

Abstract
Neurotrypsin is a multidomain protein that serves as a brain-specific serine protease. Here we report the NMR structure of its kringle domain, NT/K. The data analysis was performed with the BACUS (Bayesian analysis of coupled unassigned spins) algorithm. This study presents the first application of BACUS to the structure determination of a 13C unenriched protein for which no prior experimental 3D structure was available. NT/K adopts the kringle fold, consisting of an antiparallel β-sheet bridged by an overlapping pair of disulfides. The structure reveals the presence of a surface-exposed left-handed polyproline II helix that is closely packed to the core β-structure. This feature distinguishes NT/K from other members of the kringle fold and points toward a novel functional role for a kringle domain. Functional divergence among kringle domains is discussed on the basis of their surface and electrostatic characteristics.
The kringle fold occurs in combination with other folds in multidomain proteins, often as a part of tandem domains (1). The most frequent number of homologous repeats is 2, and the maximum number of consecutive kringle domains is 37 in apolipoprotein A (2). The kringle family has a broad repertoire of domain partners. For example, the hyaluronan-binding protein is the combination of a kringle domain with three epidermal growth factor domains and a serine protease. These proteins perform a variety of functions and can act as proteases (plasminogen and prothrombin) (3), protease activators (urokinase, tissue plasminogen activator, and hepatocyte growth factor activator) (3, 4), growth factors (hepatocyte growth factor and macrophage-stimulating protein) (5), and lipid transporters (apolipoprotein A) (2).
Human neurotrypsin is a multidomain protein of 875 amino acids, composed of a proline-rich basic segment at the N-terminus, followed by a kringle domain, four scavenger receptor cysteine-rich repeats, and a trypsin-like serine protease domain at the C-terminus (6, 7). Neurotrypsin is predominantly expressed in neurons of the cerebral cortex, hippocampus, and amygdala (8, 9). Mutational analysis established a relationship between neurotrypsin and autosomal recessive mental retardation (9). Electron microscopic mapping of the subcellular distribution of neurotrypsin in the adult human brain demonstrated that this protein is present in presynaptic terminals, close to the membrane lining the synaptic cleft (9). Neurotrypsin is released into the synaptic cleft by exocytosis in association with neuronal activity (10). Externalized neurotrypsin remains at the synapse for minutes possibly due to transient binding to extracellular synaptic components (10). The underlying protein–protein binding interactions may be mediated by the kringle domain of neurotrypsin, NT/K. As a first step toward an understanding of the molecular mechanism of the biological function of neurotrypsin, we have determined the three-dimensional solution structure of its kringle domain by NMR spectroscopy. The assignment of the NOESY1 cross-peaks was performed with the Bayesian analysis of coupled unassigned spins (BACUS) algorithm (11) and is the first application of this automated approach to the structure determination of a 13C unenriched protein for which no prior experimental 3D structure was available.
MATERIALS AND METHODS
Sample Preparation
A 77 amino acid recombinant neurotrypsin kringle domain from Rattus norvegicus was expressed in Escherichia coli, purified as described previously (12), and characterized by sequence analysis and mass spectrometry. N-terminal sequence analysis showed the predicted sequence and truncated amino acid sequences (<10%) due to trypsin cleavage. The mass spectrum of a 15N-labeled NT/K sample was recorded on a VG-Platform mass spectrometer (Micromass) with a precision of ±0.01% (±1 Da/10 kDa). ESIMS showed a major peak at 8634.50 ± 0.68 Da attributed to 15N-labeled NT/K and an additional peak at 8532.63 ± 0.85 (<20%) due to the natural 14N isotopic abundance species. The NMR sample contained 1 mM protein (extinction coefficient ε280 30855 M−1 cm−1) in 90/10% H2O/D2O (pH 5.2) and 0.02% NaN3.
NMR Spectroscopy
NMR spectra were acquired at 300 K on a 600 MHz Bruker Avance DRX spectrometer. Chemical shifts were referenced to 1,4-dioxane at 3.75 ppm (13). Data were processed with FELIX (MSI, San Diego, CA) and analyzed using the CcpNmr Analysis program (14). Backbone and side chains were assigned on the basis of homonuclear COSY, TOCSY (mixing times τmix = 40 and 70 ms), (1H–1H)-NOESY (τmix = 100 ms), and heteronuclear experiments: (1H–15N)-HSQC, (1H–15N)-HMBC, natural isotopic abundance (1H–13C)-HSQC, 3D HNHA, 3D HNHB, 3D TOCSY-(1H–15N)-HSQC (τmix = 70 ms), and 3D NOESY-(1H–15N)-HSQC (τmix = 200 ms).
Coupling constants (3JHN–Hα) were measured, using the diagonal-peak to cross-peak intensity ratio in the HNHA experiment (15). The 3JHN–Hα values were used to derive 31 ϕ torsion angle restraints for non-Gly residues. In addition, 9 (ϕ, ψ)i and 44 ( χ1, χ2)i angles were estimated on the basis of the observed NOE data and rotamer libraries for backbone and side chain conformations (16, 17).
Sequential Assignments of Spin Systems
Grouping of spectral peaks into J-connected spin systems was achieved through the analysis of homonuclear and 15N-edited heteronuclear NMR spectra, both manually and with the help of the program SPI (18). The residue types of the spin systems were identified by the program TYPESYST (19) on the basis of the correlations of 1H, 13C, and 15N chemical shifts with the type of amino acid, deduced from Biological Magnetic Resonance Bank (BMRB) statistics (20).
BACUS (11) is an iterative algorithm for determining the probabilities of peak-to-resonance assignments for a set of NOESY cross-peaks, by considering both the proximity of the observed peaks to chemical shifts and the J-connectivity of resonances gleaned from COSY and TOCSY experiments. The covalent connectivity information has been described in terms of the reporter peak concept. In essence, a reporter peak is a cross-peak which would be expected to be present, with a given probability, if a resonance assignment hypothesis for another peak is correct. The idea stems from the fact that proton resonances within a J-connected spin system are necessarily close in space. Accordingly, when considering two neighboring spins, if an NOE peak is assignable to a pair of proton resonances, one from each spin system, then the likelihood of other peaks, representing resonance pairs from the same two spin systems, is increased.
Spin systems have been assigned to specific residues of the primary amino acid sequence using the program LINKMAP (19) on the basis of the probabilities generated by BACUS for the identities of the NOE peaks, using a Metropolis-based algorithm (21).
NOESY Assignment and Structure Calculations
Once the sequence-specific assignments were obtained, BACUS was rerun to determine medium- and long-range distance restraints. The BACUS analysis was carried out independently for 2D (1H–1H)-NOESY and 3D NOESY-(1H–15N)-HSQC spectra, and each spectrum was calibrated separately (see below). The chemical shift tolerances were set to 0.02, 0.04, and 0.25 ppm for the direct, 1H indirect, and 15N dimensions, respectively. The NOESY cross-peak assignments were subsequently used to generate distance restraints for the structure determination. The NOE distance restraints were supplemented with the dihedral angle (see above) and hydrogen bond (see below) restraints.
