified from a specially constructed expression host (a ΔsodA deletion strain of E. coli BW25113 created by phage λ Red recombineering methods (23,24)) to preclude contamination with wild-type MnSOD. The pBAD2sodA expression vector has proven to be very efficient, routinely yielding >250 mg purified EC MnSOD protein from 15 g cells in 1 L expression cultures under L-arabinose induction. The recombinant protein was purified by ion exchange and chomatofocussing chromatography.
Nonreducing denaturing electrophoresis (SDS-PAGE) was routinely used to evaluate the disulfide status of the protein, taking advantage of the distinct electrophoretic mobilities of thiol- and disulfide-containing forms. Subunit-bridging disulfides result in the denatured protein migrating at approximately twice the monomer mass, while more subtle changes are produced by intra-chain disulfides. Linkage isomers of interchain disulfides also produce subtle variations in electrophoretic mobility that can be used to assign the site of the cross-link. For each sample, thiol quantitation by DTNB assay was performed to determine the free sulfhydryl content of the purified protein. Most of the Cys variants were isolated as reduced, thiol-containing species, requiring oxidation to form the disulfide.
Iodosobenzoic acid (IBA) (35) was used to convert the thiol-containing protein to the disulfide form, as described in the Methods. Depending on the specific variant (see below), IBA was added to either the metal-containing holoprotein, or to the apoprotein following metal extraction. The progress of the oxidation reaction was monitored by nonreducing SDS-PAGE. Following oxidation, excess IBA reagent was removed by desalting and the protein characterized by SDS-PAGE, and DTNB analysis. The fluorimetric metal uptake assay (19) (see Methods) was used to evaluate the metal binding behavior of the variants (Fig. 6).
Figure 6.

Representative metal uptake timecourses for apo-MnSOD variants. Co2+ was added to apo-MnSOD (50 μg/mL in 20 mM MOPS) at t = 0 and binding was monitored fluorimetrically as described in the Methods. Reaction timecourses for mutational variants are presented as a stack plot. Four distinct kinetic patterns are distinguished: (A) biphasic, indistinguishable from WT; (B) biphasic, distinct from WT; (C) monophasic, fast-reacting; (D) monophasic, slow-reacting. Individual traces: (1) WT apo-MnSOD, 37°C, pH 7.6; (2) apo-MnSOD (R72C/D174C/bBm), 37°C, pH 7.6; (3) apo-MnSOD (H17C/N190C) (-SS-), 37°C, pH 7.6; (4) apo-MnSOD H30A, 33°C, pH 7.0; (5) apo-MnSOD Y174F, 37°C, pH 7.6; (6) apo-MnSOD Y34A, 31°C, pH 7.0; (7) apo-MnSOD Y34F, 37°C, pH 7.6; (8) apo-MnSOD S126C (-SS-), 37°C, pH 8.8; (9) apo-MnSOD S126C (-SH), 25°C, pH 7.6; (10) apo-MnSOD (S126C/Y174C) 2×(-SS-), 37°C, pH 7.6; (11) apo-MnSOD E170A, 37°C, pH 7.0; (12) apo-MnSOD E170C (-SS-), 37°C, pH 7.6;; (13) apo-MnSOD Y174C (-SS-), 37°C, pH 7.6; (14) apo-MnSOD H171A, 37°C, pH 7.0.
A. (R72C/D147C)
The domain separation model for MnSOD metal uptake was based on the observation that the metal binding site lies on the domain interface, and that the contact surface between the two domains might permit them to open, making the binding site accessible to metal ions in solution. As noted above, screening Cβ separations did not identify any pairs of residues appropriate for introducing a disulfide bond at this domain interface. However, two residues that form a “clasp” salt bridge between the domains (Arg72 and Asp147, for which a Cβ separation of 7.2 Å is predicted) were selected as a starting point for constraining this region of the protein structure (Fig. 5). Substitution of both of these residues with Cys yielded the MnSOD (R72C/D147C) variant, which, as expected, only formed disulfides under forcing conditions that resulted in polymerization of the protein. However, the predicted separation between SG sulfhydryls in the (R72C/D147C) variant (approximately 4.7 Å) is within the span predicted for a bifunctional sulfhydryl reagent, bis-bromobimane (bBm) (36) (3.17 - 6.61 Å (48)). Formation of a covalent bridge between the Cys residues was monitored fluorimetrically, and the product was characterized by mass spectrometry (Table 2), non-reducing SDS-PAGE and DTNB assay (Table 3), confirming the presence of the cross-link and the absence of intermolecular cross-links. Metal uptake by apo-MnSOD (R72C/D147C/bBm) (Fig. 6, trace 2) was essentially indistinguishable from that observed for WT protein (Fig. 6, trace 1) demonstrating that the clasp region of the domain interface is not directly involved in the metal uptake process. The high specific activity of the Mn derivative containing the bBm bridge (Table 3) also shows that the modification has little effect on the active site.
Table 2.
Mass analysis of MnSOD R72C/D147C variant.
| Molecular Species | Mass Shift Relative to WT MnSODa (amu) | |
|---|---|---|
| predicted | observed | |
| R72C/D147C 2×(-SH) | -66 | -65 |
| R72C/D147C bBBm adductb | +103 | +101 |
Determined by ESI-MS of the protein as described in the Methods.
