

Abstract
Tumor necrosis factor (TNF) plays an important role in the pathogenesis of inflammatory bone loss through stimulation of osteoclastic bone resorption and inhibition of osteoblastic bone formation. Compared with the well established role of TNF in osteoclastogenesis, mechanisms by which TNF inhibits osteoblast function have not been fully determined. Runx2 is an osteoblast-specific transcription factor whose steady-state protein levels are regulated by proteasomal degradation, mediated by the E3 ubiquitin ligases, Smurf1 and Smurf2. We hypothesized that TNF inhibits osteoblast function through Smurf-mediated Runx2 degradation. We treated C2C12 and 2T3 osteoblast precursor cell lines and primary osteoblasts with TNF and found that TNF, but not interleukin-1, significantly increased Smurf1 and Smurf2 expression. TNF increased the degradation of endogenous or transfected Runx2 protein, which was blocked by treating cells with a proteasomal inhibitor or by infecting cells with small interfering (si)RNA against Smurf1 or Smurf2. TNF inhibited the expression of bone morphogenetic protein and transforming growth factor-β signaling reporter constructs, and the inhibition of each was blocked by Smurf1 siRNA and Smurf2 siRNA, respectively. Overexpression of Smurf1 and/or Smurf2 siRNAs prevented the inhibitory effect of TNF on Runx2 reporter. Consistent with these in vitro findings, bones from TNF transgenic mice or TNF-injected wild type mice had increased Smurf1 and decreased Runx2 protein levels. We propose that one of the mechanisms by which TNF inhibits bone formation in inflammatory bone disorders is by promoting Runx2 proteasomal degradation through up-regulation of Smurf1 and Smurf2 expression.
Tumor necrosis factor (TNF)2 is a major contributor to pathologic bone loss through stimulation of osteoclastic bone resorption and inhibition of osteoblastic bone formation. In patients with rheumatoid arthritis, TNF and other cytokines are overproduced in inflamed joints by various cells infiltrating the synovial membrane. This leads to severe local erosion of cartilage and bone, periarticular osteopenia, as well as systemic osteoporosis (1, 2). Under these conditions, osteoblasts do not catch up with the accelerated bone resorption, indicating impaired osteoblast function (3). The inhibitory effects of TNF on bone formation in vitro were first described in 1987 in neonatal rat calvarial organ cultures (4). Subsequent studies demonstrated that TNF inhibits recruitment of osteoblast progenitors, reduces expression of genes produced by mature osteoblasts, and promotes osteoblast apoptosis through nuclear factor-κB signaling pathway (5-9). However, compared with our understanding of the role of TNF in osteoclast biology, little is known of the molecular mechanisms that mediate the effect of TNF on osteoblast inhibition.
To date, the most important mechanistic finding of TNF-mediated osteoblast inhibition was the demonstration that TNF decreases Runt-related gene 2 (Runx2) expression and its DNA binding activity in osteoblasts (10). This is partially through suppression of Runx2 gene transcription and destabilization of Runx2 mRNA through the TNF receptor 1 signaling pathway (10-12). However, because TNF-induced reduction in nuclear Runx2 protein (more than 90%) was greater than expected, compared with the decrease in total Runx2 mRNA (50%), it has been predicted that TNF may also have post-transcriptional effects. Furthermore, pharmacological inhibitors of the cell survival-promoting kinases, Akt, phosphatidylinositol 3-kinase, and extracellular signal-regulated kinases, fail to reverse the inhibitory effects of TNF on osteoblast differentiation in vitro (11), suggesting that other signal pathways may be involved.
In the past several years, ubiquitin-mediated proteasomal degradation has been implicated in the regulation of bone morphogenetic protein (BMP)-2 and transforming growth factor-β (TGF-β) signaling pathways in various cell types (13, 14). We and others have demonstrated that the E3 ubiquitin ligase, Smad ubiquitin regulatory factor (Smurf)1, regulates osteoblast differentiation by promoting proteasomal degradation of the BMP signaling protein, Smad1 and Smad5, and of the osteoblast transcription factor, Runx2 (15-18). Smurf2, a closely related homolog of Smurf1, was shown to reduce the steady-state protein levels of Smad1 and 2, but not Smad3 and 4, in Smurf2-transfected cells (19, 20). Ectopic expression of Smurf1 in 2T3 osteoblast precursors and C2C12 myoblast/osteoblast precursors induces the proteasomal degradation of Smad1 and Runx2 proteins, leading to inhibition of osteoblast differentiation (17). Smurf1 blocks BMP-induced osteogenic conversion of C2C12 cells and facilitates their myogenic differentiation by inducing degradation of Smad5 (16). In vitro, Smurf1 also targets a member of the Rho family of small GTPases, RhoA, for ubiquitination and degradation (21, 22). In vivo, overexpression of Smurf1 in osteoblasts by the osteoblast-specific type I collagen (Col1a1) promoter leads to reduction in osteoblast proliferation and activity. Col1a1-Smurf1 transgenic mice have decreased bone formation rates and decreased Runx2 protein expression in osteoblasts (18). Smurf1 knock-out (Smurf1−/−) mice were generated recently. Although they survive to adulthood, they exhibit an age-dependent increase in bone mass (23). Interestingly, osteoblasts from Smurf1−/− mice have normal levels of the BMP receptors, Smad1, 2, 3, and 5, and Runx2, all of which have been defined previously as targets of Smurf1. Because Smurf1 and Smurf2 possess overlapping functions, it is possible that Smurf2 compensates for the loss of Smurf1 in these knock-out animals (19, 20, 24). Currently, the role of Smurf2 in osteoblast function in vivo and the role of Smurf E3 ligases in the pathogenesis of bone diseases remain poorly understood. Additionally, there is little information on the regulation of Smurfs expression under physiological and pathological conditions.
To explore the molecular mechanisms of TNF-mediated osteoblast inhibition, we tested the hypothesis that TNF inhibits osteoblastic bone formation by up-regulating Smurf E3 ligases that degrade Runx2 protein. We found that TNF increased Smurf1 and Smurf2 expression in osteoblasts, leading to enhanced ubiquitination and degradation of Runx2 protein. This TNF-induced Runx2 degradation was reversed by proteasome inhibitors and by knocking down endogenous Smurf1 or Smurf2 using small interfering RNA (siRNA) against Smurf1 or Smurf2. Bones from TNF-overexpressing mice exhibited increased Smurf1 and decreased Runx2 protein levels. Taken together, our findings point to a novel molecular mechanism of TNF inhibition of osteoblasts, which involves post-transcriptional regulation of protein function through Smurf E3 ligase-mediated proteasomal degradation.
MATERIALS AND METHODS
Animals
TNF transgenic (Tg) mice in a CBA × C57BL/6 background (3647 TNF-Tg line) were obtained from Dr. G. Kollias. C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME). The Institutional Animal Care and Use Committee approved all animal studies.