The structures were calculated with a simulated annealing protocol, using the crystallography and NMR system program (CNS 1.1) (22) adapted to incorporate a backbone–backbone hydrogen-bonding potential (23). The cycles of distance restraint verification and simulated annealing were performed iteratively until the structures satisfy the experimental restraints, with no distance violations larger than 0.5 Å and no dihedral angle violations larger than 5°.
NOE-derived interproton distances involving nonstereospecifically assigned methyl, methylene, and aromatic protons were represented by the r−6 average (24). The target function to be minimized included the following terms: soft square-well potentials for distance restraints, flat-bottom harmonic potentials for torsion angle restraints, a hydrogen-bonding potential of mean force (23), harmonic terms for the covalent geometry (bonds, angles, improper torsions), and a quartic repulsion-only term for the nonbonded interactions. The force constants used for the terms in the target function were as follows: 5 kcal mol−1 Å−2 for the experimental NOE distance restraints, 20 kcal mol−1 rad−2 for the torsion angles, 50 kcal mol−1 Å−2 for the hydrogen bond restraints, 1000 kcal mol−1 Å−2 for bond lengths, 500 kcal mol−1 rad−2 for angles and improper torsions, and 4 kcal mol−1 Å−4 for the quartic nonbonded repulsion term. The refinement protocol consisted of Cartesian molecular dynamics simulated annealing followed by slow cooling and a final energy minimization stage. The temperature in the annealing procedure was decreased from 2001 to 1 K in steps of 10 K. The molecular dynamics calculation at each temperature consisted of 1000 steps with a step size of 0.002 ps.
The 18 hydrogen bonds between the backbone amide and carbonyl atoms were progressively identified in the calculated structures, based on the criteria (25) O• • •HN < 3.0 Å and N–H• • •O > 90°. During later refinement stages, six hydrogen bonds involving side-chain donors and acceptors were recognized as well. Hydrogen bond restraints were introduced as two unambiguous distance restraints: O• • •HN = 1.90 ± 0.20 Å and O• • •N = 2.90 ± 0.20 Å.
NOE Distance Calibration
Peak picking and integration of NOESY spectra were carried out using the “box sum” method of CcpNmr Analysis (14). The reciprocally transposed (symmetry related) peaks picked from a WATERGATE (1H–1H)-NOESY spectrum (26) were detected via a symmetrization procedure, and those with the maximum volume values were accepted within a tolerance of 0.012 ppm in both 1H dimensions.
The NOE cross-peak volumes (Vij) were corrected for the WATERGATE excitation profile and converted into distances (rij) using rij = kVij−α. Methylene pairs and aromatic ring protons served as references for the distances. Relative errors in the evaluated distances were estimated from the ratio of standard deviation of the calculated distances to the mean distance. Calibration yields k = 575 and 304, α = 0.258 and 0.234, and error estimates 0.25 and 0.40 for 2D (1H–1H)-NOESY (τmix = 100 ms) and 3D NOESY-(1H–15N)-HSQC (τmix = 200 ms) spectra, respectively. The lower bound of the distances was set equal to the sum of the van der Waals radii of two 1H atoms, 1.8 Å, and the upper bound for the NOE calibration was taken at 7 Å. Distance restraints involving methyl, backbone amides HN, side-chain Trp Hε1, Arg Hε, and Asn/Gln NH2δ,ε protons were corrected for multiplicity and deuteration using the correction factor Z = [ni × nj × (1 – nexc)i × (1 – nexc)j]α, where ni and nj are the numbers of equivalent spins in the groups corresponding to resonances i and j and (1 – nexc)i and (1 – nexc)j are the fractional occupancies for the nondeuterated sites. The correction scales the distance restraints up by ∼30% and ∼2% for methyl and exchangeable protons in ∼10% D2O, respectively.
Final Refinement Protocol
Final structure refinement in explicit water was performed with the ARIA 2.0 (ambiguous restraints for iterative assignment) program (27), using fixed unambiguous distance restraints and a 3D structure, both obtained with BACUS/CNS (see above), and restraints on dihedral angles and hydrogen bonds. The lower and upper bounds for the NOE distance calibrations were set to 1.8 and 7.0 Å, respectively. The chemical shift tolerances for the 2D (1H–1H)-NOESY spectrum were set to 0.02 ppm, and those for the 3D NOESY-(15N–1H)-HSQC spectrum to 0.02, 0.04, and 0.25 ppm for the direct, 1H indirect, and 15N dimensions, respectively. The ARIA protocol performed nine iterative cycles, each consisting of NOESY assignments and structure calculations. Each cycle generated 50 structures of which the 25 lowest energy structures were passed to the subsequent cycle as the initial ensemble. In the ninth and final iteration, 150 structures were calculated and the 70 lowest energy structures were refined in explicit TIP3P water. From the final ensemble of 70 calculated structures a subset of the 30 best structures was selected based on PROCHECK-NMR (28) scores.
Conservation Scores for Residues in Multiple Sequence Alignments
A conservation score was calculated for each column of the multiple alignments using Scorecons (29). The scores range from 0 to 1, in which columns with all identities are assigned the highest value and the others get a score according to the degree of conservancy as inferred from the multiple alignments.
Electrostatic Potential Calculation
Electrostatic potential calculations were carried out by numerically solving the linearized Poisson–Boltzmann equation, using the DelPhi program (30). A standard set of formal charges was assigned to the titratable residues (e.g., Arg, +1e, Lys, +1e, Asp, −1e, and Glu, −1e). Histidine residues were modeled as electrically neutral entities. N-terminal amino and C-terminal carboxyl groups were considered with a charge of ±1. The van der Waals radii and charges were taken from the PARSE parameter set (parameters for solvation energy). The dielectric boundary of the protein was defined by the solvent-excluded surface, using the 1.4 Å probe radius for water. The dielectric constant was equal to 4 in the protein interior and 80 in the surrounding solvent. The ionic strength was set to the physiological value (150 mM NaCl) and the ion exclusion radius to 2 Å. The DelPhi potentials were calculated, using a three-step focussing protocol. In the first step the potential at the boundary was approximated by the sum of the Debye–Hückel potentials of all charges, and the size and spacing of the grid were taken as 1653 and 0.84 Å. In the second and third runs the potential was refined, using focussing boundary conditions with the same size and spacings of 0.42 Å and 0.28 Å, respectively. The adopted spacing values corresponded to grid “perfils” of 30%, 60%, and 90%. The electrostatic potential was mapped onto the molecular surface of the protein using GRASP (30).