Prepared as described in the Methods.
Table 3.
Properties of EC MnSOD cysteine variants
| Variant | Free SHa(mol SH/ mol protein) | Thiol Status | Metallation state | Specific Activity (U/mg) | Quaternary Structure | Mn Content | Metal Uptakeh |
|---|---|---|---|---|---|---|---|
| S126C | 0.99 | SH | Apob | - | Monomerf | + | |
| Mnc | 4815±232 (66%)e | Dimerf | 0.89±0.01 | ||||
| S126C | 0.02 | -SS- | Apob | - | Dimerf,g | + | |
| Mnc | 4741±82 (65%)e | Dimerf,g | 0.95±0.04 | ||||
| Y174C | 0.02 | -SS- | Apob | - | Dimerg | + | |
| Mnc | 1123±20 (28%)e | n.d. | 0.99±0.01 | ||||
| S126C/Y174C | 0.07 | 2×(-SS-) | Apob | - | Dimerg | + | |
| Mnc | 103±1 (2%)e | n.d. | 0.20±0.01 | ||||
| Mnd | - | n.d. | 0.05±0.02 | ||||
| E170C | 0.02 | (-SS-) | Apob | - | Dimerg | + | |
| Mnc | 44±2 (0.6%) | n.d. | 0.48±0.005 | ||||
| R72C/D147C-bBm | 0.01 | Apob | - | n.d. | + | ||
| Mnc | 6287±70 (86%) | n.d. | 0.89±0.03 | ||||
| H17C/N190C | 0.02 | (-SS-) | Apob | - | n.d. | + | |
| Mnc | 8267±232(113%) | n.d. | 1.02±0.05 |
Based on DTNB assay
Before metal uptake experiment.
After metal reconstitution.
After denaturation/renaturation/reconstitution in presence of Mn2+.
Relative to WT MnSOD (7328±216 U/mg)
Based on size exclusion chromatography
Based on nonreducing SDS-PAGE
Evaluated by Co2+ quenching in the fluorimetric metal uptake assay.
B. (H17C/N190C)
A second domain interface that might be involved in metal binding is formed by the contact between first α-helix of the N-terminal domain (Helix-1) and the C-terminal domain. Helix-1 donates one of the metal ligands (His26) as well as the two substrate gateway residues (His30, Tyr34) to the active site structure, and separation between Helix-1 from the C-terminal domain would open a path into the active site. In order to test the possible involvement of Helix-1 motion in the metal uptake process, the helix was constrained by introducing Cys residues in the preceding turn (H17C) as well as in an adjacent C-terminal helix (N190C) (Fig. 5). The relatively large Cβ separation for this pair of residues (4.5 Å) makes it likely that a disulfide formed between them will be in an extended conformation, preventing separation of these structural elements. The apo-MnSOD (H17C/N190C) disulfide performed a metal uptake reaction indistinguishable from WT protein (Fig. 6, trace 3) and the Mn complex is fully active (Table 3). The absence of any detectable perturbation of metal uptake by constraints restricting the domain interface at either end of the molecule provide a clear indication that domain separation is not required for metal binding by apo-MnSOD.
C. S126C
In WT MnSOD, Ser126 lies on the dimer interface close to the molecular 2-fold symmetry axis, the hydroxyl group forming a hydrogen bond with the corresponding residue in the opposing subunit (Fig. 5). The Cys substitution variant EC MnSOD S126C was isolated as the free thiol form, which SEC shows is monomeric in solution (Table 3). Ionization of the opposing cysteine thiols on the subunit interface of this variant will generate a pair of thiolate anions, contributing to electrostatic destabilization of the dimer and possibly accounting for dissociation of the subunits. Metal uptake by apo-MnSOD S126C (SH) is very fast (Fig. 6, trace 9), similar to apo-MnSOD E170A (Fig. 6, trace 11) (see below), which is also monomeric (26). The product (Mn2-MnSOD S126C (SH)) is predominantly dimeric in solution, and has approximately 65% WT SOD activity (Table 3).
Oxidation of apo-MnSOD S126C by IBA proceeds smoothly yielding the disulfide-crosslinked dimer. The homologous Mycobacterium tuberculosis FeSOD S123C variant, which has recently been reported (49), was also expressed in E. coli as a thiol form, although in that case the quaternary structure was not investigated. The thiol form of M. tuberculosis FeSOD S123C spontaneously oxidized to the disulfide during months-long crystallization in air, and X-ray crystallography confirmed that the presence of the Cys123 disulfide did not perturb the structure of the protein.
Metal uptake kinetics in the fluorimetric assay shows a distinctly WT-like biphasic reaction progress curve for apo-MnSOD S126C (-SS-) (Fig. 6, trace 8). Manganese is also stably bound, and the Mn2-MnSOD S126C (-SS-) exhibits 66% of WT catalytic activity (Table 3). The Cys126 disulfide is a covalent cross-link that prevents separation of the subunits and constrains motion of the lower part of the subunit interface (Fig. 5). Thus, metal uptake by apo-MnSOD S126C (-SS-) excludes complete dissociation of the subunits as a requirement for metal binding. However, in principle, partial separation of the subunit interface could still occur in this single-disulfide variant, with the disulfide serving as a pivot point.