Antibodies
Monoclonal antibodies specific for FLAG and β-actin were purchased from Sigma. Anti-Runx2 monoclonal antibody was from MBL (Woburn, MA). Anti-ubiquitin monoclonal antibody was from Santa Cruz (Santa Cruz, CA). Anti-Smurf1 polyclonal antibody was from Abgent (San Diego, CA).
Cell Culture and Transfection Conditions
C2C12 myoblast/osteoblast precursors were cultured in Dulbecco's modified Eagle's medium, and 2T3 osteoblast precursors were cultured in α-minimal essential medium supplemented with 1% penicillin-streptomycin (all from Invitrogen) and 10% fetal calf serum (JRH Biosciences, Lenexa, KS). When cells were grown to 90% confluence, the cDNA expression plasmid, pCMV-FLAG-tagged Runx2 (F-Runx2) and/or a pCMV-Myc-tagged Smurf1 (M-Smurf1) were transiently transfected into the cells using Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacturer's instructions. Total amounts of transfected plasmids in each group were equalized by the addition of empty vector. After transfection (24 h), the cells were cultured further in the presence and absence of murine TNF (R&D Systems) and subjected to reverse transcription (RT)-PCR or Western blot analysis.
Bone Nodule Formation
Bone marrow cells were flushed from the tibiae and femur of wild type (wt) and TNF-Tg mice, and the cells were seeded at a density of 2 × 106/ml. The cells were cultured in 37 °C with a humidified 5% CO2 atmosphere. When the cells reached confluence (day 0), the medium was changed to an osteoblast-inducing medium (α-minimal essential medium-supplemented 10% fetal calf serum with 100 μg/ml l-ascorbic acid and 5 mm β-glycerophosphate) with or without 40 ng/ml BMP-2, and the medium was changed twice a week. After an 18-day incubation, the cells were fixed with 10% formalin and stained by the von-Kossa method. The area of mineralized bone nodules was determined under light microscopy by point counting, as described previously (25).
Quantitative Real Time RT-PCR
Cells were homogenized using 1 ml of TRIzol reagent (Invitrogen), and total RNA was extracted according to the manufacturer's protocol. cDNA was synthesized using 20 μl of reverse transcription reaction solution containing 1 μg of total RNA, 10 mm Tris-HCl buffer (pH 8.3), 50 mm KCl, 5 mm MgCl2, 1 mm deoxynucleoside triphosphates, 2.5 μm random hexamers, 20 units RNase inhibitor, and 50 units of Moloney murine leukemia virus reverse transcriptase (all from Roche Applied Science). Quantitative real time PCR amplifications were performed in an iCycler real time PCR machine using iQ SYBR Green supermix (both from Bio-Rad Laboratories) according to the manufacturer's instruction. The sequences of primer sets for Smurf1, Smurf2, alkaline phosphatase (ALP), osteocalcin (OC), and β-actin mRNAs, target sites on mRNAs and product sizes by PCR are shown in Table 1. To minimize the background of products amplified from genomic DNAs, these primers were designed to exist on two different exons. The quantity of Smurf1, Smurf2, ALP, and OC mRNA in each sample was normalized using the CT (threshold cycle) value obtained for the β-actin mRNA amplifications.
TABLE 1.
Sequences of primers used in the real time PCR
| Genes | GenBank™ accession number | Sequences of primersa | Target sites on genes | Product sizes |
|---|---|---|---|---|
| bp | ||||
| Smurf1 | NM029438 | F: 5′–AGTTCGTGGCCAAATAGTGG–3′ R: 5′–GTTCCTTCGTTCTCCAGCAG–3′ |
687–785 | 99 |
| Smurf2 | NM 025481 | F: 5′–GTGAAGAGCTCGGTCCTTTG–3′ R: 5′–AGAGCCGGGGATCTGTAAAT–3′ |
1041–1154 | 114 |
| ALP | AF285233 | F: 5′–CGGGACTGGTACTCGGATAA–3′ R: 5′–ATTCCACGTCGGTTCTGTTC–3′ |
550–706 | 157 |
| OC | AH004426 | F: 5′–CTTGGTGCACACCTAGCAGA–3′ R: 5′–CTCCCTCATGTGTTGTCCCT–3′ |
638–824 | 186 |
| β-actin | NM 001101 | F: 5′–AGATGTGGATCAGCAAGCAG–3′ R: 5′–GCGCAAGTTAGGTTTTGTCA–3′ |
1134–1251 | 118 |
F, forward primer; R, reverse primer.
Western Blot Analysis
Cells were washed with cold phosphate-buffered saline (PBS), and whole cell lysates were prepared by the addition of M-PER mammalian protein extraction reagent (Pierce) containing a protease inhibitor mixture (Roche Applied Science). Twenty μg of protein was loaded per lane and separated on a 10% polyacrylamide gel, followed by transfer to a nitrocellulose membrane (Bio-Rad) by electro-blotting. Membrane was blocked for nonspecific binding in 3% nonfat dry milk and followed by incubation with an antibody at 4 °C. After membrane was washed, the blots were probed with a horseradish peroxidase-conjugated secondary antibody (Bio-Rad) and visualized by an enhanced chemiluminescence system (Amersham Biosciences) according to the manufacturer's instructions.
Ubiquitination of Runx2
2T3 cells were incubated in medium containing 7.5 ng/ml TNF for 72 h in the presence of PBS or 0.1 mm MG132 (Calbiochem) for the last 12 h of TNF treatment. For the immunoprecipitation, cell lysate was incubated with anti-Runx2 antibody overnight at 4 °C followed by the addition of protein G-agarose (Roche Applied Science) overnight at 4 °C. The immunoprecipitates were washed with 50 mm Tris-HCl buffer (pH 8.0), containing 150 mm NaCl, 1% Nonidet P-40, 0.05% deoxycholate, and 0.1% SDS, resuspended in 1 × reducing sample buffer, and subjected to Western blot analysis with an anti-ubiquitin antibody. The same membrane was stripped and reprobed for Runx2.
siRNA and Virus Infection
Platinum-E cells were transfected with a retrovirus vector (pRetro-H1G) that encodes Smurf1 or Smurf2 siRNA, or an empty vector using FuGENE 6 reagent (Roche Applied Science). The sequences of Smurf1 and Smurf2 siRNAs are shown in Table 2. After 2 days, viral supernatants were harvested and filtered using a 0.45-μm membrane filter. 2T3 cells were infected with virus supernatant in the presence of Polybrene. After 4 h, 2 ml of α-minimal essential medium was added to the cells to dilute Polybrene. The cells were cultured for an additional 48 h in α-minimal essential medium containing 10% fetal bovine serum. The cells were then used for RNA extraction or Western blot analysis.
TABLE 2.
Sequences of siRNA used in the infection
| Target genesa | Sequences of siRNAb | Target sites on mRNA |
|---|---|---|
| Smurf1 (NM 029438) | 5′–GATTCGAACCTTGCAAAGAAAGAC ttcaagagaGTCTTTCTTTGCAAGGTTCTTTTTTC–3′ |
372–390 |
| Smurf2 (NM 025481) | 5′–GATTCGACCAACAGCAACAGCAAG ttcaagagaCTTGCTGTTGCTGTTGGTCTTTTTTC–3′ |
1199–1217 |
Parentheses indicate GenBank™ accession numbers.
siRNAs are designated to encode two complementary sequences of 19 nucleotides homologous to a segment of Smurf1 or Smurf2 mRNA (underlined) separated by a nine-nucleotide space (small characters), and have a terminator signal (TTTTTTC) at a 3′ terminus.