Affinity Chromatography
HPLC was performed on a Biocad perfusion chromatography workstation (PerSeptive Biosystems, Flamingham, MA). Analytical scale separations were carried out on a heparin affinity Poros 20 HE column (100 × 4.6 mm2, 20 μm particle size). The Poros 20 HE was packed with cross-linked poly(styrene-divinylbenzene) flow-through particles coated with a polyhydroxylated polymer conjugated with heparin. Binding runs were performed with 20 mM Tris buffer at pH 7 and 8. Prior to each run the column was equilibrated with buffer; the protein sample was eluted with a linear gradient 0–1 M KCl in the same buffer at a flow rate of 5 mL/min. Total run time was 12 min, and the elution was monitored via UV absorbance at 280 nm.
Preparation of Unilamellar Vesicles (ULVs)
Ten milligrams of POPC, 1-palmitoyl-2-oleoyl-sn-glycero-phosphatidylcholine, containing a double bond on the sn-2 hydrocarbon chain phospholipid (16:0,18:1PC), was mixed with 1000 μL of buffer, and multilamellar vesicles were prepared, using three repetitive cycles of heavy vortexing at room temperature for 5 min and freezing at −20 °C for 10 min as described elsewhere (31). This suspension was passed 25 times through two polycarbonate filters of 500 Å above the main phase transition temperature, using a mini extruder (Avanti Inc.). According to literature report (31), this protocol yields a population of ULVs with an average diameter of 600 Å. The vesicle solutions were stored at room temperature for no longer than 1 week.
RESULTS AND DISCUSSION
Sequential Assignments of Spin Systems
The NMR study presented here was performed on a 13C unenriched sample. The spin systems of all residues were identified, with the exception of Pro7 and Pro56 for which no resonances were observed. Analysis of the natural isotopic abundance (1H–13C)-HSQC spectrum of NT/K, on the basis of the uniquely assigned 1H resonances, resulted in the identification of 28 Cα, 24 Cβ, 15 Cγ, and 13 Cδ resonances. Unique typing was established for 65 spin systems (87%) and ambiguous typing (cysteine or aspartic acid) for the 10 remaining spin systems. However, the typing probabilities PCys,i and PAsp,i (i = 1,…, 10) gave the averages and and clearly favored one of the two residues. Cysteine had the maximum probability in 5 of the 10 ambiguous cases. This result implies that one cysteine was misidentified as an aspartate because the primary NT/K sequence has 6 cysteine residues. Indeed, given the statistics, there is a 89% probability that in a population of 10 ambiguous cases at least one of the ambiguously identified residues (Cys18) occurs with the minimum probability, Min(PCys,i,PAsp,i). It is reassuring that as many as 74 out of the 75 maximum probability based TYPE-SYST identifications were in agreement with the manual assignments.
The spin systems were assigned to specific residues of the amino acid sequence as described in Materials and Methods. The validity of the automated sequence-specific assignment was verified by manual analysis (32). Unique sequence-specific assignments were obtained for 64 backbone spin systems (85.3%) and ambiguous positions for 9 spin systems (12%) with an ambiguity of 2 positions per residue and average maximum probability 0.82. The only assignment error was a switch between Pro32 and Pro33, owing to degeneracy of the Hα chemical shifts for these residues. Thus, LINKMAP discounts false positive hits by producing low assignment scores, which demonstrates that our method can reliably assign spin systems even based on fairly ambiguous 2D NOESY data and chemical shifts that include 28 13Cα, 24 13Cβ, 15 13Cγ, and 13 13Cδ unambiguous resonances from the natural isotopic abundance (1H–13C)-HSQC spectrum.
NOESY Assignment and Structure Calculations
About 90% of the cross-peaks were assigned uniquely in both 2D 1H NOESY and in 3D NOESY-(1H–15N)-HSQC spectra. The cysteine β-methylene protons were stereospecifically assigned to staggered conformations with χ1 of −60° for Cys18, Cys47, and Cys72 and 60° for Cys58, on the basis of NOE intensities of cross-peaks involving the HN,Hα, and Hβ protons. The conformations for the disulfide bridges were obtained as a mixture of the right-handed and left-handed forms, i.e., with positive and negative values for the central torsion angle χ3, without affecting the backbone structure. In the final structure refinement, left-handed conformers (χ3= −90°) have been enforced in order to be consistent with cystine conformations in known X-ray kringle fold structures (33-35). A full account of the dihedral angles and atom distances in the disulfides will be presented in a separate paper.
Solution Structure of NT/K
Figure 1A shows best fit superpositions of the backbone atoms for 30 NT/K structures. A stereo ribbon diagram of a representative conformer is displayed in Figure 1B. The structural statistics of the ensemble is given in Table 1. The RMS deviations of covalent bonds from ideal peptide geometry are ∼0.005 Å. The structures violate on the average as little as 0.3 out of 1342 interproton distance restraints (threshold 0.5 Å) and 0.2 out of 70 dihedral angle restraints (threshold 5°). The active NOE restraints are satisfied with a precision of 0.04 Å in the refined structure. Excluding disordered N- and C-terminal segments 1–7 and 73–77, the RMS deviations of the 30 structures from the mean coordinate positions are 0.96 Å for the backbone atoms and 1.44 Å for all atoms. The two inner disulfides (backbone RMSD 0.26 Å) are the best defined structural elements of the protein.
FIGURE 1.

(A) Superposition of the backbone atoms (N, Cα, C′) of residues 8–74 in the ensemble of 30 NT/K structures. (B) Stereoview of ribbon diagram showing the secondary structure elements in NT/K. The disulfides (yellow) are labeled a, b, and c; the PPII-helices (blue) are designated 1 (residues 17–20), 2 (residues 24–26), and 3 (residues 31–33); β-strands (residues 57–62, 66–71) and a single bridge (residues 48–57) are shown in orange; green loop corresponds to residues 47–56; N- and C-termini are in red and light blue, respectively.
Table 1.
Structural Statistics for the Ensemble of 30 NT/K Structures Using BACUS Distance Restraints
| Data for Structural Calculations | |
| total unambiguous NOEs | 1342 |
| intraresidue unambiguous NOEs [(i, j) i = j] | 613 |
| sequential unambiguous NOEs [(i, j) |i − j| = 1] | 313 |
| medium-range unambiguous NOEs [(i, j) 2 ≤ |i − j| ≤ 4] | 119 |
| long-range unambiguous NOEs [(i, j) |i − j| ≥ 5] | 295 |
| pairs of hydrogen bond restraints | 24 |
| dihedral angle restraints | 40 ϕ, 9 ψ, 48 χ |
| Calculation Statistics | |
| RMSDa from experimental restraints | |
| distance (Å) | 0.04 (1)b |
| dihedral (deg) | 0.90 (15) |
| RMSD from idealized covalent geometry | |
| bonds (Å) | 0.0048 (3) |
| angles (deg) | 0.59 (3) |
| impropers (deg) | 1.86 (15) |
| Structural Parameters | |
| RMSD (Å) from the mean for backbone atoms (N, Cα, C′) | |
| residues 8–72 | 0.96 (20) |
| residues 8–72, all heavy atoms | 1.44 (23) |
| cystine b and cystine c | 0.26 (13) |
| cystine b and cystine c,c all heavy atoms | 0.33 (16) |
| β-secondary structured | 0.49 (12) |
| residues 10–19 (gray loop)e | 0.68 (27) |
| residues 20–46 (gray loop) | 0.56 (19) |
| residues 47–56 (green loop) | 1.17 (69) |
| residues 57–70 (orange loop) | 0.53 (22) |
| Average Number of Restraint Violations per Structure | |
| distance (>0.5 Å) | 0.3 |
| dihedral (>5°) | 0.2 |
| hydrogen bonds | 0 |
| Ramachandran Analysis | |
| residues in most favored regions (%) | 76.2 (3) |
| residues in additional allowed regions (%) | 21.1 (3) |
| residues in generously allowed regions (%) | 1.4 (1) |
| residues in disallowed regions (%) | 1.4 (1) |
Root-mean-square deviations.