D. Y174C
Apo-MnSOD Y174C (-SS-) disulfide, formed by metal extraction from purified EC MnSOD Y174C (SH), refolding and oxidization by IBA, covalently cross-links the dimer across the subunit interface, at the opposite edge from the Cys126 disulfide (Fig. 5). Replacement of Tyr174 by Cys also removes a residue that forms a conserved hydrogen bond to one of the gateway residues (His30) in WT MnSOD (Fig. 1). Metal uptake by apo-MnSOD Y174C (-SS-) is monophasic and relatively slow (Fig. 6, trace 13), indicating that metal binding is significantly perturbed in this variant, but still proceeds and the protein binds and retains the full complement of metal ion (Table 3). Metal uptake by the covalently cross-linked dimeric protein demonstrates that complete dissociation of the subunits is not required for metal binding. However, some restricted motion of the subunit interface may occur in the MnSOD Y174C disulfide, as noted above for the MnSOD S126C disulfide.
E. (S126C/Y174C)
A dicysteine-variant was constructed to more completely constrain motion at the subunit interface, and exclude the possibility of a “butterfly” motion of the protein involving partial separation of the subunits. The two cysteines in this variant (Cys126, Cys174) form a pair of disulfides with the corresponding residues in the opposing subunit, constraining the interface at opposite sides of the molecule (Fig. 5).
MnSOD (S126C/Y174C) was treated with IBA to form the double disulfide, from which metal extraction and refolding afforded apo-MnSOD (S126C/Y174C) 2×(-SS-). The fluorimetric metal uptake assay demonstrates that this variant is able to bind Co2+ ion (Fig. 6, trace 10) although the metal ion is relatively weakly bound, and is lost during desalting in the presence of EDTA, and denaturation-renaturation in the presence of metal ion (Table 3). The progress curve for the metal uptake reaction is consistent with predominantly “open” form for this variant, and the inability of the protein to retain the metal ion might indicate that conversion to the “closed” metal complex is blocked.
F. E170C
Previous studies have demonstrated the importance of a conserved outer-sphere glutamate (Glu170) in the structure and function of the EC MnSOD active site (26). The salt bridge cross-link formed by Glu170 with the coordinating His171 in the opposing subunit is an important element stabilizing the dimeric organization of the protein, and replacement by alanine (MnSOD E170A) results in dissociation of the metallated protein into monomers, and also appears to alter the metal binding selectivity of the apoprotein in vivo (26). Like Ser126 and Tyr174, Glu170 spans the midpoint of the subunit interface close to the molecular two-fold symmetry axis, and replacement by Cys would be expected to stabilize the dimeric structure, forming a covalent disulfide cross-link between the Cys170 residues arising from opposing subunits (Fig. 5).
MnSOD (E170C) was isolated as the metallated disulfide form following purification. Metal extraction and refolding afforded apo-MnSOD (E170C) (-SS-) disulfide. Metal binding is observed in the fluorimetric metal uptake assay, with essentially monophasic kinetics (Fig. 6, trace 12) lacking pH dependence (-m = -0.04), suggesting that the site responsible for the proton sensitivity is severely perturbed in this variant. Approximately half of the bound metal appears to be lost during desalting in the presence of EDTA (Table 3). The relative stability of the half-apo complex (compared to holo-) may reflect the role of Glu170 as a charge-compensating outer sphere counterion in the active site. Summing charges on the buried metal ion and its coordinating ligands leads to a +1 overall charge on the metal complex in SOD, which is normally balanced by the -1 charge provided by the Glu170 carboxylate in the outer sphere. However, in MnSOD E170C disulfide, this charge compensation is absent, and the two metal cations in adjacent subunits experience electrostatic repulsion when the sites are fully occupied, which is relieved when one of the metal ions dissociates.
Disulfide engineering thus provides clear evidence excluding subunit dissociation as the basis for apo-MnSOD metal binding, and also excludes several domain interfaces from contributing to the process. However, the dramatic effect of several of the inter-subunit cross-links on the metal binding reaction suggests that some motion at the subunit interface may be required for metal uptake to occur.
3. Active site variants
Residues surrounding the active site of EC MnSOD form an extensive hydrogen-bonding web whose role in catalysis has previously been investigated by mutagenesis (26-29). Mutations have targeted the “substrate gateway” residues His30 and Tyr34 (H30A, Y34F, Y34A), as well as the interface-spanning residue Tyr174 (Y174F). X-ray crystal structures have been solved for Mn complexes of several of these variants. In order to refine the apo-MnSOD metal binding mechanism and elucidate the structural basis for the conformational gating transition, the metal uptake behavior of these variants has been explored.
