

Abstract
Background and Purpose
Astrocytes, neurons, and microvessels together form a neurovascular unit allowing blood flow to match neuronal activity. Adenosine diphosphate (ADP) is an important signaling molecule in the brain, and dilation in response to ADP is astrocyte-dependent in rats and newborn pigs. Carbon monoxide (CO), produced endogenously by catabolism of heme to CO, iron, and biliverdin via heme oxygenase (HO), is an important cell signaling molecule in the neonatal cerebral circulation. We hypothesize ADP stimulates CO production by glia limitans astrocytes and that this CO causes pial arteriolar dilation.
Methods
Experiments were performed using anesthetized piglet with closed cranial windows, and freshly isolated piglet astrocytes and microvessels. Astrocyte injury was caused by topical application of L-2-alpha aminoadipic acid (2 mM, 5 h). Cerebrospinal fluid (CSF) was collected from under the cranial windows for measurement of ADP-stimulated CO production. CO was measured by gas chromatography-mass spectroscopy analysis.
Results
Before, but not after, astrocyte injury in vivo, topical ADP stimulated both CO production and dilation of pial arterioles. Astrocyte injury did not block dilation to isoproterenol or bradykinin. Chromium mesoporphyrin, an inhibitor of HO, also prevented the ADP-induced increase in CSF CO and pial arteriolar dilation caused by ADP but not dilation to sodium nitroprusside. ADP also increased CO production by freshly isolated piglet astrocytes and cerebral microvessels, although the increase was smaller in the microvessels.
Conclusions
These data suggest that glia limitans astrocytes employ CO as a gasotransmitter to cause pial arteriolar dilation in response to ADP.
Keywords: carbon monoxide, glia, toxin, newborn, cerebral circulation
INTRODUCTION
Cerebrovascular smooth muscle tone and thus cerebrovascular resistance is regulated by a variety of signals coming from nerves, endothelium, pericytes, and astrocytes, which together with the vascular smooth muscle form a neurovascular unit 1. Astrocytes can respond to synaptic activity and signals from other neurovascular unit components by releasing vasoactive compounds 2, 3.
Astrocytes outnumber neurons in the brain and are remarkably multifunctional cells 4. A typical astrocyte can send processes to hundreds of synapses. Most astrocytes also extend at least one process that contacts a blood vessel 5, 6. Thus, astrocytes are in a unique position for sensing neuronal activity, integrating that information, and communicating with cerebral blood vessels 4,5,7.
Endogenous carbon monoxide (CO) is a gasotransmitter that can be related to neural function 8 and blood flow regulation in the brain 9-12. CO is produced physiologically by catabolism of heme via heme oxygenase (HO), with oxidation of NADPH, to CO, iron, and biliverdin 13. Heme oxygenase-2 (HO-2) is constitutively highly expressed in astrocytes, neurons, and cerebral microvessels 14. CO production can be controlled by regulation of substrate availability to HO-2 or the effective catalytic activity of HO-2 15. In the newborn pigs, CO is involved in pial arteriolar responses to neuronal activity, hypoxia and changing blood pressure 9, 10, 12, 16. In vivo, topical application of CO dilates pial arterioles in newborn pigs 10. The vasodilatory effect of CO is caused by activation of Ca2+-activated K+ (KCa) channels 17.
Adenosine diphosphate (ADP) is a regulator of cerebral blood flow 18. ADP can produce endothelium-dependent cerebral vasodilation 19, which may be mediated in part by nitric oxide (NO) and an endothelium-derived hyperpolarizing factor (EDHF) 20, 21. In adult rats and newborn pigs, ADP-dependent pial arteriolar dilation is also astrocyte dependent 11, 22. Astrocytes and endothelium dependence are consistent with astrocyte-derived CO as a final signal because, in newborn pig5, NO, prostanoids and cGMP are important as permissive factors enabling vascular responses to CO 23, 24.
The goal of the present study was to address the hypothesis that ADP stimulates production of CO by glia limitans astrocytes and that this CO causes pial arteriolar dilation in the newborn piglet brain.
MATERIALS AND METHODS
Methods
The animal protocols conform to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were reviewed and approved by the Animal Care and Use Committee of the University of Tennessee Health Science Center. Newborn pigs (1-3 days old; 1-2.5 kg) were anesthetized with ketamine hydrochloride (33 mg/kg, intramuscularly) and acepromazine (3.3 mg/kg, intramuscularly) and maintained on α-chloralose (50 mg/kg, intravenously). A catheter was inserted into a femoral artery to monitor blood pressure and to collect blood for measurement of PO2, PCO2 and pH. A second catheter was placed in a femoral vein for anesthetic and fluid administration. The animals were intubated, mechanically ventilated and, if needed, supplemented with O2 to maintain arterial pH, PO2 and PCO2 within the normal range. A heating pad was used to maintain the animals at 37.5-38.5°C, which was monitored with a rectal probe.
Materials
Water-soluble adenosine 5’-diphosphate monosodium salt was purchased from Calbiochem (Gibbstown, NJ). The HO inhibitor chromium mesoporphyrin (CrMP) was purchased from Frontier Scientific (Logan, UT). All other reagents were purchased from Sigma Chemicals (St. Louis, MO) unless otherwise stated.