The numbers in parentheses are the standard deviations in the least significant digits.
Cystine b, Cys18–Cys58, and cystine c, Cys47–Cys72.
Residues 10–12, 16–18, 19, 48, 57–62, 66–71.
Color code refers to Figure 1B.
The NT/K secondary structure elements are given in Table 2. The structure comprises an antiparallel β-sheet (36), formed by hydrogen bonding between the β-strands Trp57-Asn62 and Lys66-Tyr71 that are connected by β-hairpin Asn62-Gly65 (orange loop in Figure 1B). In addition, there is an antiparallel β-bridge (36), Arg48-Trp57, at “narrow-pair” hydrogen-bonding positions which are linked via β-hairpin Asn51-Gly54 (green loop in Figure 1B). NT/K has three disulfide bridges. The outer disulfide bridge Cys2-Cys74 (cystine a) tethers residues near the disordered Nand C-termini of the protein and is poorly defined. The two inner disulfide bridges, Cys18-Cys58 (cystine b) and Cys47-Cys72 (cystine c), are essential for the kringle domain topology. The residues of cystine b are incorporated in adjacent β-strands, while the residues of cystine c are located just beyond the ends of nonadjacent β-strands.
Table 2.
Secondary Structure Elements of the NT/K
| secondary structure | residues |
|---|---|
| β-ladders | Thr12 N–Val16 O |
| Cys18 N–Asn10 O | |
| Leu19 N–Phe59 O | |
| Arg61 N–Pro17 O | |
| Arg48 N–Trp57 O | |
| Trp57 N–Arg48 O | |
| Cys58 N–Gly70 O | |
| Gly70 N–Cys58 O | |
| Tyr60 N–Asp68 O | |
| Asp68 N–Tyr60 O | |
| Asn62 N–Lys66 O | |
| Lys66 N–Asn62 O | |
| Cys72 N–Pro56 O | |
| helical segments | |
| PPII | Pro17–His20 |
| PPII | Val24–Pro26 |
| PPII | Ser31–Pro33 |
| 310 | Ala37–Leu39 |
| β-hairpins | |
| type 1 | Thr12–Gly15 |
| type 1 | Asp51–Gly54 |
| type 1 | Asn62–Gly65 |
There are nine proline-containing segments in NT/K. Three of them form left-handed polyproline II (PPII) helices: Pro17-Cys18-Leu19-His20 (PPII 1), Val24-Pro25-Pro26 (PPII 2) and Ser31-Pro32-Pro33 (PPII 3). The residues in these segments adopt the typical ϕ and ψ angles (−75° and +145°, respectively) of PPII-helices (37). PPII 1 overlaps with the antiparallel “wide-pair” bridges Ala11-Pro17 and Cys18-Tyr60.
Hydrogen bonds R30HN-D68Oδ1 (supported by NOE R30HND68Hα) and V67HN- E29Oε1 (supported by NOEs E29HNV67HN, E29HN-V67Hγ2*, E29Hγ*-V67HN) bring the outer edge of the core β-sheet in close proximity with residues Glu29 and Arg30. The NT/K structure also exhibits the H-bond W69Hε1-S31O (supported by the NOEs S31HN-W69Hε1, S31HN-W69Hδ1, S31Hα-W69Hε1, S31Hβ*-W69Hξ2). These H-bonds accommodate the side chain of Pro33 between those of Trp57 and Trp69 (Figure 2). The positioning of Pro33 inside the groove lined by residues Trp57 and Trp69 is consistent with the observed NOEs (P33Hβ*-W57Hε1, P33Hγ*-W57Hε1, P33Hγ*-W57Hδ1, P33Hγ*-W57Hε3, P33Hγ*-W57Hξ2, P33Hδ*-W57Hξ2, P33Hδ*-W57Hη2, P33Hγ*-W69Hε1, P33Hγ*-W69Hξ2, P33Hγ*-W69Hη2, P33Hδ*-W69Hη2) and is validated by its 1H chemical shifts. Ring currents in the surrounding aromatic residues cause the large differences (∼2 ppm) between the observed chemical shifts (−0.098 and –0.176 ppm for P33Hγ* and 0.284 ppm for P33Hδ*) and the BMRB averages (1.93 ppm for ProHγ* and 3.64 ppm for ProHδ*) (20). The positioning of Pro33 in NT/K is unique in the kringle family (see below).
FIGURE 2.
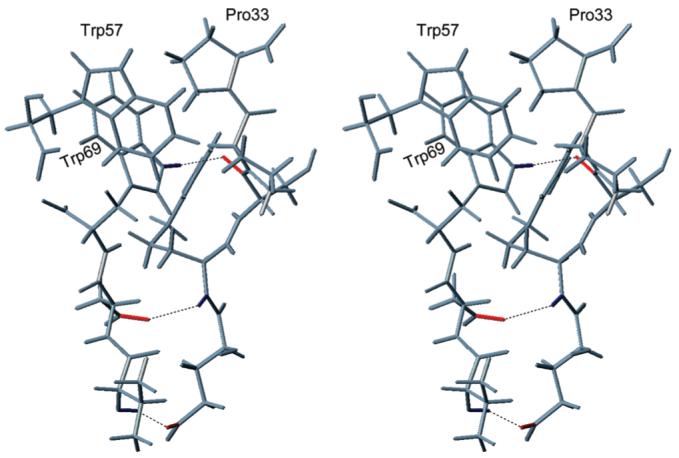
Stereoview of fragment of NT/K containing the hydrogen bonds 30→68, 67→29, and 69→31 which accommodate Pro33 between the side chains of Trp57 and Trp69.