A. H30A
His30 forms half of the substrate gateway (Fig. 1) which is thought to restrict access to the active site metal ion in SOD, conferring substrate selectivity and possibly facilitating proton transfer steps in the redox turnover mechanism (28). The substrate gateway is also a barrier to metal binding, so displacement of the gateway residues might provide a pathway for metal entry into the active site. In addition to its role as a gateway residue, His30 is also involved in a π-stacking interaction with His171, and forms a hydrogen bond with Tyr174. Replacement of His30 by alanine (MnSOD H30A) removes the steric bulk of the His side chain and at the same time disrupts interactions with His171 and Tyr174. The structure of the metallated Mn2-MnSOD H30A protein has previously been reported (PDB ID 1io8) (28).
Apo-MnSOD H30A exhibits rapid metal uptake at 37°C, characteristic of the “open” state behavior of WT apo-MnSOD. However, at lower temperature the biphasic metal uptake kinetics are restored (Fig. 6, trace 4), associated with a sigmoidal temperature dependence (Tm = 30°C) (Fig. 7, curve 3). It is clear that while H30A substitution significantly perturbs the structural elements involved in the conformational gating process (shifting the closed-open equilibrium in favor of the open form) those features are still present in this variant. The pH dependence of the gating transition is retained but similarly shifted, with an apparent pKa of 7.7 at 25°C, and a log-linear rate dependence slope (-m = +0.7) (data not shown), indicating that the group responsible for the pH sensitivity of metal uptake is still present in this variant.
Figure 7.

Temperature dependence of metal uptake for active site variants of MnSOD. Apoprotein (50 μg/mL) in 20 mM MOPS pH 7 was analyzed by the fluorimetric metal uptake assay as described in the Methods. Kinetic time courses were analyzed to extract the gating fraction in the fast phase (A0/ATOT) for each temperature and the temperature dependence fit to a two-state thermal melting curve. (1) apo-MnSOD (Y34F), Tm = 24°C; (2) apo-MnSOD (Y34A), Tm = 28°C; (3) apo-MnSOD (H30A), Tm = 30°C; (4) apo-MnSOD (Y174F), Tm = 39°C; (5) apo-MnSOD (H171A), Tm = 43°C; (6) WT apo-MnSOD, Tm = 45°C.
B. Y34A
Tyr34 combines with H30 to form the substrate gateway in MnSOD. As with MnSOD H30A, replacement of Tyr34 with alanine (MnSOD Y34A) is predicted to remove a significant steric barrier to access to the metal binding site, mimicking displacement of the side chain. However, Y34A substitution will also disrupt the extensive hydrogen bonding web surrounding the active site.
As observed for apo-MnSOD H30A, apo-MnSOD Y34A exhibits perturbed biphasic metal binding reaction (Fig. 6, trace 6), with the midpoint for the temperature-dependent metal uptake profile shifted to 28°C(Fig. 7, curve 2). Again, while strongly perturbed, the characteristic features of the gated metal uptake process appear to be preserved in this variant.
C. Y34F
Conservative replacement of Tyr34 by phenylalanine retains most of the steric bulk of the side chain, and the X-ray crystal structure of this variant (PDB ID 1en5) shows that the phenylalanine ring position is essentially superimposable with the Tyr34 ring it replaces, but elimination of the Tyr34 hydroxyl group significantly disrupts the hydrogen bonding web around the active site (27). The metal uptake behavior of apo-MnSOD Y34F resembles that observed for other substrate gateway variants (H30A, Y34A), in that the biphasic conformational gating signature is preserved (Fig. 6, trace 7), but shifted even more dramatically (Tm = 24°C) (Fig. 7, curve 1). This suggests the conformational gating transition is sensitive to the hydrogen bonding character of the substrate gateway residues, rather than their steric bulk. As with apo-MnSOD H30A, the pH dependence of the gating transition of apo-MnSOD Y34F is shifted to a lower pH range with an apparent pKa of 6.8 at 25°C, although preserving the kinetic sensitivity to pH (-m = +0.6) (data not shown).
All three mutational variants (H30A, Y34A, Y34F) retain the metal cofactor after reconstitution, and exhibit activity (data not shown) similar to the as-isolated metal complex reported previously (27-29). Interestingly, these variants appear to have a greater effect on metal binding than on catalysis, suggesting that conservation of these residues may be driven to a significant extent by their contribution to the metal uptake process.
D. Y174F
Replacement of the subunit interface-spanning residue Tyr174 by Cys disulfide permits metal uptake to occur, although the uptake kinetics are significantly perturbed by the substitution (see above). Substitution of this residue by phenylalanine (MnSOD Y174F) has been shown to affect the structure of the protein at the subunit interface, as a result of eliminating a hydrogen bond with His30. The previously reported crystal structure of Mn2-MnsSOD Y174F (PDB ID 1i0h, 1ixB, 1ix9) indicates that Y174F substitution alters the orientation of the H30 ring and results in a local reorganization of the dimer interface (29). However, metal uptake by apo-MnSOD Y174F still exhibits conformational gating (Fig. 6, trace 5), and is actually less dramatically perturbed compared to the other active site variants, with Tm = 39°C (Fig. 7, curve 4).
E. E170A
Removing the subunit interface-spanning Glu170 residue destabilizes the dimeric structure of EC MnsSOD, resulting in extensive dissociation into monomers (26) (Fig. 4). Disruption of the dimer dramatically alters metal uptake, and this variant is characterized by rapid, monophasic metal binding kinetics (Fig. 6, trace 11), similar to those observed for apo-MnSOD S126C (-SH).