Luciferase Assay
2T3 cells were transfected with the BMP, 12×SBE-OC-Luc (17) or the TGF-β, p3TP-Lux (18), signaling reporter construct using Lipofectamine 2000 transfection reagent. After 6 h, cells were treated with 10 ng/ml TNF for 48 h, followed by a 24-h incubation in the presence or absence of 50 ng/ml BMP-2 or 2 ng/ml TGF-β (both from R&D Systems). For determination of the effect of TNF on Runx2 expression, 2T3 cells were cotransfected with a Runx2 expression vector, F-Runx2, and the Runx2 reporter construct, 6×OSE2-OC-pGL3 (17) followed by a 48-h TNF treatment. Cell lysates were extracted, and luciferase activity was measured using a Dual Luciferase Reporter Assay System (Promega) and normalized by Renilla luciferase activity.
Caspase-3 Assay
2T3 cells were treated with TNF (2.5, 5, and 7.5 ng/ml) for 24, 48, and 72 h and then lysed in a buffer containing 1% Nonidet P-40, 200 mm NaCl, 20 mm Tris-HCl (pH 7.4), 10 μg/ml leupeptin, and aprotinin (0.27 mm trypsin inhibitor/ml). Caspase-3 activity was determined by incubation of cell lysate (containing 25 μg of total protein) with 50 μm fluorogenic substrate, N-acetyl-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin (Calbiochem) in 200 μl of 10 mm HEPES (pH 7.4), containing 220 mm mannitol, 68 mm sucrose, 2 mm NaCl, 2.5 mm KH2PO4, 0.5 mm EGTA, 2 mm MgCl2, 5 mm pyruvate, 0.1 mm phenylmethylsulfonyl fluoride, and 1 mm dithiothreitol. The release of fluorescent 7-amino-4-methylcoumarin was measured by spectrofluorometry (excitation/emission, 499/521 nm).
MTT Cell Viability Assay
Cell viability was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma) dye reduction assay according to the method of Green et al. (26). Briefly, after cells in 96-well plates were treated with various concentrations of TNF for 24, 48, or 96 h, 10 μl of MTT was added for 3 h, and the absorbance was read at 540 nm. Cell viability was calculated as the ratio of optical densities in wells with and without TNF.
Statistics Analysis
All data are represented as the mean ± S.E. Comparisons of results were performed by paired Student's t tests, accepting p < 0.05 as the criterion of significance. All experiments were repeated at least twice with similar results.
RESULTS
TNF-Tg Mice Have Reduced Osteoblast Function
The TNF transgenic mouse is a well established animal model of rheumatoid arthritis, which exhibits polyarthritis because of chronic exposure to low levels of TNF (27). At 3–4 months of age, TNF-Tg mice develop a moderate to severe form of rheumatoid arthritis-like joint inflammation and destruction. This is characterized by chronic inflammation, local bone and cartilage erosion, and increased circulating TNF levels (27, 28). Apart from these well described features, animals develop general osteoporosis as show in Fig. 1A. Trabecular bone is markedly reduced in the metaphysis of long bones of TNF-Tg mice, compared with wt littermates (Fig. 1B). To examine whether osteoblast function is altered in TNF-Tg mice, bone marrow stromal cells were isolated from 4-month-old TNF-Tg mice and wt littermates. Cells were cultured in osteoblast differentiation medium to form mineralized bone nodules. Compared with wt mice, cells from TNF-Tg mice formed significantly fewer and smaller nodules under basal conditions and in the presence of BMP-2 (Fig. 1, C and D), indicating reduced osteoblast function.
FIGURE 1. Decreased bone volume and osteoblast functions in TNF-Tg mice.
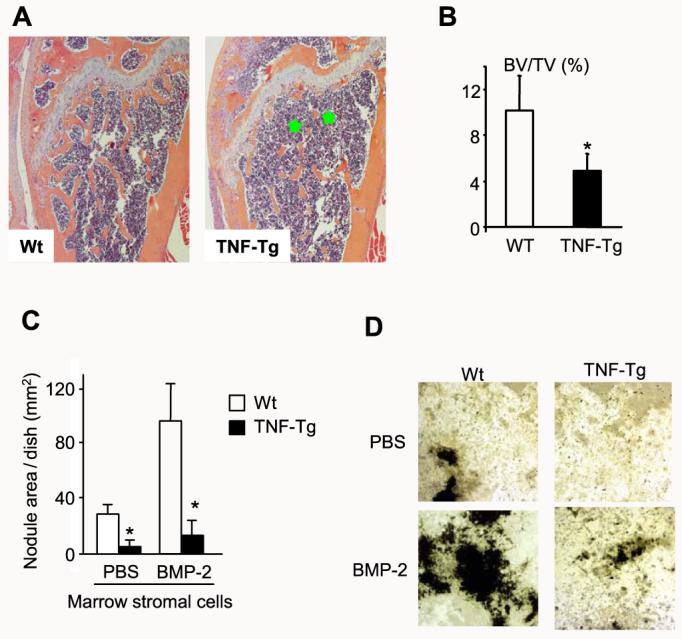
A, femur from a 4-month-old TNF-Tg mouse and a wt littermate were fixed and processed. Paraffin-embedded sections were stained with hematoxylin and eosin. Lower bone volume in the metaphysis of the femur (green stars) is present in the TNF-Tg mouse (magnification, ×4). B, the distal femoral trabecular bone volume was measured as described previously (38). The values are the mean ± S.E. of eight or nine mice. *,p < 0.05 versus wt mice. C, bone marrow stromal cells were isolated from 4-month-old TNF-Tg mice and wt littermates and cultured in osteoblast differentiation medium containing 100 μg/ml l-ascorbic acid and 5 mm β-glycerophosphate in the presence or absence of 40 ng/ml BMP-2 for 2 weeks. The cells were fixed, and mineralized bone nodules were identified by vonKossa staining. The area of bone nodules was measured under light microscopy using point counting. The values are the mean ± S.E. of three wells. *,p < 0.05 versus wt cells. The same results were obtained from three pairs of TNF-Tg mice and wt littermates. D, mineralized bone nodules from one representative pair of TNF-Tg mice and wt littermates (magnification, ×2).