Comparison of NT/K with Proteins of the Kringle Family
Multiple sequence alignments show that members of the kringle (K) family, including NT/K, share a sequence identity of ∼30%, and the cysteines occur at identical positions in all sequences and rank among the most conserved residues in the K family (Figure 3). The loop lengths are fairly constant across the K family (Table S1) while the N- and C-termini of NT/K are five and three residues shorter, respectively, than in other kringles (Figure 3). The following structural features are conserved throughout the K family: (i) a core consisting of the antiparallel β-sheet (residues 57–62 and 66–71) bridged by an overlapping pair of disulfides (cystine b and cystine c) and (ii) an overlapping pattern of a PPII-helix (residues 17–20, containing a conserved half-cystine at position 18), with two antiparallel wide-pair bridges (residues 11–17, 18–60, comprising conserved residues at positions 18, 59, and 60).
FIGURE 3.

Multiple sequence alignments of the kringle family: NT/K (this work), PGN/K1-K3 (code 1ki0), PGN/K4 (code 1pk4), PGN/K5 (code 5hpg), tPA/K2 (code 1tpk), uPA/K (code 2fd6), apoA/K4 (code 1i71), HGF/K1 (code 1bht), PTN/K1 (code 1nl1), and PTN/K2 (code 2hpp). The numbering in the top row refers to the sequential numbering of NT/K. The columns are shaded according to the conservation scores given in the second row from the bottom (e.g., 6 corresponds to a rounded score of 0.6, 7 to a rounded score of 0.7, etc., and 0 to the highest score of 1.0). Positions in which the conservation score is lower than 0.5 are indicated with a dot.
The superpositions of NT/K with plasminogen kringle 4 (33) and the urokinase plasminogen activator kringle (35), based on the alignment of cystines b and c, are presented in Figure S3. Average RMSDs obtained from pairwise alignments of inner disulfides (Table S2) and of core residues (Table S3) within the K family are 0.22 ± 0.09 Å (0.20 ± 0.05 Å without NT/K) and 0.40 ± 0.14 Å (0.36 ± 0.14 Å without NT/K), respectively. The RMSDs for comparisons of NT/K (an NMR structure) are systematically larger than the RMSDs for the comparisons between the other structures (all X-ray structures).
There is, however, a distinction in the terminal regions. An uncertainty in comparing these areas is that they show considerable conformational variability (disorder) as a result of solvent exposure. Thus, with the caveat that the RMSDs for the N- and C-termini are high (1.85 ± 0.48 Å for residues 1–7 and 1.14 ± 0.38 Å for residues 73-77) due to a paucity of long-range NOE restraints, it appears that cystine a in NT/K (RMSD of 0.92 ± 0.29 Å) is displaced with respect to the outer disulfide in other kringle structures (see Figure S3). Obviously, displacement is likely, considering that NT/K has shorter N- and C-termini than other kringles (Figure 3). Another consequence of the deletion appears to be that the loop of residues 47–56 in NT/K (RMSD of 1.17 ± 0.69 Å) (green in Figure 1B) adopts a different orientation than in other kringle structures (Figure S3), in spite of a fair conservation in loop length and sequence (Table S1 and Figure 3). The difference may originate from loss of hydrogen bonding or other van der Waals interactions between the loop and the N-terminal segment, which are farther apart in NT/K than in other kringle structures (see Figure S3). For example, in plasminogen kringle 4 (33) the homologous residues in the N-terminus form two hydrogen bonds to the conserved residues Cys47 and Arg48, whereas the NT/K structure has the backbone hydrogen bond C47HN-G9O but lacks the side-chain hydrogen bond to Arg48.
The loops in kringle domains contain divergent patches that confer the affinities and specificities of the interactions with different substrates and binding partners. Thus, notwithstanding their high structural similarity of the core, kringle domains show significant diversity in sequence and function. The molecular surface and electrostatic surface potential of NT/K are illustrated in Figure 4 and compared with those for the plasminogen kringle 4 (33) (PGN/K4), the prothrombin kringle 2 (34) (PTN/K2), and the urokinase plasminogen activator kringle (35) (uPA/K). The electrostatic potentials were calculated using DelPhi (30) (see Materials and Methods), mapped onto the molecular surfaces, and depicted in the 180° orientation relative to the ribbon representation of the folding of NT/K shown in Figure 1B. The conserved residue Trp57 (Trp62 in the numbering based on the plasminogen kringle 5 (38)) appears at the center of all protein faces of Figure 4, left.
FIGURE 4.
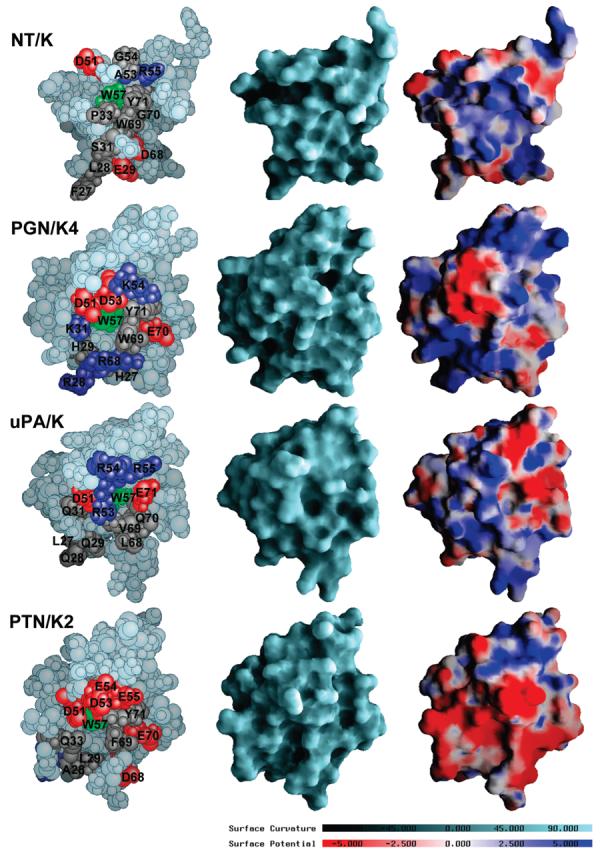
Comparison of molecular surface features of NT/K (this work), PGN/K4 (code 1pk4) (33), uPA/K (code 2fd6) (35) and PTN/K2 (code 2hpp) (34). The structures were superimposed using cystines b and c and depicted in the 180° orientation relative to the ribbon representation of the folding of NT/K shown in Figure 1B. The views on the left are space-filling atomic models. Residues discussed in the text have been highlighted: positively charged residues (Arg and Lys) are in dark blue, negatively charged residues (Asp and Glu) are in red, neutral residues are in gray, and the conserved residue W57 is in green. The views in the center are representations of the accessible molecular surfaces. The surfaces are color coded according to curvature with convex areas in aquamarine, the planar regions in light gray, and concave areas in dark gray. The views on the right are depictions of the electrostatic surface potentials. The potentials were calculated at 150 mM NaCl using DelPhi (30), interpolated onto the molecular surfaces, and visualized with GRASP (30), using a scale from −5 kBT/e (dark red) to +5 kBT/e (dark blue), where kB is the Boltzmann constant, T is ambient temperature, and e is the unit charge.