F. H171A
His171 is the partner to Glu170 in the dimer-stabilizing salt bridge, and serves as a ligand in the metal complex. Replacement by alanine (MnSOD H171A) does not have as dramatic effect on quaternary structure as E170A substitution, since Glu170 also makes hydrogen bonding contacts with a backbone peptide in the opposing subunit, and MnSOD H171A is experimentally found to be dimeric in solution. Metal uptake by apo-MnSOD H171A is perturbed, but the kinetics are distinct from those observed for the other active site variants described above. The fast phase amplitude of the conformational gating transition is very small at 37°C, yielding essentially monophasic metal uptake kinetics (Fig. 6, trace 14) that are virtually independent of pH (-m = +0.15). In contrast to the pH dependence of metal uptake by WT apo-protein and H30A and Y34F variants, where a proton molecularity approaches unity (-m= +1.1, +0.7, and +0.6, respectively), analysis of the pH dependence for apo-MnSOD H171A reflects loss of the proton ionization step in the metal uptake reaction of the variant protein, as is also seen for MnSOD E170C (-SS-)(see above). On the other hand, the temperature-dependence of metal uptake is nearly identical to WT apo-MnSOD (Fig. 7, curve 5). The H171A mutation thus appears to uncouple thermal and pH effects on metal binding by apo-MnSOD.
The properties of these active site variants allow the structural basis for the gating transition in apo-MnSOD to be more clearly defined. While the substrate gateway residues (His30,Tyr34) represent the most obvious barrier to metal binding in the outer sphere of the metal binding site, their replacement with less bulky residues only perturbs - but does not eliminate - the characteristic biphasic kinetics and pH sensitivity of metal uptake by apo-MnSOD. The effects of these substrate gateway mutations is most likely indirect, acting through π-stacking and hydrogen-bonding networks in the active site environment. Only apo-MnSOD E170C (-SS-) and apo-MnSOD H171A exhibit metal uptake kinetics lacking pH sensitivity. As discussed above (Section 1), the pH dependence implicates a salt bridge in the protein in the conformational gating process, and the observed pKa is consistent with deprotonation of a histidinecarboxylate ion pair in the closed→open transition, focusing attention on the interaction between Glu170 and His171.
4. A model for the conformational gating transition
The evidence accumulated from these studies suggests that the salt bridge between Glu170 and His171 plays a key role in the conformational gating transition of apo-MnSOD. However, if these residues are responsible for gating metal uptake, they must rearrange in the “open” state since they block access to the active site region in the metallated protein (Fig. 1) that corresponds to the “closed” form of the apoprotein. Analysis of the metal uptake behavior of apo-MnSOD variants demonstrates that subunit dissociation is not required for metal binding, although some motion of the subunit interface appears to be involved in the gating transition, associated with a more subtle reorganization of the subunit interface. Inspection of the crystal structure of the metallated protein (PDB 1vew) suggests an alternative conformation that might represent the “open” form of the complex, generated by simple side chain torsions for Glu170 and His171 without any change in the peptide backbone (Fig. 8). Rotation of the Glu170 side chain allows the carboxylate headgroup to ion pair with a pair of residues (Tyr173, Arg181) which are highly conserved over Mn,Fe-SOD sequences, yet have no recognized function. Reorientation of His171 can then proceed through simple rotation around the Cα-Cβ bond, forming a hydrogen bond between the imidazole ring and the Glu170 carboxylate. While this alternative conformation has never been observed in any metallated SOD crystal structure, it is consistent with the evidence for a critical role for Glu170 and His171 in the conformational gating process, as well as the requirement for motion on the subunit interface for the gating transition to occur. Other residues in the active site environment (including His30 and Tyr34) are clearly important in determining the relative stability of the alternative structures.
Figure 8.

Proposed structural basis for the conformational gating transition. (Top) Closed state model, stabilized by a hydrogen bond salt bridge between the interface-spanning glutamate (Glu170B) and the active site residue (His171A) within the opposing subunit. Based on the structure of the metallated MnSOD (PDB ID 1vew). Side chain dihedral rotations that generate the open state structure are indicated by the curved arrows. (Bottom) Hypothetical model for the open state structure of apo-MnSOD. Reorientation of Glu170B glutamate within the conserved inter-subunit channel is facilitated by formation of a salt bridge with the arginine residue (Arg181B) and a hydrogen bond with tyrosine (Tyr173B). Rotation of the His171A side chain, in turn, permits it to form a hydrogen bond with the carboxylate of Glu170B. In the open conformation, a continuous channel is formed leading into the metal binding site from solution (Figure 9, Bottom).