TNF Increases Smurf1 Expression, Runx2 Degradation, and Ubiquitination of Runx2 Protein
Smurf1 is a negative regulator of the BMP signaling pathway and inhibits osteoblast function by promoting Runx2 degradation (17). To determine whether TNF affects Smurf1 expression, C2C12 myoblast/osteoblast precursor cells, 2T3 osteoblast precursors, and primary wt calvarial preosteoblasts were treated with PBS or 2.5–7.5 ng/ml TNF. Smurf1 expression was measured by real time RT-PCR at 24, 48, and 72 h. The highest dose of TNF (7.5 ng/ml) significantly increased Smurf1 mRNA levels at 24 h in both cell lines, with the highest indication being observed at 72 h. At this time, the lowest dose of TNF (2.5 ng/ml) also had increased Smurf1 mRNA expression (Fig. 2A). TNF-induced Smurf1 expression was similar in 2T3 and C2C12 cells. No significant increase in Smurf1 level was observed when the cells were treated with 7.5 ng/ml TNF for 2, 4, 8, and 12 h (data not shown). Consistent with these mRNA results, TNF increased Smurf1 protein expression in a dose-dependent manner after 24 h (Fig. 2B). Similarly, TNF also increased Smurf1 expression in primary preosteoblasts. TNF increased Smurf1 mRNA levels in these cells by 4–5-fold over the PBS-treated cells at 48 h (the ratio of Smurf1/β-actin in TNF-treated group versus that from PBS group: 4.7 ± 0.14, p < 0.002). To determine the specificity of TNF for Smurf1, C2C12 and 2T3 cells were treated with IL-1 or receptor activator NF-κB ligand (RANKL) that activate intracellular signaling pathways similar to those activated by TNF. They did not alter Smurf1 mRNA abundance (Fig. 2C). These doses of IL-1 and RANKL (10 ng/ml) stimulate osteoclast formation from osteoclast precursors (data not shown).
FIGURE 2. TNF increases Smurf1 expression and induces Runx2 degradation.

A, C2C12 or 2T3 cells were treated with PBS or TNF (2.5–7.5 ng/ml) for 24, 48, and 72 h. Smurf1 and β-actin mRNA levels were measured by real time RT-PCR. The relative expression level of Smurf1 was normalized to β-actin in the same sample. The -fold increase was calculated as follows: (relative Smurf1 level in TNF-treated sample)/(relative Smurf1 level in PBS-treated sample). The values are the mean ± S.E. of three dishes. *, p < 0.05 versus the PBS-treated group. B, cells were treated with 2.5–7.5 ng/ml TNF for 72 h. Smurf1 and β-actin protein levels were examined by Western blot analysis. C, cells were treated with various doses of IL-1 and RANKL and with 7.5 ng/ml TNF for 72 h. The relative expression levels of Smurf1 mRNA were determined by real time RT-PCR, as described in A. The values are the mean ± S.E. of three dishes. *, p < 0.05 versus the PBS-treated group. D, cells were treated with 2.5–7.5 ng/ml TNF for 24, 48, and 72 h. Cell lysates were used for measuring caspase-3 activity or MTT assay. The values are the mean ± S.E. of three dishes. *, p<0.05 versus the PBS-treated group. E, cells were transfected with M-Smurf1 and/or F-Runx2 expression plasmids or empty vector for 24 h and cultured for 72 h in the presence of PBS or TNF (2.5–7.5 ng/ml). F-Runx2 expression was detected by Western blot using anti-FLAG antibody.
To examine whether TNF-induced apoptosis is associated with increased Smurf1 expression, 2T3 cells were treated with various doses of TNF for 24, 48, and 72 h. Apoptosis was determined by measuring caspase-3 activity and cell viability by MTT assay. Smurf1 mRNA expression was examined by real time PCR in the same samples. At the doses (2.5–7.5 ng/ml) that TNF increased Smurf1 expression (data not shown), osteoblasts were morphological normal with normal caspase-3 activity (Fig. 2D). In contrast, cells treated with 10 and 20 ng/ml TNF induced cell apoptosis, and dead cells were detached from the culture plates (data not shown).
To determine whether TNF induces Runx2 degradation, C2C12 or 2T3 cells were cotransfected with FLAG-tagged-Runx2 (F-Runx2) and/or Myc-tagged-Smurf1 (M-Smurf1) expression vectors or an empty vector in the presence of TNF. F-Runx2 expression was detected by Western blot analysis using an anti-FLAG antibody. As a positive control, Smurf1 overexpression decreased F-Runx2 protein levels. Similar to Smurf1 overexpression, TNF significantly reduced F-Runx2 protein levels in a dose-dependent manner (Fig. 2E).
Smurf1 induces Runx2 degradation by increasing its ubiquitination (17). If TNF-induced Runx2 degradation is mediated by Smurf1, we should be able to detect increased ubiquitinated-Runx2 complexes in TNF-treated cells. To test this hypothesis, 2T3 cells were treated with TNF in the presence and absence of the proteasomal inhibitor MG132, and endogenous Runx2 protein was immunoprecipitated with an anti-Runx2 antibody and followed by Western blot analysis using an anti-ubiquitin antibody. MG132 treatment revealed small amounts of ubiquitinated Runx2 in vehicle-treated cells. These were greatly increased in the presence of TNF (Fig. 3), indicating that TNF induces ubiquitination of Runx2 protein, leading to its rapid breakdown through proteasomal degradation.
FIGURE 3. TNF induces ubiquitination of Runx2 protein.
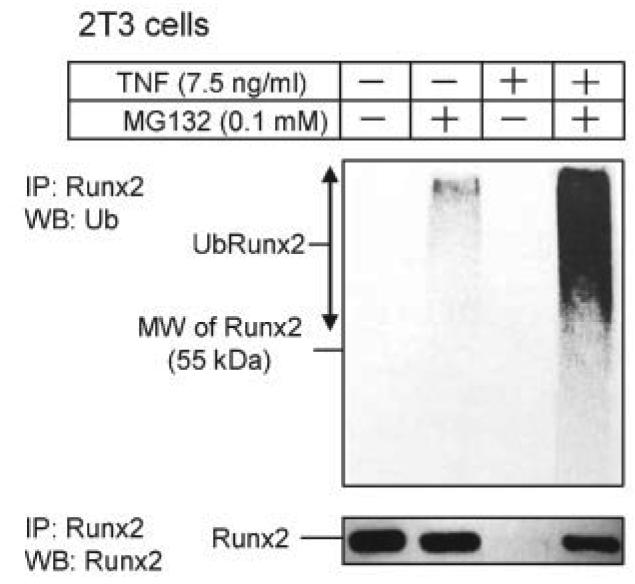
2T3 cells were treated with 7.5 ng/ml TNF for 72 h in the presence or absence of 0.1 mM MG132 for the last 12 h of TNF treatment, and endogenous Runx2 was immunoprecipitated (IP) by anti-Runx2 antibody. Ubiquitinated Runx2 (Ub-Runx2) protein ladders were detected by anti-ubiquitin antibody (upper panel). After stripping the antibody, total un-ubiquitinated Runx2 protein levels were determined by anti-Runx2 antibody (lower panel). MW, molecular mass.