Cranial Window Placement
The scalp was surgically retracted and a 2-cm diameter craniotomy was made over the parietal cortex. The dura was cut and all cut edges were retracted over the bone so that the periarachnoid space was not exposed to damaged bone or damaged membranes. A stainless steel and glass window was implanted into the hole and cemented sequentially with bone wax and dental acrylic. The window consisted of three parts: a stainless steel ring, a circular glass coverslip, and three ports consisting of 17-gauge hypodermic needles attached to three precut holes in the stainless steel ring. The space under the window was filled with artificial cerebrospinal fluid (aCSF) equilibrated with 6% CO2 and 6% O2 that produced gases and pH within the normal range for CSF (pH = 7.33-7.40, PCO2 = 42-46 mm Hg, and PO2 = 43-50 mm Hg). Fluid under the cranial window was exchanged through the needles oN the sides of the window. Pial arterioles were observed with a dissecting microscope. Diameters were measured with a video micrometer and monitor.
In all experiments, the physiological variables were within normal limits. PaO2, PaCO2, pH, and mean arterial pressure in these groups did not show any significant differences when initial and final values over the course of the experiments were compared.
In Vivo Experiments
After implantation of the cranial window, at least 30 min were allowed before experimentation was begun. Isoproterenol (ISO 10-7 M), ADP (10-6 M, 10-5 M, 10-4 M), bradykinin (10-6 M), and sodium nitroprusside (10-6 M) were applied directly to pial arterioles; the maximum diameter attained during a 5-min period was used for measurement because the onset of dilation after topical application of these agonists is rapid, with maximum diameter typically attained within 3 min. The cranial window was flushed with aCSF between experiments, and pial arterioles were allowed to return to control diameter before the next agonist was applied. Control responses were compared with the same treatments after astrocyte injury or HO inhibition.
Glia-limitans astrocyte injury was produced by exposing the superficial cortical glia limitans under the cranial window to the selective glia toxin, L-2-α-aminoadipic acid L-2αAAA (2 mM for 5 h) 11, 22, 26, 27. The cellular specificity of L-2αAAA results from the rapid uptake of the toxin by the cysteine-glutamate antiporter expressed by glia but not other cells 28, 29. The precise mechanism underlying the gliotoxicity caused by cellular loading with L-2αAAA is not known. For these experiments, we modified the method from the one developed to produce removal of the influence of glia limitans astrocyte signals on pial arteriolar responses in adult rats 22, 27. The inactive isomer, D-2α aminoadipic acid (D-2αAAA) (2 mM for 5h) was used as control.
To investigate the contribution of CO produced endogenously by HO to vascular responses, the brain surface under the window was exposed to a HO inhibitor, CrMP (2 × 10-5 M) 10, 29. CrMP was topically applied and because of its photosensitivity, lights were turned off between measurements.
Cerebral CO Production
Collections of CSF from under the cranial window were made under control conditions and during subsequent ADP treatment before and after treatment with L-2αAAA. Collections were made after the aCSF had been under the window for 5 min. In order to obtain CSF from under the window, fresh aCSF was injected into one of the needle ports on the cranial window and 400 μl of displaced CSF was collected in a 2-ml glass bottle using a metal spout on another port. We have previously shown that this collection method produces results after collection from under the window that equate to known concentrations 12. The total end volume was increased to 1.7 ml, 31CO standard was added, and the vial was sealed with a rubber and Teflon cap. CO in the headspace gas was measured by gas chromatography-mass spectrometry and quantified by comparison to the 31CO standard as described before 30.
Astrocyte and Microvessel Collection
Microvessels and astrocytes were prepared as described before 11, 25. Briefly, the brain was removed and placed in ice-cold Krebs solution. The brain cortex tissue was minced and gently homogenized in a Dounce homogenizer with a loose pestle. The homogenate was passed through a 300-μm nylon mesh screen, and the passage was refiltered through a 60-μm nylon mesh screen. The cerebral microvessels were retained on the 60-μm filters and the filtrate was the astrocyte-enriched fraction of cerebral cortex. Microvessels were washed off the screen by agitation in Krebs and both the microvessels and astrocytes were concentrated by centrifugation. The concentrated cells were resuspended in Krebs.
CO Production by Microvessels and Astrocytes
Freshly isolated cerebral microvessels and astrocytes in 1.7ml Krebs solution were placed inside 2.0-ml amber vials. For HO and P2Y1 purinergic receptor inhibition, the microvessels and astrocytes were pretreated with CrMP (2×10-5M) or the P2Y1 inhibitor, MRS-2179 (10μM), for 30 min before the experiment was started, and the inhibitors were maintained throughout. Vehicle or ADP and the internal standard (31CO) were injected into the bottom of the vial, which was immediately sealed with a rubberized Teflon-lined cap and incubated at 37°C for 30 min. The samples were placed in hot water (75°C) for 8 min. to kill the cells and inactivate HO. CO in the headspace was measured by gas chromatography-mass spectrometry as previously described 30. Protein was determined by the Lowery method.
Statistical analysis
Values are presented as means ± SE. Results were subjected to a one-way ANOVA for repeated measures with Tukey’s post hoc test to isolate differences between groups. A level of P< 0.05 was considered significant.