This model also accounts for the ready access of metal ions in solution to the metal binding site in the open state. Reorientation of the side chains opens a new channel into the buried binding site (Fig. 9, Bottom), lined by substrate gateway residues (His30,Tyr34) and metal gateway residues (Glu170,His171, Tyr173,Arg181). The charge status of the residues in this open state is also favorable for metal entry through the channel. The Glu170 carboxylate (-1) neutralizes the +1 charge of the Arg181 guanidinium group, removing an electrostatic barrier for binding a metal cation, and the His171 side chain is deprotonated to a neutral imidazole in the open form. The proposed reorientation of His171 also permits coordination of the metal at the NE2 nitrogen, which ligates the metal ion in the holoprotein, as it enters the metal entry channel. There is no obvious obstacle to this structural rearrangement, which occurs in a conserved channel that transects the dimer interface just below the Glu170-His171 salt bridge (Fig. 9, Top) and previously had no known function. While it is difficult to evaluate all of the factors contributing to the relative stability of the open and closed conformations, their interconversion under mild conditions (temperature, pH) suggests that these two states are closely matched energetically.
Figure 9.

Proposed metal entry channel for apo-manganese superoxide dismutase. (Top) Stereoview molecular surface representation (1.4 Å probe radius) of the active site environment of native, metallated E. coli MnSOD, corresponding to the closed state apoprotein structure (Figure 8, Top), with the buried metal binding site lying behind the His171A side chain. The surface color reflects the contribution of specific residues. (Bottom) Stereoview molecular surface view (1.4 Å probe radius) of the active site environment in the open state structure model (Figure 8, Bottom) from the same viewpoint as above, showing how reorientation of the side chains creates a metal entry channel leading from solution into the buried metal binding site. The surface color reflects the contribution of specific residues. Residue code: magenta, His30A; blue, Tyr34A; green, Trp169A; yellow, His171A; red, Glu170B; turquoise, Arg181B. Rendered using Chimera (Ref. 13). Based on PDB ID 1vew.
CONCLUSIONS
Conformational gating of metal uptake by apo-MnSOD is a striking feature of the metal binding process reflecting unexpected dynamic features of the protein structure. Previous models that have been proposed for the open state of apo-MnSOD required large-scale reorganization of the protein structure, involving either subunit dissociation or domain separation. Both models are now excluded by studies of metal uptake by MnSOD variants in which those interfaces are constrained by covalent disulfide cross-linking. A detailed characterization of the properties of WT protein and mutational variants demonstrates that metal uptake by apo-MnSOD can occur under physiological conditions (temperature, pH) and provides new insight into the gating mechanism, leading to a model based on a localized conformational change that opens a metal entry channel into the active site. Two residues (Glu170, His171) that lie on both domain and subunit interfaces are predicted to reorient, assisted by other residues in the active site environment, adopting an alternative conformation that removes the barriers to metal entry into the active site. The residues involved are highly conserved over the entire superfamily of Mn,Fe-SOD structures, suggesting a universal mechanism controlling metal uptake by these proteins that may contribute to metal binding selectivity in vivo.
ACKNOWLEDGMENT
The authors would like to thank Dr. Larry David, Dr. Debra McMillen and Dr. John Klimeck of the Proteomics Core Facility (funded by the Oregon Opportunity and the National Institutes of Health) for their assistance.
This work was supported by the National Institutes of Health (GM42680 to J. W. W.).
ABBREVIATIONS
- EC
Escherichia coli;
- SOD
superoxide dismutase;
- IBA
2-iodosobenzoic acid;
- bBm
bis-bromobimane [3,7-dimethyl-4,6-di(bromomethyl)-1,5-diazabicyclo[3.3.0]octa-3,6-dien-2,8-dione];
- 2-ME
2-mercaptoethanol;
- GnHCl
guanidinium hydrochloride;
- SEC
size exclusion chromatography;
- DTNB
5,5′-dithio-bis-(2-nitrobenzoic acid);
- EDTA
ethylenediamine tetraacetic acid.