TNF-induced Runx2 Degradation Is Dependent on Smurf1 and Smurf2
To determine whether TNF-induced Runx2 degradation is dependent on Smurf1, 2T3 cells were infected first with retroviral supernatant containing double-stranded siRNA specific for Smurf1 to knock down endogenous Smurf1. They were then transfected with F-Runx2 in the presence of TNF. Smurf1 siRNA decreased TNF-induced Smurf1 mRNA by 95% and reduced Smurf1 expression in PBS-treated cells to almost undetectable levels (Fig. 4A). Without changing the expression of Smurf2 mRNA (Fig. 4B), Smurf1 siRNA partially blocked TNF-mediated inhibition of ALP mRNA expression compared with the empty vector control (Fig. 4C) and reduced TNF-induced Runx2 degradation by 30% (by a densitometric analysis, Fig. 4D). Interestingly, Smurf1 siRNA alone increased ALP mRNA levels by 58%, suggesting that Smurf1 may regulate osteoblast function under basal conditions.
FIGURE 4. Smurf1 or Smurf2 siRNA blocks TNF-induced Runx2 degradation.
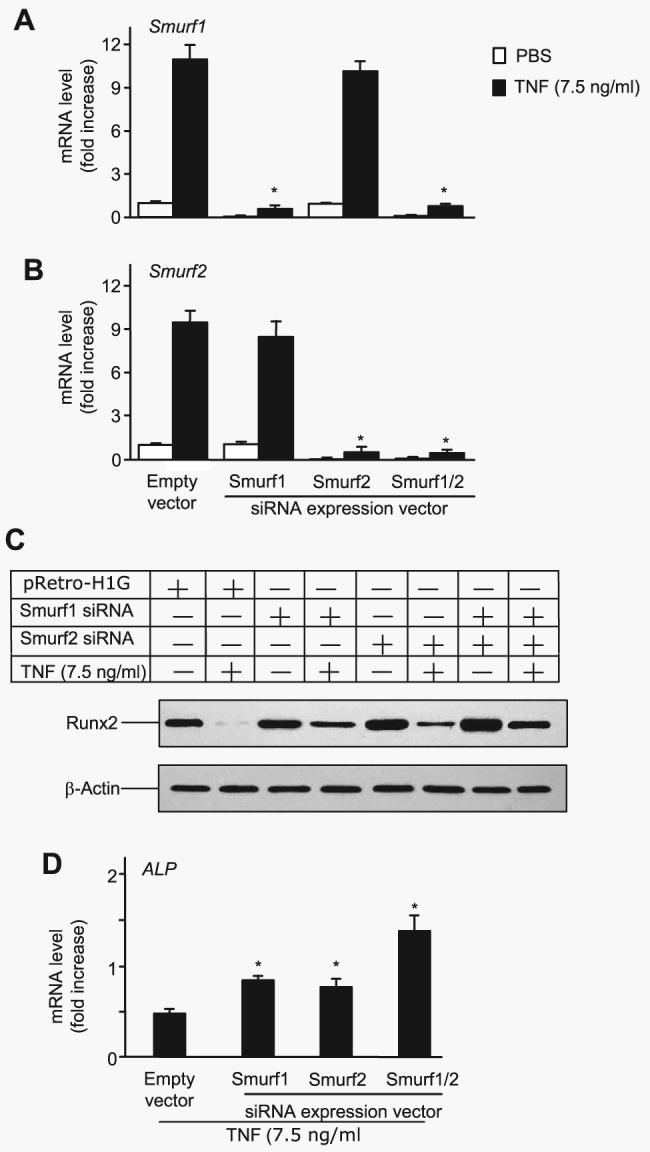
2T3 cells were infected with retroviral supernatant containing Smurf1 and/or Smurf2 siRNAs or empty vector, then cells were transfected with F-Runx2 expression plasmid (4 μg/dish) and treated with 7.5 ng/ml TNF for 72 h. Smurf1 (A) and Smurf2 (B) mRNA levels were measured by real time RT-PCR. The values are the mean ± S.E. of three dishes. The -fold increase was calculated as described in Fig. 2A. *, p<0.05 versus the empty vector-infected TNF group. C, the expression of F-Runx2 was determined by Western blot analysis using anti-FLAG antibody as described in Fig. 2E. D, ALP mRNA expression was assessed by real time RT-PCR. The values are the mean ± S.E. of three dishes. The -fold increase was calculated as described in Fig. 2A. *, p<0.05 versus empty vector-infected TNF group.
Smurf2 is another E3 ubiquitin ligase that affects osteoblast function by interfering with TGF-β signaling (20). To examine whether TNF regulates Smurf2 expression, 2T3 cells were treated with PBS or TNF (7.5 ng/ml) for 72 h, and Smurf2 mRNA levels were examined by real time RT-PCR. Similar to, but to a lesser extent than Smurf1, TNF significantly increased Smurf2 mRNA by 9.5 ± 0.88-fold (p<0.004 versus PBS-treated cells). Because there is no Smurf2 antibody available to detect mouse Smurf2, the effect of TNF on Smurf2 protein levels cannot be assessed currently. To examine whether TNF-induced Runx2 degradation is also mediated by Smurf2, we used Smurf2 siRNA. Similar to Smurf1 siRNA, blocking endogenous Smurf2 led to reduced TNF-mediated inhibition of ALP expression and TNF-induced Runx2 degradation (Fig. 4, C and D) to a similar extent. Combining Smurf1 and Smurf2 siRNAs further prevented TNF-induced Runx2 degradation by 60% (Fig. 4C).
TNF Inhibits BMP and TGF-β Signaling through Individual Smurfs, and Its Inhibition on Runx2 Is Mediated by Both Smurf1 and Smurf2
Smurf1 recognizes and directs the ubiquitination and proteosomal degradation of Smad1 and 5 (15), whereas Smurf2 acts on both Smad1 and 2, but not Smad3 (20). Therefore, Smurf1 is thought to target BMP-2 and Smurf2 to target TGF-β signaling (14). Because TNF increases Smurf1 and Smurf2, it should target the BMP-2 and/or TGF-β signaling pathways. To test this, 2T3 cells were transfected with the Runx2, BMP, or TGF-β reporter constructs and a Runx2 expression vector or treated with BMP-2 or TGF-β, in the presence or absence of TNF in a luciferase reporter assay. Smurf1 and/or Smurf2, or control siRNA was used to determine whether blockade of Smurfs affects the effect of TNF. TNF significantly inhibited a Runx2-induced increase in Runx2 reporter by 86% (Fig. 5A). Smurf1 siRNA or Smurf2 siRNA alone reduced the inhibitory effect of TNF by 30 - 40%, and a combination of Smurf1 and Smurf2 siRNA further inhibited TNF action by 60–80%, indicating an additive effect (Fig. 5A). In contrast to the partial reduction of the effect of TNF on Runx2 reporter, Smurf1 siRNA alone reduced TNF-mediated inhibition of the BMP-2 signaling reporter by 90%, whereas Smurf2 siRNA had no effect (Fig. 5B). Smurf2 siRNA blocked TNF-mediated TGF-β signaling reporter inhibition by 60%, and Smurf1 siRNA had no effect (Fig. 5C). A combination of Smurf1 and Smurf2 siRNA had no additive effect on TNF inhibition on the BMP-2 or TGF-β signaling reporter. Thus, although TNF inhibits Runx2 activation through both Smurf1 and Smurf2, its inhibition in the BMP-2 and TGF-β signaling pathways is through Smurf1 and Smurf2, respectively.