The tissue plasminogen activator kringle 2 and the plasminogen kringles, including PGN/K4, have affinity for zwitterions such as lysine, ω-aminocarboxylic acids, and trans-(aminomethyl)cyclohexanecarboxylic acid (AMCHA), which resides in residues 27–31, 51–53, 57–59, and 68–71 (31–35, 55–57, 62–64, and 71–74 in PGN/K5 numbering, Figure S1) (33, 38, 39). Anionic (Asp55 and Asp57 in PGN/K5 numbering) and cationic (Arg71 in PGN/K5 numbering) centers form salt bridges with lysine analogues. The aromatic residues Trp57 and Trp69 (Trp62 and Trp72 in PGN/K5 numbering) outline the hydrophobic pocket in PGN/K4 and provide favorable van der Waals contacts for the hydrocarbon moiety of the ligand. In addition, side chains of two histidine residues (His31 and His33 in PGN/K5 numbering) are hydrogen-bonded to the backbone carbonyls of other residues within the site: 27→60, 27→62, 29→21, and 31→29 (31→65, 31→67, 33→25, and 35→33 in PGN/K5 numbering), further enhancing the structural stability of the lysine-binding region. Some of the exposed residues have been labeled in Figure 4, left.
The surfaces in Figure 4 do not exhibit any extended patches that are conserved, even in pairwise comparisons of proteins. The surface potentials reflect the intrinsic polarities of the residues, which have been indicated in the corresponding colors: blue for positive charge and red for negative charge. The residues along the groove centered at residue Trp57 (Trp62 in PGN/K5 numbering) have been mutated in all four domains, leading to drastic changes in the electro-static and hydrophobic properties and associated functionalities. For example, the surfaces of uPA/K and PTN/K2 are unable to bind lysine-type ligands (34, 40). Indeed, our fluorescence titration experiments confirm the expected lack of affinity of NT/K for AMCHA (data not shown). In passing from NT/K to the lysine-binding kringle PGN/K4, the attendant Ala53Asp and Asp68Arg mutations alter the electrostatic profile in a way that eliminates the zwitterion-binding capability of NT/K. In addition, the positioning of the side chain of residue Pro33 between those of residues Trp57 and Trp69 hinders ligand access into the groove. In PGN/K4 residue Pro33 (not shown) is directed away from residues Trp57 and Trp69 (Trp62 and Trp72 in PGN/K5 numbering) and is located at the opposite face of the protein.
Domains such as the urokinase plasminogen activator kringle (uPA/K) and the hepatocyte growth factor kringle 1 (HGF/K1) utilize mutations of the lysine-binding region, changing the electric charges of residues 53–55 in sign and magnitude, for heparin recognition (40-42). Multiple sequence alignments of the K family reveal that none of these residues are conserved throughout the K family. As a result of sequence variations, different members of the K family utilize different contacts for binding heparin. The binding occurs at a surface with a high density of positively charged residues; for example, in uPA/K heparin binds to the cluster, consisting of four arginine residues Arg48 and Arg53-Arg55 (Arg53, Arg57-Arg59 in PGN/K5 numbering) (40, 41); in HGF/K1 the putative heparin-binding region involves residues Arg48 and Arg51 (42). In contrast, PTN/K2 uses an enlarged anionic cluster, spanning residues Asp51, Asp53–Glu55 (Asp55, Asp57–Glu59 in PGN/K5 numbering), for binding to the highly electropositive surface of thrombin at a site that has been proposed to be the heparin binding region (34). Although there are several basic residues in the solvent exposed surface of NT/K, this protein does not exhibit heparin affinity as demonstrated by affinity chromatography (see Materials and Methods).
Given the vicinity of neurotrypsin to the presynaptic membrane, it was of interest to investigate the interaction of NT/K with neutral phospholipid vesicles, using tryptophan fluorescence spectroscopy. The emission of NT/K, using excitation wavelength of 280 nm, had a maximum at 338 nm. The shape and position of the fluorescence emission peak did not change with the addition of POPC ULVs (see Materials and Methods); the only change observed was a linear decrease in the total peak intensity with increasing POPC concentration, due to Rayleigh scattering. Indeed, superposition of spectra obtained at different POPC concentrations gave after normalization identical graphs (Figure S2), indicating that NT/K did not bind to the lipid vesicles.
A distinct feature of the NT/K structure is the presence of the PPII-helix Ser31-Pro32-Pro33 (PPII 3), which is absent in other kringles. Proline-rich regions are often involved in protein–protein interactions (43). Polyproline sequences preferably form a PPII-helix (37), which provides a readily accessible flat hydrophobic interface complete with potential hydrogen-binding sites at the exposed backbone oxygens (44). The solvent-exposed position of PPII 3 in the NT/K structure raises the possibility that this site be suitable for interactions with proteins present in the extracellular matrix. While evidence for this hypothesis is currently lacking, the NT/K structure presented here provides a firm foundation for future functional assays.
Agrin, a multidomain heparan sulfate proteoglycan, is the key organizer of synapse formation at the neuromuscular junction (45). The integration of agrin into basement membranes is mediated through binding of the coiled-coil domain of laminin to the N-terminal domain of agrin (NtA) (45). Engel and co-workers (46) reported that NtA has a high structural similarity with the homologous N-terminal protease inhibition domain of the tissue inhibitor of metalloproteases-1 (TIMP-1), leading the authors (46) to propose that NtA also acts as a protease inhibitor. Independently, Sonderegger and co-workers (47) demonstrated that agrin is a highly specific substrate for neurotrypsin at the synapse. The cleavage of agrin implies that the NtA domain is clearly not an inhibitor of proteolysis by neurotrypsin. Alternatively, whether laminin binding to agrin inhibits cleavage by neurotrypsin is unknown.
Concluding Remarks
The NT/K structure provides insights into the lack of affinity of NT/K for heparin and lysine analogues. The presence of the surface-exposed PPII-helix and its close packing to the core β-structure are features that distinguish NT/K from other members of the kringle fold and point toward a novel functional role for a kringle domain.
ACKNOWLEDGMENT
We thank Virgil Simplaceanu (Carnegie Mellon University) for technical assistance with the NMR measurements, Dr. Johann Schaller (University of Bern, Bern, Switzerland) for the ESI-MS analysis of the NT/K sample, and Dr. Stephanie Tristram-Nagle (Carnegie Mellon University) for help with the unilamellar vesicle preparation.
Footnotes
This work was sponsored by NIH Grant HL-29409 to M.L., and E.L.B. acknowledges financial support from NSF Grant MCB-0424494.
Coordinates for the ensemble of 29 NT/K structures (accession code 2k4r) and a representative conformer (accession code 2k51) have been deposited in the RCSB Protein Data Bank. Experimental NMR restraints and 1H and 15N chemical shift assignments (accession code 15806) have been deposited in the Biological Magnetic Resonance Bank.