REFERENCES
- 1.Kuchar J, Hausinger RP. Biosynthesis of metal sites. Chem. Rev. 2004;104:509–525. doi: 10.1021/cr020613p. [DOI] [PubMed] [Google Scholar]
- 2.Rosenzweig AC. Metallochaperones: bind and deliver. Chem. Biol. 2002;9:673–677. doi: 10.1016/s1074-5521(02)00156-4. [DOI] [PubMed] [Google Scholar]
- 3.Culotta VC, Yang M, O'Halloran TV. Activation of superoxide dismutases: putting the metal to the pedal. Biochim. Biophys. Acta. 2006;1763:747–758. doi: 10.1016/j.bbamcr.2006.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blokesch M, Paschos A, Theodoratou E, Bauer A, Hube M, Huth S, Böck A. Metal insertion into NiFe-hydrogenases. Biochem. Soc. Trans. 2002;30:674–680. doi: 10.1042/bst0300674. [DOI] [PubMed] [Google Scholar]
- 5.Dos Santos PC, Dean DR, Hu Y, Ribbe MW. Formation and insertion of the nitrogenase iron-molybdenum cofactor. Chem. Rev. 2004;104:1159–1173. doi: 10.1021/cr020608l. [DOI] [PubMed] [Google Scholar]
- 6.Gifford JL, Walsh MP, Vogel HJ. Structures and metal-ion-binding properties of the Ca2+-binding helix-loop-helix EF-hand motifs. Biochem. J. 2007;405:199–221. doi: 10.1042/BJ20070255. [DOI] [PubMed] [Google Scholar]
- 7.McCord JM. Oxygen-derived free radicals. New Horiz. 1993;1:70–76. [PubMed] [Google Scholar]
- 8.Fridovich I. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 1995;64:97–112. doi: 10.1146/annurev.bi.64.070195.000525. [DOI] [PubMed] [Google Scholar]
- 9.Parker MW, Blake CC. Iron- and manganese-containing superoxide dismutases can be distinguished by analysis of their primary structures. FEBS Lett. 1988;229:377–382. doi: 10.1016/0014-5793(88)81160-8. [DOI] [PubMed] [Google Scholar]
- 10.Yamakura F, Kobayashi K, Furukawa S, Suzuki Y. In vitro preparation of iron substituted human manganese superoxide dismutase: possible toxic properties for mitochondria. Free Radical Biol. Med. 2007;43:423–430. doi: 10.1016/j.freeradbiomed.2007.04.024. [DOI] [PubMed] [Google Scholar]
- 11.Edwards RA, Baker HM, Whittaker MM, Whittaker JW, Jameson GB, Baker EN. Crystal structure of Escherichia coli manganese superoxide dismutase at 2.1 Å resolution. J. Biol. Inorg. Chem. 1998;3:161–171. [Google Scholar]
- 12.Lah MS, Dixon MM, Pattridge KA, Stallings WC, Fee JA, Ludwig ML. Structure-function in Escherichia coli iron superoxide dismutase: comparisons with the manganese enzyme from Thermus thermophilus. Biochemistry. 1995;34:1646–1660. doi: 10.1021/bi00005a021. [DOI] [PubMed] [Google Scholar]
- 13.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera - A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 14.Mizuno K, Whittaker MM, Bächinger HP, Whittaker JW. Calorimetric studies on the tight binding metal interactions of Escherichia coli manganese superoxide dismutase. J. Biol. Chem. 2004;279:27339–27344. doi: 10.1074/jbc.M400813200. [DOI] [PubMed] [Google Scholar]
- 15.Ose DE, Fridovich I. Manganese-containing superoxide dismutase from Escherichia coli: reversible resolution and metal replacements. Arch. Biochem. Biophys. 1979;194:360–364. doi: 10.1016/0003-9861(79)90628-3. [DOI] [PubMed] [Google Scholar]
- 16.Beyer WF, Jr., Fridovich I. In vivo competition between iron and manganese for occupancy of the active site region of the manganese-superoxide dismutase of Escherichia coli. J. Biol. Chem. 1991;266:303–308. [PubMed] [Google Scholar]
- 17.Whittaker MM, Whittaker JW. Thermally triggered metal binding by recombinant Thermus thermophilus manganese superoxide dismutase, expressed as the apo-enzyme. J. Biol. Chem. 1999;274:34751–34757. doi: 10.1074/jbc.274.49.34751. [DOI] [PubMed] [Google Scholar]
- 18.Whittaker MM, Whittaker JW. Recombinant superoxide dismutase from a hyperthermophilic archaeon, Pyrobaculum aerophilum. J. Biol. Inorg. Chem. 2000;5:402–408. [PubMed] [Google Scholar]
- 19.Whittaker MM, Mizuno K, Bächinger HP, Whittaker JW. Kinetic analysis of the metal binding mechanism of Escherichia coli manganese superoxide dismutase. Biophys. J. 2006;90:598–607. doi: 10.1529/biophysj.105.071308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jameson GB, Adams JJ, Hempstead PD, Anderson BF, Morgenstern-Badarau I, Whittaker JW, Baker EN. Superoxide dismutases from hyperthermophiles: clues to metal-ion specificity. J. Inorg. Biochem. 2003;96:70. [Google Scholar]
- 21.Lee S, Sawaya MR, Eisenberg D. Structure of superoxide dismutase from Pyrobaculum aerophilum presents a challenging case in molecular replacement with multiple molecules, pseudo-symmetry and twinning. Acta Cryst. 2003;D59:2191–2199. doi: 10.1107/s0907444903019942. [DOI] [PubMed] [Google Scholar]
- 22.Miller EM, Nickoloff JA. Escherichia coli electrotransformation. Methods Mol. Biol. 1995;47:105–13. doi: 10.1385/0-89603-310-4:105. [DOI] [PubMed] [Google Scholar]
- 23.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13:476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao B, Flores SC, Bose SK, McCord JM. A novel Escherichia coli vector for oxygen-inducible high level expression of foreign genes. Gene. 1996;176:269–270. doi: 10.1016/0378-1119(96)00246-6. [DOI] [PubMed] [Google Scholar]