RESULTS
Figure 1 shows effects of topically applied gliotoxin L-2αAAA (2μM, 5h), on isoproterenol (10-7 M)- and ADP (10-4 M)-induced pial arteriolar dilation in vivo. After astrocyte injury, dilation to ADP was blocked, whereas dilation in response to isoproterenol (10-7 M), which increases cAMP through vascular smooth muscle β-adrenergic receptors, was unaltered (Fig. 1). Similarly, dilations to lower doses of ADP were blocked completely following L-2αAAA treatment (61 ± 5, 76 ± 7*, and 83 ± 7* μm at 0, 10-6, 10-5 M ADP prior to and 60 ± 5, 61 ± 5, and 62 ± 5 μm following L-2αAAA treatment (N = 6 piglets, *P<0.05 compared to 0 ADP). Conversely, the inactive amino acid, D-2αAAA, had no effect on pial arteriolar dilation to ADP (10-4 M): 53 ± 2μm to 65 ± 3μm before and 52 ± 3μm to 63 ± 4μm after D-2αAAA (n=9)
Fig. 1.
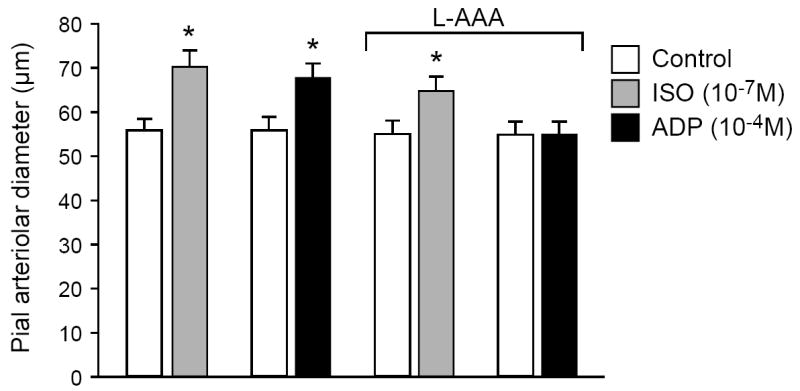
Pial arteriolar dilation in response to isoproterenol (ISO; 10-7 M) and to adenosine diphosphate (ADP; 10-4 M) before and after treatment with L-2-α-aminoadipic acid (L-2αAAA; 5 h, 2 mM). Values are means ± SEM. N = 14. *P < 0.05 compared with preceding bar.
The effects of ADP on aCSF CO concentration before and after treatment with L-2αAAA (5 h, 2 mM) are shown in Fig. 2. CO production by the brain surface was detected in the aCSF collected from beneath the cranial window. CO production was increased by ADP. This increase was completely blocked following L-2αAAA-induced astrocyte injury.
Fig. 2.
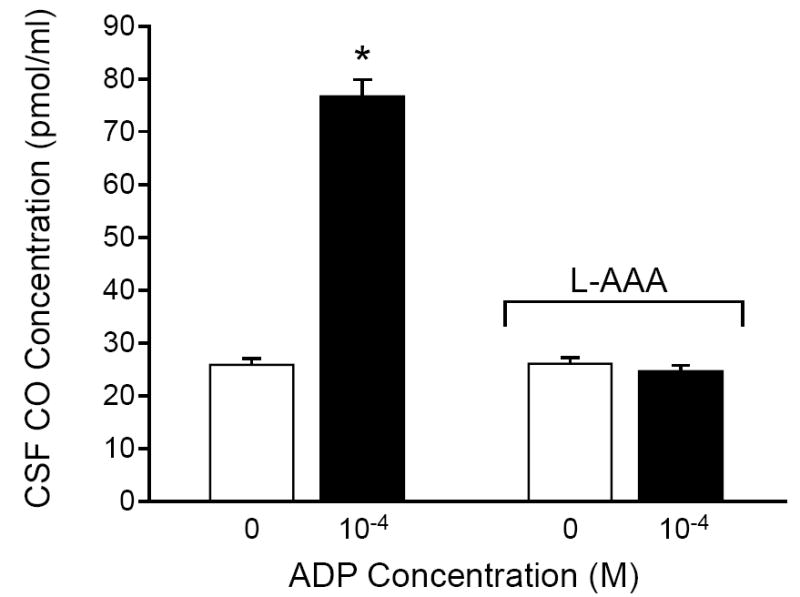
Effect of adenosine diphosphate (ADP) on CO concentration in aCSF collected from under the cranial window before and after treatment with L-2-α-aminoadipic acid (L-2αAAA; 5 h, 2 mM). Values are means ± SEM. N = 6. *P < 0.05 compared with control.
Both SNP (2 × 10-7 M) and ADP (10-5 M, 10-4 M) caused increases in pial arteriolar diameter (Fig. 3). The dilation to ADP was blocked by CrMP, the metal porphyrin inhibitor of HO (Fig. 3). CrMP did not inhibit the vasodilation in response to SNP that dilates by increasing vascular smooth muscle cGMP.
Fig. 3.
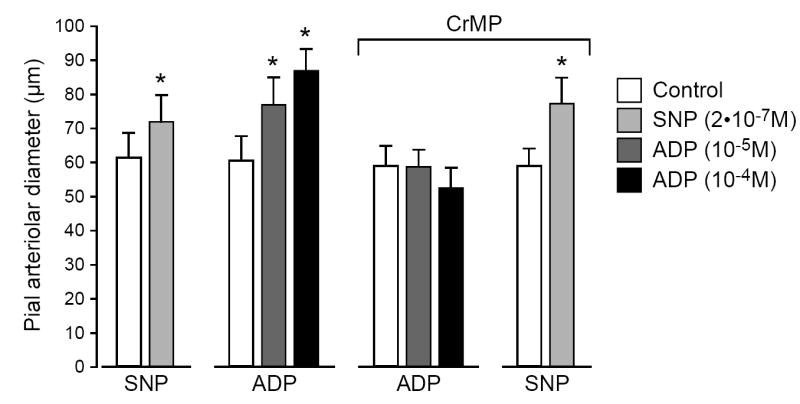
Effects of sodium nitroprusside (SNP; 2 × 10-7 M) and adenosine diphosphate (ADP; 10-5 M, 10-4 M) on pial arteriolar diameters before and in the presence of CrMP (2 × 10-5 M). Values are means ± SEM. N = 6. *P < 0.05 compared to preceding control.