FIGURE 5. TNF targets both BMP and TGF-β signaling pathways in osteoblasts.
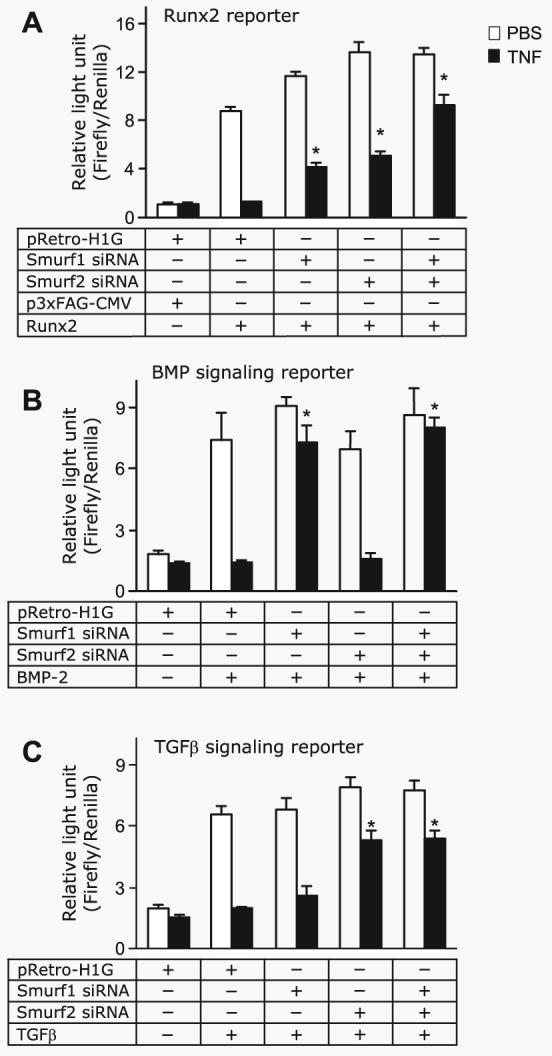
A, 2T3 cells were infected with retroviral supernatant containing Smurf1 and/or Smurf2 siRNAs or empty vector, then cells were cotransfected with the Runx2 expression vector (Runx2), the empty expression vector (p3×FAG-CMV), and the Runx2 reporter vector (6×OSE2-OC-Luc) for 8 h and then treated with 7.5 ng/ml TNF for 48 h. B and C, 2T3 cells were infected with retroviral supernatant containing Smurf1 and/or Smurf2 siRNA or empty vector, then the BMP-2 and TGF-β signaling reporter constructs, 12×SBE-OC-Luc (B) or p3TP-Lux (C) were transfected into cells for 8 h. The cells were treated with 7.5 ng/ml TNF for 48 h and then stimulated with 50 ng/ml BMP-2 (B) or 2 ng/ml TGF-β (C) for 24 h in the presence of PBS or 10 ng/ml TNF. The values are the mean × S.E. of three dishes. *, p<0.05 versus empty vector-infected TNF group.
TNF-overexpressing Mice Have Increased Smurf1 and Decreased Runx2 Expression
If Smurfs are responsible for reduced osteoblast function in conditions in which TNF is overexpressed, we should be able to detect elevated Smurf levels in TNF-Tg mice or mice receiving TNF treatment. To examine this, total RNA was extracted from the metaphysis of 4-month-old TNF-Tg mice and wt littermates, and the expression of Smurf1, Smurf2, ALP, and OC mRNA was examined by real time RT-PCR. Compared with that of wt mice, the expression of Smurf1 was significantly increased, and ALP and OC expression was decreased in TNF-Tg mice (Fig. 6A). No change of Smurf2 mRNA expression was observed (data not shown). Western blot analysis revealed that Smurf1 protein was increased, and Runx2 protein was decreased (Fig. 6A). To determine the acute effect of TNF on Smurf and osteoblast marker gene expression, wt mice were injected with TNF (0.25 μg/injection, three times/day × 3 days) or PBS over the calvarial bones. The expression of Smurf1, Smurf2, ALP, and OC mRNA was assessed. Similar to the results obtained from TNF-Tg mice, the expression of Smurf1 was increased at both mRNA and protein levels. Runx2 protein and ALP and OC mRNA levels were decreased in TNF-treated wt mice (Fig. 6B). These results suggest that TNF may inhibit osteoblast function in vivo by promoting Runx2 degradation through up-regulation of Smurf E3 ligases.
FIGURE 6. TNF increases Smurf1 expression in vivo.
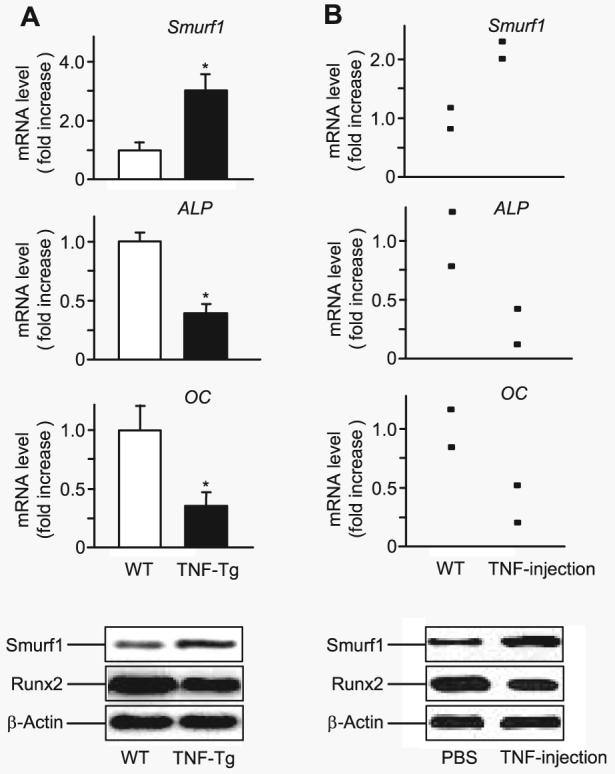
A, RNA and protein were extracted directly from the metaphyseal region of 4-month-old TNF-Tg mice and wt littermates. The relative expression levels of Smurf1, ALP, and OC were measured by real time RT-PCR (upper panels), as described in Fig. 2. The values are the mean ± S.E. of four pairs of TNF-Tg and wt mice. *, p < 0.05 versus wt mice. Smurf1 and β-actin protein levels were determined by Western blot analysis (lower panels). The data are representative of three pairs of TNF-Tg or wt mice. B, TNF (0.25 μg/injection, three times/day × 3 days, n = 2) or PBS was injected over the calvarial bones of wt mice. RNA and protein were extracted directly from calvarial bones. Expression levels of Smurf1, ALP, and OC mRNA and Smurf1 and β-actin protein were examined as described in A. The data show the values of individual mice.