Abbreviations: AMCHA, trans-(aminomethyl)cyclo-hexanecarboxylic acid; apoA/K4, apolipoprotein A kringle 4; ARIA, ambiguous restraints for iterative assignment; BACUS, Bayesian analysis of coupled unassigned spins; BMRB, Biological Magnetic Resonance Bank; CcpNmr, collaborative computing project for NMR; CNS, crystallography and NMR system; coK, conserved residues in the kringle domain; COSY, correlation spectroscopy; 2D, 3D, two, three dimensional; ESIMS, electrospray ionization mass spectrum; HGF/K1, hepatocyte growth factor kringle 1; K, kringle domain; HMBC, heteronuclear multiple bond correlation; HPLC, high-performance liquid chromatography; HSQC, heteronuclear single quantum coherence; NMJ, neuromuscular junction; NMR, nuclear magnetic resonance; NOE, nuclear Overhauser effect; NOESY, nuclear Overhauser effect spectroscopy; NT/K, neurotrypsin kringle; PGN/K4, plasminogen kringle 4; PDB, Protein Data Bank; POPC, 1-palmitoyl-2-oleoyl-sn-glycero-phosphatidylcholine; PPII, left-handed polyproline II helix; PTN/K2, prothrombin kringle 2; TIMP-1, tissue inhibitor of metalloprotease-1; TOCSY, total correlation spectroscopy; tPA/K2, tissue plasminogen activator kringle 2; RMSD, root-mean-square deviation; uPA/K, urokinase-type plasminogen activator kringle; ULVs, unilamellar vesicles.
SUPPORTING INFORMATION AVAILABLE
Table S1 listing the numbers of residues in the loops of proteins from the kringle family, Tables S2 and S3 listing RMSDs obtained from pairwise alignments of inner disulfides and of core residues of proteins in the K family, Figure S1 giving the sequence alignment of Figure 3, but now using the plasminogen kringle 5 domain residue numbering as a reference, Figure S2 showing the overlay of fluorescence spectra obtained at different POPC concentrations after normalization, and Figure S3 showing the superposition of NT/K with PGN/K4 and uPA/K. This material is available free of charge via the Internet at http://pubs.acs.org. The BACUS program has been integrated in CcpNmr Analysis that is available from the following Web site: http://www.ccpn.ac.uk/ccpn/software.
REFERENCES
- 1.Murzin AG, Brenner SE, Hubbard TJP, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 2.McLean JW, Tomlinson JE, Kuang WJ, Eaton DL, Chen EY, Fless GM, Scanu AM, Lawn RM. cDNA sequence of human apolipoprotein A is homologous to plasminogen. Nature. 1987;330:132–137. doi: 10.1038/330132a0. [DOI] [PubMed] [Google Scholar]
- 3.Waisman DM, editor. Plasminogen: Structure, Activation, and Regulation. Springer-Verlag; New York: 2003. [Google Scholar]
- 4.Blasi F, Carmeliet P. uPAR: A versatile signalling orchestrator. Nat. Rev. Mol. Cell Biol. 2002;3:932–943. doi: 10.1038/nrm977. [DOI] [PubMed] [Google Scholar]
- 5.Bock GR, Goode JA, editors. Ciba Foundation Symposium Series. Wiley; New York: 1998. Plasminogen-Related Growth Factors. [Google Scholar]
- 6.Gschwend TP, Krueger SR, Kozlov SV, Wolfer DP, Sonderegger P. Neurotrypsin, a novel multidomain serine protease expressed in the nervous system. Mol. Cell. Neurosci. 1997;9:207–219. doi: 10.1006/mcne.1997.0616. [DOI] [PubMed] [Google Scholar]
- 7.Yamamura Y, Yamashiro K, Tsuruoka N, Nakazato H, Tsujimura A, Yamaguchi N. Molecular cloning of a novel brain-specific serine protease with a kringle-like structure and three scavenger receptor cysteine-rich motifs. Biochem. Biophys. Res. Commun. 1997;239:386–392. doi: 10.1006/bbrc.1997.7417. [DOI] [PubMed] [Google Scholar]
- 8.Wolfer DP, Lang R, Cinelli P, Madani R, Sonderegger P. Multiple roles of neurotrypsin in tissue morphogenesis and nervous system development suggested by the mRNA expression pattern. Mol. Cell. Neurosci. 2001;18:407–433. doi: 10.1006/mcne.2001.1029. [DOI] [PubMed] [Google Scholar]
- 9.Molinari F, Rio M, Meskenaite V, Encha-Razavi F, Auge J, Bacq D, Briault S, Vekemans M, Munnich A, Attie-Bitach T, Sonderegger P, Colleaux L. Truncating neurotrypsin mutation in autosomal recessive nonsyndromic mental retardation. Science. 2002;298:1779–1781. doi: 10.1126/science.1076521. [DOI] [PubMed] [Google Scholar]
- 10.Frischknecht R, Fejtova A, Viesti M, Stephan A, Sonderegger P. Activity-induced synaptic capture and exocytosis of the neuronal serine protease neurotrypsin. J. Neurosci. 2008;28:1568–1579. doi: 10.1523/JNEUROSCI.3398-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grishaev A, Llinás M. BACUS: A Bayesian protocol for the identification of protein NOESY spectra via unassigned spin systems. J. Biomol. NMR. 2004;28:1–10. doi: 10.1023/B:JNMR.0000012846.56763.f7. [DOI] [PubMed] [Google Scholar]
- 12.Ozhogina OA, Trexler M, Banyai L, Llinás M, Patthy L. Origin of fibronectin type II (FN2) modules: Structural analyses of distantly-related members of the kringle family identify the kringle domain of neurotrypsin as a potential link between FN2 domains and kringles. Protein Sci. 2001;10:2114–2122. doi: 10.1110/ps.15801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Marco A. pH dependence of internal references. J. Magn. Reson. 1977;26:527–528. [Google Scholar]
- 14.Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinás M, Ulrich EL, Markley JL, Ionides J, Laue ED. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins: Struct., Funct., Bioinf. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 15.Vuister GW, Bax A. Quantitative J correlation: a new approach for measuring homonuclear three-bond J(HNHα) coupling constants in 15N-enriched proteins. J. Am. Chem. Soc. 1993;115:7772–7777. [Google Scholar]
- 16.Lovell SC, Word JM, Richardson JS, Richardson DC. The penultimate rotamer library. Proteins: Struct., Funct., Genet. 2000;40:389–408. [PubMed] [Google Scholar]
- 17.Lovell SC, Davis IW, Adrendall WB, de Bakker PIW, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by C alpha geometry: phi, psi and C beta deviation. Proteins: Struct., Funct., Genet. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 18.Grishaev A, Llinás M. Sorting signals from protein NMR spectra: SPI, a Bayesian protocol for uncovering spin systems. J. Biomol. NMR. 2002;24:203–213. doi: 10.1023/a:1021660608913. [DOI] [PubMed] [Google Scholar]