To investigate the possibility that astrocyte-derived CO may play a permissive role in enabling an EDRF-mediated response to ADP, the CO concentration in CSF was elevated by adding 10-7M CO to the aCSF following L-AAA injury and the responses to ADP determined. Following L-2αAAA, dilation to ADP was absent (59±6, 61±6, and 61±6μm at 0, 10-6M, 10-5M ADP, n=5 piglets), but dilations to bradykinin (60±6 and 69±7*μm with 0 and 10-6M bradykinin (n=5 piglets, *P<0.05) and isoproterenol (60±6 and 75±8*μm with 0 and 10-7M isoproterenol (n=5 piglets, *P<0.05) remained. Addition of CO (10-7M) to aCSF following L-2αAAA did not enhance responses to ADP (75±7, 75±6, and 75±7μm at 0, 10-6M, 10-5M, ADP, n=5 piglets).
Treatment with ADP dose-dependently increased CO production in freshly isolated piglet astrocytes (Fig. 4) and cerebral microvessels (Fig. 5). CrMP blocked ADP-induced increases in both astrocytes and microvessels. Although ADP increased CO production by both astrocytes and microvessels, the increases caused in astrocytes (59 ± 7 and 116± 9% at 10-5M and 10-4M ADP) were greater than in microvessels (29 ± 11 and 80± 13% at 10-5M and 10-4M ADP). The ADP-induced CO production by astrocytes appears to involve activation of P2Y1 receptor because the elevation of CO is blocked by MRS2179 (31±7**,+ and 7±5 pmol/mg protein without and with MRS2179, respectively. *P<0.05 compared to zero. +P<0.05 compared to with MRS2179 (n=7).
Fig. 4.
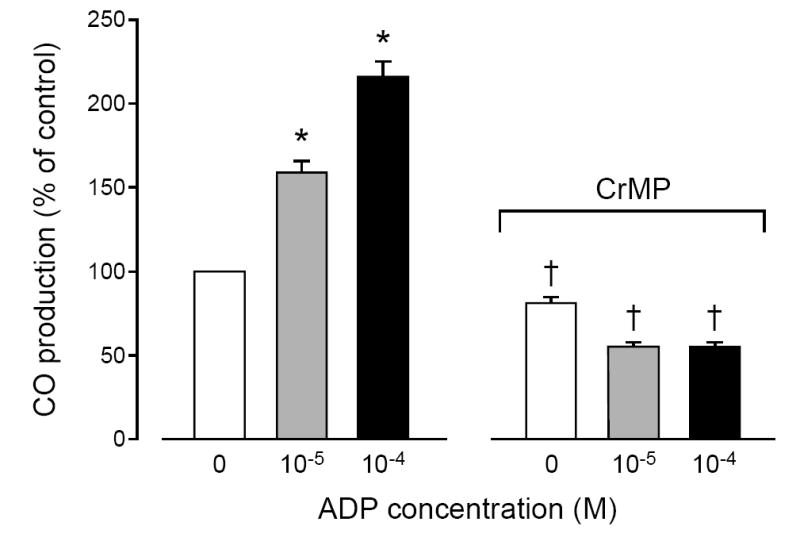
Effect of adenosine diphosphate (ADP; 10-5 M, 10-4 M) on CO production by piglet astrocytes in the absence and in the presence of CrMP (2 × 10-5 M). Mean ± SEM relative to control. N = 6 experiments. *P < 0.05 compared with no ADP.† = P < 0.05 compared to before CrMP.
Fig. 5.
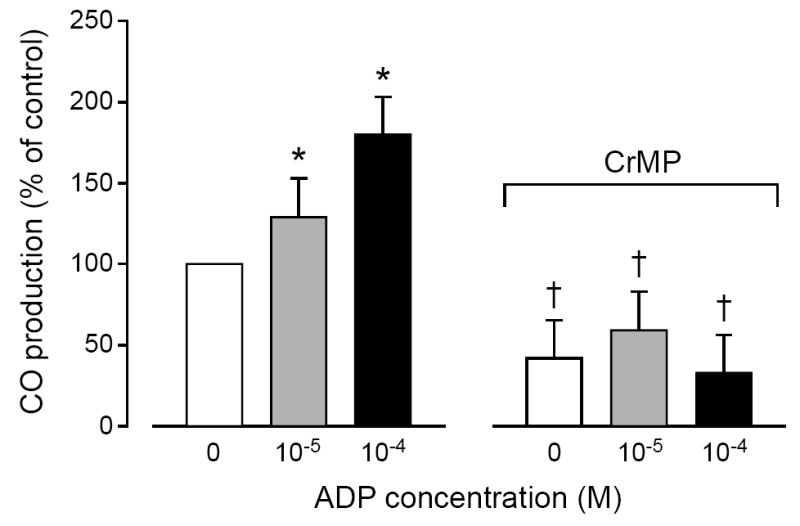
Effect of adenosine diphosphate (ADP; 10-5 M, 10-4 M) on CO production by piglet cerebral microvessels in the absence and in the presence of CrMP (2 × 10-5 M). Values are means ± SEM relative to control. N = 6. *P < 0.05 compared with no ADP.† = P < 0.05 compared to before CrMP.