DISCUSSION
TNF is a central proinflammatory cytokine that contributes to local and systemic bone loss in inflammatory bone diseases, such as rheumatoid arthritis. Under these conditions, osteoblast-mediated bone formation cannot compensate for accelerated osteoclastic bone resorption, suggesting a direct inhibitory effect of TNF on osteoblasts. Here we provide evidence for the regulation of osteoblast-specific transcription factor Runx2 stability by TNF through the E3 ligases, Smurf1 and Smurf2. Although overexpression of Smurfs has been known to promote Runx2 degradation in vitro, our findings provide the first report that TNF inhibits osteoblast function by controlling the ubiquitination status of Runx2 protein through Smurfs. We have several lines of evidence implicating Smurfs in the regulation of TNF-induced Runx2 ubiquitination and degradation: (i) TNF promoted ubiquitination and degradation of transfected and endogenous Runx2; (ii) it increased the expression levels of Smurf1 and Smurf2; (iii) TNF-induced ubiquitination and degradation of Runx2 were attenuated in cells overexpressing Smurf1 and Smurf2 siRNA; (iv) expression of Smurf1 and Smurf2 siRNA rescued the inhibitory effect of TNF on Runx2 reporter; (v) bones of mice that have elevated TNF levels have increased Smurf1 and decreased Runx2 protein expression. In 2002, Gilbert et al. (10) reported that TNF reduced nuclear Runx2 protein in osteoblasts by 90%, but it inhibited Runx2 transcription only by 40–50%. They proposed that there may be other mechanism(s) accounting for the 90% reduction in Runx2 protein. Here, we found that overexpression of Smurf1 and Smurf2 siRNA can reverse TNF-induced down-regulation of Runx2 protein by 50–60% (Fig. 4D). We suspect that there may also be transcriptional inhibition of Runx2 to complement this TNF-induced reduction in protein expression. Thus, our data complement Gilbert's results and demonstrate that TNF regulates Runx2 by two distinct mechanisms: it inhibits Runx2 gene transcription and promotes Runx2 protein degradation through Smurf E3 ligases.
TNF has been implicated in mediating protein degradation through several E3 ligases in other cell types. For example, TNF induces TNF receptor-associated factor (TRAF)2 degradation in HeLa cells and mouse embryonic fibroblasts through a RING-type ubiquitin ligase, Siah2 (29). It stimulates expression of the gene for a F-box E3 ligase, atrogin1/MAFbx (30), which is involved in muscle atrophy (31) and sepsis (32). Interestingly, TNF-induced TRAF2 degradation or up-regulation of atrogin1/MAFbx occurs within 4 h, and this is within the same time frame that treatment of bone marrow cells with RANKL and interferon-γ resulted in degradation of TRAF6 (33). In contrast, TNF-induced Smurf up-regulation and Runx2 degradation take 24–72 h (Fig. 2), suggesting an indirect effect. We have attempted to dissect the possible requirements of transcription or translation for TNF-increased Smurf expression using pharmacological blockers. Unfortunately, blocking of transcription or translation by cycloheximide or actinomycin D in the presence of TNF causes massive apoptosis of cells, and thus we could not determine whether newly synthesized proteins are required for TNF-induced Smurf expression. Nevertheless, given that TNF induces various E3 ligases at different times, it is possible that TNF uses different types of E3 ligases to regulate the stability of specific targeting proteins in response to various stimuli. For example, TNF stimulates Siah2 and atrogin1/MAFbx E3 ligases in response to early signaling events during acute stress conditions and increases Smurf expression to regulate protein degradation in chronic conditions, such as rheumatoid arthritis and cancer (34).
TNF binds to TNF receptors and transduces the signals through TRAFs and various kinases to activate transcription factors, such as NF-κB and AP-1 family members. Activation of the IL-1 receptor via IL-1 or RANK through RANKL results in the activation of similar signaling pathways. Our finding that IL-1 and RANKL do not affect Smurf1 expression is interesting. There are several possible explanations: (i) osteoblasts do not express RANK, the receptor for RANKL, and thus they cannot respond to RANKL treatment. Whether osteoblasts express functional RANK is an issue of debate. It has been reported that glutathione S-transferase-RANKL stimulates osteoblast bone forming activity in vitro and in vivo (35). However, others argued that RANKL has no effect on osteoblasts because they do not express RANK. We have been unable to detect any effect of RANKL on osteoblast function in various assays,3 suggesting that RANKL may not play an important role in this cell type. (ii) IL-1 may affect Smurfs in experimental conditions that differ from ours, given the fact that in some reports IL-1 has an inhibitory effect on osteoblast function. (iii) TNF may use a unique mechanism to regulate Smurf expression. Thus, characterization of the Smurf1 and Smurf2 promoters should have a great impact on our understanding of how Smurfs are regulated by TNF.
Unlike other cytokines, TNF is a strong apoptosis inducer (36, 37), and thus increased Smurf expression could be associated with TNF-induced cell death. Because Smurf1-overexpressing 2T3 cells and osteoblasts in Col1a1-Smurf1 transgenic mice have rates of cell survival comparable with those of empty vector-overexpressing or wt cells (17, 18), elevated Smurf1 appears not to result in osteoblast apoptosis. Our finding that at the doses that TNF induced Smurf 1 expression, osteoblasts are normal suggests that increased Smurf1 is not associated with cell apoptosis (Fig. 2).
It is of interest to note that even though only increased Smurf1 expression was detected in bones of TNF transgenic mice or TNF-injected wt mice (Fig. 6), TNF increased both Smurf1 and Smurf2 expression in vitro (Figs. 2 and 4). One explanation is that Smurf1 and Smurf2 may have different response thresholds to TNF in vivo. Supporting this, our preliminary results showed that in joints of TNF-Tg mice where TNF levels were extremely high (40-fold over wt mice), both Smurf1 and Smurf2 levels were increased although the increase in Smurf2 was half of that of Smurf1. This is important because cells from Smurf1-null mice exhibit normal levels of Runx2 and BMP-Smads, which is explained by elevated Smurf2 expression in the absence of Smurf1 (23). By increasing both Smurf1 and Smurf2 in osteoblasts, TNF can overcome this compensatory effect between Smurf1 and Smurf2 to induce Runx2 degradation in vivo (Fig. 6). However, the real significance of Smurf1-mediated protein degradation in TNF-induced osteoblast inhibition needs to wait for in vivo data from TNF-Tg/Smurf1 knockout hybrid mice.
It is unlikely that increasing protein breakdown is the only mechanism by which TNF executes its effect in bone cells. We have found that TNF increased Smurf1 expression in several types of osteoblast precursors, including MC3T3E1 (data not shown), 2T3, and primary calvarial cells. Interestingly, TNF also increases Smurf1 expression in C2C12 cells (Fig. 2). Because untreated C2C12 cells have multiple potency to differentiate to other cell types, we do not know whether TNF-induced Smurf1/2 expression represents a generalized feature of connective tissue cell signaling response to TNF. However, TNF had no effect on Smurf1 expression in osteoclast precursors derived from bone marrow, spleen, and peripheral blood, or in ST2 murine stromal cells under the same experimental conditions (data not shown). Thus, TNF-induced Smurf expression does not appear as a common phenomenon for all cell types. Whether Smurfs have different target proteins in different cells needs to be investigated further.