- 19.Grishaev A, Steren CA, Wu B, Pineda-Lucena A, Arrow-smith C, Llinás M. ABACUS, a direct method for protein NMR structure computation via assembly of fragments. Proteins: Struct., Funct., Bioinf. 2005;61:36–43. doi: 10.1002/prot.20457. [DOI] [PubMed] [Google Scholar]
- 20.Seavey BR, Farr EA, Westler WM, Markley JL. A relational database for sequence-specific protein NMR data. J. Biomol. NMR. 1991;1:217–236. doi: 10.1007/BF01875516. [DOI] [PubMed] [Google Scholar]
- 21.Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller AH, Teller E. Equation of state calculations by fast computing machines. J. Chem. Phys. 1953;21:1087–1092. [Google Scholar]
- 22.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 23.Grishaev A, Bax A. An empirical backbone-backbone hydrogen-bonding potential in proteins and its applications to NMR structure refinement and validation. J. Am. Chem. Soc. 2004;126:7281–7292. doi: 10.1021/ja0319994. [DOI] [PubMed] [Google Scholar]
- 24.Neuhaus D, Williamson MP. Nuclear Overhauser Effect in Structural and Conformation Analysis. Wiley-VCH; New York: 2000. p. 656. [Google Scholar]
- 25.Baker EN, Hubbard RE. Hydrogen bonding in globular proteins. Prog. Biophys. Mol. Biol. 1984;44:97–179. doi: 10.1016/0079-6107(84)90007-5. [DOI] [PubMed] [Google Scholar]
- 26.Piotto M, Saudek V, Sklenar V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR. 1992;2:661–665. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 27.Linge JP, O'Donoghue SI, Nilges M. Automated assignment of ambiguous nuclear overhauser effects with ARIA. Nucl. Magn. Reson. Biol. Macromol., Part B. 2001;339:71–90. doi: 10.1016/s0076-6879(01)39310-2. [DOI] [PubMed] [Google Scholar]
- 28.Laskowski RA, Rullmann JAC, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 29.Valdar WSJ, Thornton JM. Protein-protein interfaces: Analysis of amino acid conservation in homodimers. Proteins: Struct., Funct., Genet. 2001;42:108–124. [PubMed] [Google Scholar]
- 30.Honig B, Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 31.Kucerka N, Tristram-Nagle S, Nagle JF. Structure of fully hydrated fluid phase lipid bilayers with monounsaturated chains. J. Membr. Biol. 2006;208:193–202. doi: 10.1007/s00232-005-7006-8. [DOI] [PubMed] [Google Scholar]
- 32.Wüthrich K. NMR of proteins and nucleic acids. Wiley; New York: 1986. [Google Scholar]
- 33.Mulichak AM, Tulinsky A, Ravichandran KG. Crystal and molecular structure of human plasminogen kringle 4 refined at 1.9 Å resolution. Biochemistry. 1991;30:10576–10588. doi: 10.1021/bi00107a029. [DOI] [PubMed] [Google Scholar]
- 34.Arni RK, Padmanabhan K, Padmanabhan KP, Wu TP, Tulinsky A. Structures of the noncovalent complexes of human and bovine prothrombin fragment-2 with human PPACKthrombin. Biochemistry. 1993;32:4727–4737. doi: 10.1021/bi00069a006. [DOI] [PubMed] [Google Scholar]
- 35.Huai Q, Mazar AP, Kuo A, Parry GC, Shaw DE, Callahan J, Li YD, Yuan C, Bian CB, Chen LQ, Furie B, Furie BC, Cines DB, Huang MD. Structure of human urokinase plasminogen activator in complex with its receptor. Science. 2006;311:656–659. doi: 10.1126/science.1121143. [DOI] [PubMed] [Google Scholar]
- 36.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 37.Adzhubei AA, Sternberg MJE. Left-handed polyproline II helices commonly occur in globular proteins. J. Mol. Biol. 1993;229:472–493. doi: 10.1006/jmbi.1993.1047. [DOI] [PubMed] [Google Scholar]
- 38.Tulinsky A, Park CH, Mao B, Llinás M. Lysine fibrin binding sites of kringles modeled after the structure of kringle 1 of prothrombin. Proteins: Struct., Funct., Genet. 1988;3:85–96. doi: 10.1002/prot.340030203. [DOI] [PubMed] [Google Scholar]
- 39.Marti DN, Hu CK, An SSA, von Haller P, Schaller J, Llinás M. Ligand preferences of kringle 2 and homologous domains of human plasminogen: Canvassing weak, intermediate, and high-affinity binding sites by 1H NMR. Biochemistry. 1997;36:11591–11604. doi: 10.1021/bi971316v. [DOI] [PubMed] [Google Scholar]
- 40.Li X, Bokman AM, Llinás M, Smith RAG, Dobson CM. Solution structure of the kringle domain from urokinase-type plasminogen-activator. J. Mol. Biol. 1994;235:1548–1559. doi: 10.1006/jmbi.1994.1106. [DOI] [PubMed] [Google Scholar]
- 41.Stephens RW, Bokman AM, Myohanen HT, Reisberg T, Tapiovaara H, Pedersen N, Grondahlhansen J, Llinás M, Vaheri A. Heparin binding to the urokinase kringle domain. Biochemistry. 1992;31:7572–7579. doi: 10.1021/bi00148a019. [DOI] [PubMed] [Google Scholar]
- 42.Lietha D, Chirgadze DY, Mulloy B, Blundell TL, Gherardi E. Crystal structures of NK1-heparin complexes reveal the basis for NK1 activity and enable engineering of potent agonists of the MET receptor. EMBO J. 2001;20:5543–5555. doi: 10.1093/emboj/20.20.5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Williamson MP. The structure and function of proline-rich regions in proteins. Biochem. J. 1994;297:249–260. doi: 10.1042/bj2970249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kay BK, Williamson MP, Sudol P. The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 2000;14:231–241. [PubMed] [Google Scholar]
- 45.Bezakova G, Ruegg MA. New insights into the roles of agrin. Nat. Rev. Mol. Cell Biol. 2003;4:295–308. doi: 10.1038/nrm1074. [DOI] [PubMed] [Google Scholar]
- 46.Stetefeld J, Jenny M, Schulthess T, Landwehr R, Schumacher B, Frank S, Ruegg MA, Engel J, Kammerer RA. The laminin-binding domain of agrin is structurally related to N-TIMP-1. Nat. Struct. Biol. 2001;8:705–709. doi: 10.1038/90422. [DOI] [PubMed] [Google Scholar]
- 47.Stephan A, Mateos JM, Kozlov SV, Cinelli P, Kistler AD, Hettwer S, Rulicke T, Streit P, Kunz B, Sonderegger P. Neurotrypsin cleaves agrin locally at the synapse. FASEB J. 2008;22:1861–1873. doi: 10.1096/fj.07-100008. [DOI] [PubMed] [Google Scholar]