DISCUSSION
The major findings in newborn pigs are: 1) treatment with the astrocyte toxin, L-2αAAA, and/or, the HO inhibitor, CrMP, block pial arteriolar dilation to ADP, but not to isoproterenol, bradykinin or sodium nitroprusside, 2) ADP increases brain CO production and this increase is blocked by the astrocyte toxin or inhibition of HO, and 3) ADP increases CO production by astrocytes, and to a lesser extent cerebral microvessels. These data, coupled with previous results showing CO dilates pial arterioles in vivo, suggest CO is an astrocyte-derived mediator of ADP-induced pial arteriolar dilation in piglets.
ADP can produce endothelium-dependent cerebral vasodilation 19, which may be mediated in part by NO and EDHF in adult rats 20, 21, 30. In endothelium-denuded control arteries from rat brain, ADP also produced dose-dependent relaxation, but this relaxation was lower than that found in intact control arteries 31. In adult rats, ADP-induced pial arteriolar dilation involves the additive effects of an endothelium-dependent and an astrocyte-dependent component 22.
If the astrocyte component is CO, the response would be endothelial dependent in piglets. Thus, the absence of an effect of L-2αAAA on pial arteriolar dilation to CO itself11, the insensitivity of cerebrovascular endothelial cells to L-2αAAA in vitro,11 and dilation to the endothelial-dependent dilator, bradykinin32, following L-2αAAA strongly suggest the effects of L-2αAAA do not result from endothelial injury. CO exhibits its vasoactive actions in conjunction with endothelium-derived relaxing factors (EDRF) that act as permissive enablers 24, 33. In the piglet cerebral circulation these permissive enablers include both NO and prostacyclin 24, 25. The mechanism by which prostacyclin and NO permit the dilatory response to CO appears to be largely attributable to activity of PKG 34. However, the ability of NO to partially restore the dilatory response to CO even when guanylyl cyclase is blocked suggests that NO may have actions independent of cGMP 23, 34.
It is also conceivable that astrocyte CO could function as a permissive enabler of responses to an EDRF. Thus, if the astrocyte-derived CO were required for EDRF to cause smooth muscle relaxation, astrocyte injury would block the response even if the final mediator was produced by ADP stimulating EDRF release by endothelium. However, in the case of ADP in newborn pigs such a permissive role for CO appears unlikely because the addition of CO to the aCSF did not restore dilation to ADP following L-2αAAA.
In other vascular beds, CO can induce vasodilation that is independent of endothelium. Endothelium-independent vasodilatory mechanisms have been shown in the vasodilatory effect of CO in the rat tail artery 35, rat thoracic aorta 36, porcine coronary artery and the vein 37, and dog carotid and coronary arteries 38.
Astrocytes are critical players in the regulation of cerebral arteriolar diameter including dilation in response to neuronal activity. There are several molecular pathways through which astrocytes can elicit dilation 39, 40. Locally secreted substances such as adenosine, ADP or K+, act on neighboring blood vessels to cause vasodilation 41, 42. Piglet glia limitans astrocytes use CO as a messenger to cause cerebral pial arteriole dilation in response to glutamate, which would enhance local blood flow to match increased glutamatergic neuronal activity 11, 43. In adult rats, NMDA receptor activation of postsynaptic neurons leads to the stimulation of nitric oxide synthase (NOS) to produce NO, which causes vascular smooth muscle relaxation 44.
In the newborn pig cerebral circulation, topical ADP stimulated both CO production and vasodilation of pial arterioles. The inhibitor of HO, CrMP, prevented the ADP-induced increase in CSF CO and the pial arteriolar dilation caused by ADP, but did not block the dilation to sodium nitroprusside. Of note, CrMP did not block pial arteriolar dilation in control adult rats, but did inhibit ADP-induced dilation in rats transfused with cell-free hemoblobin45. Clearly, mechanisms of cerebrovascular circulatory dilation may differ with respect to species and/or age. The ADP-evoked increases in cerebral arteriolar diameter and in CO production in aCSF were abolished after treatment with L-2αAAA (2mM, 5h). L-2αAAA is a gliotoxin that can be used as a tool to ablate astrocytes in vitro or in vivo 11, 26, 27. In the present study, the absence of any direct actions of L-2αAAA on pial vascular smooth muscle function was shown by the fact that no changes in the response to isoproterenol or SNP were observed in pial arterioles in the presence of L-2αAAA. ADP also stimulated CO production by isolated astrocytes that was blocked by MRS2179, suggesting involvement to P2Y1 receptors that also cause ADP-induced, astrocyte dependent, pial arteriolar dilation in adult rats22. All these data suggest that astrocytes could deploy CO as a gasotransmitter resulting in cerebrovasodilation in response to ADP.