- 26.Whittaker MM, Whittaker JW. A glutamate bridge is essential for dimer stability and metal selectivity in manganese superoxide dismutase. J. Biol. Chem. 1998;273:22188–22193. doi: 10.1074/jbc.273.35.22188. [DOI] [PubMed] [Google Scholar]
- 27.Whittaker MM, Whittaker JW. Mutagenesis of a proton linkage pathway in Escherichia coli manganese superoxide dismutase. Biochemistry. 1997;36:8923–8931. doi: 10.1021/bi9704212. [DOI] [PubMed] [Google Scholar]
- 28.Edwards RA, Whittaker MM, Whittaker JW, Baker EN, Jameson GB. Outer sphere mutations perturb metal reactivity in manganese superoxide dismutase. Biochemistry. 2001;40:15–27. doi: 10.1021/bi0018943. [DOI] [PubMed] [Google Scholar]
- 29.Edwards RA, Whittaker MM, Whittaker JW, Baker EN, Jameson GB. Removing a hydrogen bond in the dimer interface of Escherichia coli manganese superoxide dismutase alters structure and reactivity. Biochemistry. 2001;40:4622–4632. doi: 10.1021/bi002403h. [DOI] [PubMed] [Google Scholar]
- 30.Quijano C, Hernandez-Saavedra D, Castro L, McCord JM, Freeman BA, Radi R. Reaction of peroxynitrite with Mn-superoxide dismutase. Role of the metal center in decomposition kinetics and nitration. J. Biol. Chem. 2001;276:11631–11638. doi: 10.1074/jbc.M009429200. [DOI] [PubMed] [Google Scholar]
- 31.Beyer WF, Jr., Reynolds JA, Fridovich I. Differences between the manganeseand the iron-containing superoxide dismutases of Escherichia coli detected through sedimentation equilibrium, hydrodynamic, and spectroscopic studies. Biochemistry. 1989;28:4403–4409. doi: 10.1021/bi00436a042. [DOI] [PubMed] [Google Scholar]
- 32.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 33.McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) J. Biol. Chem. 1969;244:6049–55. [PubMed] [Google Scholar]
- 34.Riddles PW, Blakeley RL, Zerner B. Reassessment of Ellman's reagent. Meth. Enzymol. 1983;91:49–60. doi: 10.1016/s0076-6879(83)91010-8. [DOI] [PubMed] [Google Scholar]
- 35.Means GE, Feeney RE. Chemical Modification of Proteins. Holden-Day; New York: 1971. pp. 157–158. [Google Scholar]
- 36.Kim J-S, Raines RT. Dibromobimane as a fluorescent cross-linking reagent. Anal. Biochem. 1995;225:174–176. doi: 10.1006/abio.1995.1131. [DOI] [PubMed] [Google Scholar]
- 37.Markley JL. Observation of histidine residues in proteins by nuclear magnetic resonance spectroscopy. Accounts Chem. Res. 1975;8:70–80. [Google Scholar]
- 38.Loudon GM. Mechanistic interpretation of pH-rate profiles. J. Chem. Ed. 1991;68:973–984. [Google Scholar]
- 39.Thurlkill RL, Grimsley GR, Scholtz JM, Pace CN. pK values of the ionizable groups in proteins. Protein Sci. 2006;15:1214–1218. doi: 10.1110/ps.051840806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anderson DE, Becktel WJ, Dahlquist FW. pH-induced denaturation of proteins: A single salt bridge contributes 3-5 kcal/mol to the free energy of folding of T4 lysozyme. Biochemistry. 1990;29:2403–2408. doi: 10.1021/bi00461a025. [DOI] [PubMed] [Google Scholar]
- 41.Frey PA, Whitt SA, Tobin JB. A low-barrier hydrogen bond in the catalytic triad of serine proteases. Science. 1994;264:1927–1930. doi: 10.1126/science.7661899. [DOI] [PubMed] [Google Scholar]
- 42.Matsumura M, Matthews BW. Stabilization of functional proteins by introduction of multiple disulfide bonds. Methods Enzymol. 1991;202:336–356. doi: 10.1016/0076-6879(91)02018-5. [DOI] [PubMed] [Google Scholar]
- 43.Tomishige M, Vale RD. Controlling kinesin by reversible disulfide cross-linking: identifying the motility-producing conformational change. J. Cell Biol. 2000;151:1081–1092. doi: 10.1083/jcb.151.5.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schultz-Heienbrok R, Maier T, Sträter N. Trapping a 96° domain rotation in two distinct conformations by engineered disulfide bridges. Prot. Sci. 2004;13:1811–1822. doi: 10.1110/ps.04629604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thornton JM. Disulfide bridges in globular proteins. J. Mol. Biol. 1981;151:261–287. doi: 10.1016/0022-2836(81)90515-5. [DOI] [PubMed] [Google Scholar]
- 46.Richardson JS. The anatomy and taxonomy of protein structures. Adv. Prot. Chem. 1981;34:167–339. doi: 10.1016/s0065-3233(08)60520-3. [DOI] [PubMed] [Google Scholar]
- 47.Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose pBAD promoter. J. Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Green NS, Reisler E, Houk KN. Quantitative evaluation of the lengths of homobifunctional protein cross-linking reagents used as molecular rulers. Protein Sci. 2001;10:1293–1304. doi: 10.1110/ps.51201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bunting KA, Cooper JB, Tickle IJ, Young DB. Engineering of an intersubunit disulfide bridge in the iron superoxide dismutase from Mycobacterium tuberculosis. Arch. Biochem. Biophys. 2002;397:69–76. doi: 10.1006/abbi.2001.2635. [DOI] [PubMed] [Google Scholar]