Of note, we could not detect any L-2αAAA-induced change in basal CO in the present or previous study11. In addition to astrocytes, neurons, endothelial cells and vascular smooth muscle cells would contribute to the cortical CSF CO concentration 46. These data suggest the fractional contribution of astrocytes to basal CSF CO concentration is sufficiently small as to not be detectable following astrocyte injury.
ADP also increases production of CO in freshly isolated cerebral microvessels but less so than in astrocytes. The microvessels are coated with astrocytes and their processes 11, so it is uncertain whether the increase in CO is from the vessels, the adhering astrocyte processes, or both.
The vasodilator effect of CO has been attributed to both the cGMP/protein kinase G signaling pathway 47, 48 and activation of KCa channels 10, 17, 35. In newborn pig pial arterioles, dilation to CO can be attributed solely to KCa channels 46. CO causes smooth muscle hyperpolarization via activation of large-conductance KCa channels 17, 49. The binding of CO to heme on the KCa channels of arteriolar smooth muscle cells 50 increases the Ca sensitivity of KCa channels 49. KCa channels are stimulated by increased local Ca concentrations produced by Ca sparks, and CO increases Ca sparks and the coupling of Ca sparks to KCa channel openings 17.
In summary, we show that topical ADP increases cerebral production of CO in vivo, dilates pial arterioles, and stimulates CO production by isolated astrocytes. Both ADP-induced production of CO and vasodilation were blocked by astrocyte injury and HO inhibition. These data are consistent with the hypothesis that glia limitans astrocytes employ CO as a signaling messenger by which ADP dilates pial arterioles and enhances local blood flow in newborn pigs.
Acknowledgments
GRANTS Research was supported by the National Heart, Lung, and Blood Institute/National Institutes of Health (NHLBI/NIH). Dr. Kanu was supported by a training grant from NHLBI/NIH.
Footnotes
DISCLOSURES None
References
- 1.Girouard H, Iadecola C. Neurovascular coupling in the normal brain and in hypertension, stroke, and alzheimer disease. J Appl Physiol. 2006;100:328–335. doi: 10.1152/japplphysiol.00966.2005. [DOI] [PubMed] [Google Scholar]
- 2.Tamayo-Orrego L, Duque-Parra JE. The metabolic regulation of cerebral microcirculation. Rev Neurol. 2007;44:415–425. [PubMed] [Google Scholar]
- 3.Paspalas CD, Papadopoulos GC. Ultrastructural evidence for combined action of noradrenaline and vasoactive intestinal polypeptide upon neurons, astrocytes, and blood vessels of the rat cerebral cortex. Brain Res Bull. 1998;45:247–259. doi: 10.1016/s0361-9230(97)00327-4. [DOI] [PubMed] [Google Scholar]
- 4.Koehler RC, Gebremedhin D, Harder DR. Role of astrocytes in cerebrovascular regulation. J Appl Physiol. 2006;100:307–317. doi: 10.1152/japplphysiol.00938.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev. 2006;86:1009–1031. doi: 10.1152/physrev.00049.2005. [DOI] [PubMed] [Google Scholar]
- 6.Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M. Signaling at the gliovascular interface. J Neurosci. 2003;23:9254–9262. doi: 10.1523/JNEUROSCI.23-27-09254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fellin T, Carmignoto G. Neuron-to-astrocyte signalling in the brain represents a distinct multifunctional unit. J Physiol. 2004;559:3–15. doi: 10.1113/jphysiol.2004.063214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boehning D, Moon C, Sharma S, Hurt KJ, Hester LD, Ronnett GV, Shugar D, Snyder SH. Carbon monoxide neurotransmission activated by CK2 phosphorylation of heme oxygenase-2. Neuron. 2003;40:129–137. doi: 10.1016/s0896-6273(03)00596-8. [DOI] [PubMed] [Google Scholar]
- 9.Kanu A, Whitfield J, Leffler CW. Carbon monoxide contributes to hypotension-induced cerebrovascular vasodilation in piglets. Am J Physiol Heart Circ Physiol. 2006;291:H2409–2414. doi: 10.1152/ajpheart.01368.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leffler CW, Nasjletti A, Yu C, Johnson RA, Fedinec AL, Walker N. Carbon monoxide and cerebral microvascular tone in newborn pigs. Am J Physiol Heart Circ Physiol. 1999;276:H1641–1646. doi: 10.1152/ajpheart.1999.276.5.H1641. [DOI] [PubMed] [Google Scholar]
- 11.Leffler CW, Parfenova H, Fedinec AL, Basuroy S, Tcheranova D. Contributions of astrocytes and CO to pial arteriolar dilation to glutamate in newborn pigs. Am J Physiol Heart Circ Physiol. 2006;291:H2897–2904. doi: 10.1152/ajpheart.00722.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kanu A, Leffler CW. Carbon monoxide and Ca2+ -activated K+ channels in cerebral arteriolar responses to glutamate and hypoxia in newborn pigs. Am J Physiol Heart Circ Physiol. 2007;293:H3193–2000. doi: 10.1152/ajpheart.00274.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maines MD. The heme oxygenase system: A regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- 14.Scapagnini G, D’Agata V, Calabrese V, Pascale A, Colombrita C, Alkon D, Cavallaro S. Gene expression profiles of heme oxygenase isoforms in the rat brain. Brain Res. 2002;954:51–59. doi: 10.1016/s0006-8993(02)03338-3. [DOI] [PubMed] [Google Scholar]
- 15.Leffler CW, Balabanova L, Sullivan CD, Wang X, Fedinec AL, Parfenova H. Regulation of CO production in cerebral microvessels of newborn pigs. Am J Physiol Heart Circ Physiol. 2003;285:H292–297. doi: 10.1152/ajpheart.01059.2002. [DOI] [PubMed] [Google Scholar]
- 16.Parfenova H, Daley ML, Carratu P, Leffler CW. Heme oxygenase inhibition reduces neuronal activation evoked by bicuculline in newborn pigs. Brain Res. 2004;1014:87–96. doi: 10.1016/j.brainres.2004.03.052. [DOI] [PubMed] [Google Scholar]
- 17.Jaggar JH, Leffler CW, Cheranov SY, Tcheranova D, S E, Cheng X. Carbon monoxide dilates cerebral arterioles by enhancing the coupling of Ca2+ sparks to Ca2+-activated K+ channels. Circ Res. 2002;91:610–617. doi: 10.1161/01.res.0000036900.76780.95. [DOI] [PubMed] [Google Scholar]
- 18.Bryan RM., Jr Purines, purine nucleotides, and pyrimidine nucleotides. Cerebral Blood Flow and Metabolism. 2002:311–324. [Google Scholar]
- 19.Faraci FM. Regulation of the cerebral circulation by endothelium. Pharmacol Ther. 1992;56:1–22. doi: 10.1016/0163-7258(92)90035-x. [DOI] [PubMed] [Google Scholar]
- 20.Mayhan WG. Endothelium-dependent responses of cerebral arterioles to adenosine 5’-diphosphate. J Vasc Res. 1992;29:353–358. doi: 10.1159/000158951. [DOI] [PubMed] [Google Scholar]
- 21.Faraci FM, Lynch C, Lamping KG. Responses of cerebral arterioles to ADP: eNOS-dependent and eNOS-independent mechanisms. Am J Physiol Heart Circ Physiol. 2004;287:H2871–H2876. doi: 10.1152/ajpheart.00392.2004. [DOI] [PubMed] [Google Scholar]
- 22.Xu HL, Ye S, Baughman VL, Feinstein DL, Pelligrino DA. The role of the glia limitans in adp-induced pial arteriolar relaxation in intact and ovariectomized female rats. Am J Physiol Heart Circ Physiol. 2005;288:H382–388. doi: 10.1152/ajpheart.00727.2004. [DOI] [PubMed] [Google Scholar]
- 23.Koneru P, Leffler CW. Role of cGMP carbon monoxide-induced cerebral vasodilation in piglets. Am J Physiol Heart Circ Physiol. 2004;286:H304–309. doi: 10.1152/ajpheart.00810.2003. [DOI] [PubMed] [Google Scholar]
- 24.Leffler CW, Nasjletti A, Johnson RA, Fedinec AL. Contributions of prostacyclin and nitric oxide to carbon monoxide-induced cerebrovascular dilation in piglets. Am J Physiol Heart Circ Physiol. 2001;280:H1490–1495. doi: 10.1152/ajpheart.2001.280.4.H1490. [DOI] [PubMed] [Google Scholar]
- 25.Leffler CW, Balabanova L, Fedinec AL, Parfenova H. Nitric oxide increases carbon monoxide production by piglet cerebral microvessels. Am J Physiol Heart Circ Physiol. 2005;289:H1442–1447. doi: 10.1152/ajpheart.00464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khurgel M, Koo AC, Ivy GO. Selective ablation of astrocytes by intracerebral injections of alpha-aminoadipate. Glia. 1996;16:351–358. doi: 10.1002/(SICI)1098-1136(199604)16:4<351::AID-GLIA7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 27.Xu HL, Koenig HM, Ye S, Feinstein DL, Pelligrino DA. Influence of the glia limitans on pial arteriolar relaxation in the rat. Am J Physiol Heart Circ Physiol. 2004;287:H331–339. doi: 10.1152/ajpheart.00831.2003. [DOI] [PubMed] [Google Scholar]
- 28.Huck S, Grass F, Hortnagl H. The glutamate analogue alpha-aminoadipic acid is taken up by astrocytes before exerting its gliotoxic effect in vitro. J Neurosci. 1984;4:2650–2657. doi: 10.1523/JNEUROSCI.04-10-02650.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pow DV. Visualising the activity of the cystine-glutamate antiporter in glial cells using antibodies to aminoadipic acid, a selectively transported substrate. Glia. 2001;34:27–38. doi: 10.1002/glia.1037. [DOI] [PubMed] [Google Scholar]
- 30.Robinson JS, Fedinec AL, Leffler CW. Role of carbon monoxide in glutamate receptor-induced dilation of newborn pig pial arterioles. Am J Physiol Heart Circ Physiol. 2002;282:H2371–2376. doi: 10.1152/ajpheart.00911.2001. [DOI] [PubMed] [Google Scholar]
- 31.You J, Johnson TD, Childres WF, Bryan RM., Jr Endothelial-mediated dilations of rat middle cerebral arteries by ATP and ADP. Am J Physiol Heart Circ Physiol. 1997;273:H1472–1477. doi: 10.1152/ajpheart.1997.273.3.H1472. [DOI] [PubMed] [Google Scholar]
- 32.Willis AP, Leffler CW. Endothelial NO and prostanoid involvement in newborn and juvenile pig pial arteriolar vasomotor responses. Am J Physiol Heart Circ Physiol. 2001;281:H2366–2377. doi: 10.1152/ajpheart.2001.281.6.H2366. [DOI] [PubMed] [Google Scholar]
- 33.Barkoudah E, Jaggar JH, Leffler CW. The permissive role of endothelial NO in CO-induced cerebrovascular dilation. Am J Physiol Heart Circ Physiol. 2004;287:H1459–1465. doi: 10.1152/ajpheart.00369.2004. [DOI] [PubMed] [Google Scholar]
- 34.Leffler CW, Fedinec AL, Parfenova H, Jaggar JH. Permissive contributions of NO and prostacyclin in CO-induced cerebrovascular dilation in piglets. Am J Physiol Heart Circ Physiol. 2005;289:H432–438. doi: 10.1152/ajpheart.01195.2004. [DOI] [PubMed] [Google Scholar]
- 35.Wang R, Wang Z, Wu L. Carbon monoxide-induced vasorelaxation and the underlying mechanisms. Br J Pharmacol. 1997;121:927–934. doi: 10.1038/sj.bjp.0701222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin H, McGrath JJ. Vasodilating effects of carbon monoxide. Drug Chem Toxicol. 1988;11:371–385. doi: 10.3109/01480548809018109. [DOI] [PubMed] [Google Scholar]
- 37.Graser T, Vedernikov YP, Li DS. Study on the mechanism of carbon monoxide induced endothelium-independent relaxation in porcine coronary artery and vein. Biomed Biochim Acta. 1990;49:293–296. [PubMed] [Google Scholar]
- 38.Vedernikov YP, Graser T, Vanin AF. Similar endothelium-independent arterial relaxation by carbon monoxide and nitric oxide. Biomed Biochim Acta. 1989;48:601–603. [PubMed] [Google Scholar]
- 39.Gordon GR, Mulligan SJ, MacVicar BA. Astrocyte control of the cerebrovasculature. Glia. 2007;55:1214–1221. doi: 10.1002/glia.20543. [DOI] [PubMed] [Google Scholar]
- 40.Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, Carmignoto G. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci. 2003;6:43–50. doi: 10.1038/nn980. [DOI] [PubMed] [Google Scholar]
- 41.Filosa JA, Bonev AD, Straub SV, Meredith AL, Wilkerson MK, Aldrich RW, Nelson MT. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat Neurosci. 2006;9:1397–1403. doi: 10.1038/nn1779. [DOI] [PubMed] [Google Scholar]
- 42.Ngai AC, Coyne EF, Meno JR, West GA, Winn HR. Receptor subtypes mediating adenosine-induced dilation of cerebral arterioles. Am J Physiol Heart Circ Physiol. 2001;280:H2329–2335. doi: 10.1152/ajpheart.2001.280.5.H2329. [DOI] [PubMed] [Google Scholar]
- 43.Li A, Xi Q, Umstot ES, Bellner L, Schwartzman ML, Jaggar JH, Leffler CW. Astrocyte-derived CO is a diffusible messenger that mediates glutamate-induced cerebral arteriolar dilation by activating smooth muscle cell KCa channels. Circ Res. 2008;102:234–241. doi: 10.1161/CIRCRESAHA.107.164145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Iadecola C. Regulation of the cerebral microcirculation during neural activity: Is nitric oxide the missing link? Trends Neurosci. 1993;16:206–214. doi: 10.1016/0166-2236(93)90156-g. [DOI] [PubMed] [Google Scholar]
- 45.Rebel A, Cao S, Kwansa H, Dore S, Bucci E, Koehler RC. Dependence of acetylcholine and ADP dilation of pial arterioles on heme oxygenase after transfusion of cell-free polymeric hemoglobin. Am J Physiol Heart Circ Physiol. 2006;290:H1027–H1037. doi: 10.1152/ajpheart.00500.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leffler CW, Parfenova H, Jaggar JH, Wang R. Carbon monoxide and hydrogen sulfide: Gaseous messengers in cerebrovascular circulation. J Appl Physiol. 2006;100:1065–1076. doi: 10.1152/japplphysiol.00793.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Christodoulides N, Durante W, Kroll MH, Schafer AI. Vascular smooth muscle cell heme oxygenases generate guanylyl cyclase-stimulatory carbon monoxide. Circulation. 1995;91:2306–2309. doi: 10.1161/01.cir.91.9.2306. [DOI] [PubMed] [Google Scholar]
- 48.Morita T, Perrella MA, Lee ME, Kourembanas S. Smooth muscle cell-derived carbon monoxide is a regulator of vascular cGMP. Proc Natl Acad Sci U S A. 1995;92:1475–1479. doi: 10.1073/pnas.92.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xi Q, Tcheranova D, Parfenova H, Horowitz B, Leffler CW, Jaggar JH. Carbon monoxide activates KCa channels in newborn arteriole smooth muscle cells by increasing apparent Ca2+ sensitivity of alpha-subunits. Am J Physiol Heart Circ Physiol. 2004;286:H610–618. doi: 10.1152/ajpheart.00782.2003. [DOI] [PubMed] [Google Scholar]
- 50.Jaggar JH, Li A, Parfenova H, Liu J, Umstot ES, Dopico AM, Leffler CW. Heme is a carbon monoxide receptor for large-conductance Ca2+-activated K+ channels. Circ Res. 2005;97:805–812. doi: 10.1161/01.RES.0000186180.47148.7b. [DOI] [PMC free article] [PubMed] [Google Scholar]