In summary, our findings reveal a novel mechanism for TNF-induced osteoblast inhibition: through unregulation of Smurf1 and Smurf2 E3 ligases to promote the degradation of Runx2 protein. Better delineation of the role of TNF and perhaps other inflammatory cytokines in proteasomal regulation of protein function by Smurf E3 ligase in osteoblasts will enhance our understanding of the molecular mechanisms responsible for local or general bone loss in inflammatory bone diseases.
Acknowledgments
We thank Bianai Fan for technical assistance with the histological analysis.
Footnotes
This work was supported by National Institutes of Health Grants AR48697 (to L. X.), AR43510 (to B. F. B), and AR051189 (to D. C.).
The abbreviations used are: TNF, tumor necrosis factor; ALP, alkaline phosphatase; BMP, bone morphogenetic protein; CMV, cytomegalovirus; E3, ubiquitin-protein isopeptide ligase; IL-1, interleukin-1; Luc, luciferase; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; OC, osteocalcin; PBS, phosphate-buffered saline; RANKL, receptor activator NF-κB ligand; RT, reverse transcription; siRNA, small interfering RNA; Smurf, Smad ubiquitin regulatory factor; Tg, transgenic; TGF-β, transforming growth factor-β; TRAF, TNF receptor-associated factor; wt, wild type.
H. Kaneki, R. Guo, D. Chen, Z. Yao, E. M. Schwarz, Y. E. Zhang, B. F. Boyce, and L. Xing, unpublished findings.
REFERENCES
- 1.Goldring SR, Gravallese EM. Curr. Opin. Rheumatol. 2000;12:195–199. doi: 10.1097/00002281-200005000-00006. [DOI] [PubMed] [Google Scholar]
- 2.Srivastava S, Weitzmann MN, Cenci S, Ross FP, Adler S, Pacifici R. J. Clin. Invest. 1999;104:503–513. doi: 10.1172/JCI7094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nair SP, Williams RJ, Henderson B. Rheumatology. 2000;39:821–834. doi: 10.1093/rheumatology/39.8.821. [DOI] [PubMed] [Google Scholar]
- 4.Canalis E. Endocrinology. 1987;121:1596–1604. doi: 10.1210/endo-121-5-1596. [DOI] [PubMed] [Google Scholar]
- 5.Li YP, Stashenko P. J. Immunol. 1992;148:788–794. [PubMed] [Google Scholar]
- 6.Taichman RS, Hauschka PV. Inflammation. 1992;16:587–601. doi: 10.1007/BF00919342. [DOI] [PubMed] [Google Scholar]
- 7.Kitajima I, Soejima Y, Takasaki I, Beppu H, Tokioka T, Maruyama I. Bone. 1996;19:263–270. doi: 10.1016/8756-3282(96)00181-0. [DOI] [PubMed] [Google Scholar]
- 8.Jilka RL, Weinstein RS, Bellido T, Parfitt AM, Manolagas SC. J. Bone Miner. Res. 1998;13:793–802. doi: 10.1359/jbmr.1998.13.5.793. [DOI] [PubMed] [Google Scholar]
- 9.Gilbert L, He X, Farmer P, Boden S, Kozlowski M, Rubin J, Nanes MS. Endocrinology. 2000;141:3956–3964. doi: 10.1210/endo.141.11.7739. [DOI] [PubMed] [Google Scholar]
- 10.Gilbert L, He X, Farmer P, Rubin J, Drissi H, van Wijnen AJ, Lian JB, Stein GS, Nanes MS. J. Biol. Chem. 2002;277:2695–2701. doi: 10.1074/jbc.M106339200. [DOI] [PubMed] [Google Scholar]
- 11.Abbas S, Zhang YH, Clohisy JC, Abu-Amer Y. Cytokine. 2003;22:33–41. doi: 10.1016/s1043-4666(03)00106-6. [DOI] [PubMed] [Google Scholar]
- 12.Gilbert LC, Rubin J, Nanes MS. Am. J. Physiol. 2005;288:E1011–E1018. doi: 10.1152/ajpendo.00534.2004. [DOI] [PubMed] [Google Scholar]
- 13.Izzi L, Attisano L. Oncogene. 2004;23:2071–2078. doi: 10.1038/sj.onc.1207412. [DOI] [PubMed] [Google Scholar]
- 14.Datto M, Wang XF. Cell. 2005;121:2–4. doi: 10.1016/j.cell.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 15.Zhu H, Kavsak P, Abdollah S, Wrana JL, Thomsen GH. Nature. 1999;400:687–693. doi: 10.1038/23293. [DOI] [PubMed] [Google Scholar]
- 16.Ying SX, Hussain ZJ, Zhang YE. J. Biol. Chem. 2003;278:39029–39036. doi: 10.1074/jbc.M301193200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao M, Qiao M, Oyajobi BO, Mundy GR, Chen D. J. Biol. Chem. 2003;278:27939–27944. doi: 10.1074/jbc.M304132200. [DOI] [PubMed] [Google Scholar]
- 18.Zhao M, Qiao M, Harris SE, Oyajobi BO, Mundy GR, Chen D. J. Biol. Chem. 2004;279:12854–12859. doi: 10.1074/jbc.M313294200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin X, Liang M, Feng XH. J. Biol. Chem. 2000;275:36818–36822. doi: 10.1074/jbc.C000580200. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Chang C, Gehling DJ, Hemmati-Brivanlou A, Derynck R. Proc. Natl. Acad. Sci. U. S. A. 2001;98:974–979. doi: 10.1073/pnas.98.3.974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang HR, Zhang Y, Ozdamar B, Ogunjimi AA, Alexandrova E, Thomsen GH, Wrana JL. Science. 2003;302:1775–1779. doi: 10.1126/science.1090772. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Wang HR, Wrana JL. Cell Cycle. 2004;3:391–392. doi: 10.4161/cc.3.4.772. [DOI] [PubMed] [Google Scholar]
- 23.Yamashita M, Ying SX, Zhang GM, Li C, Cheng SY, Deng CX, Zhang YE. Cell. 2005;121:101–113. doi: 10.1016/j.cell.2005.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, Wrana JL. Mol. Cell. 2000;6:1365–1375. doi: 10.1016/s1097-2765(00)00134-9. [DOI] [PubMed] [Google Scholar]
- 25.Boyce BF, Aufdemorte TB, Garrett IR, Yates AJ, Mundy GR. Endocrinology. 1989;125:1142–1150. doi: 10.1210/endo-125-3-1142. [DOI] [PubMed] [Google Scholar]
- 26.Green LM, Reade JL, Ware CF. J. Immunol. Methods. 1984;70:257–268. doi: 10.1016/0022-1759(84)90190-x. [DOI] [PubMed] [Google Scholar]
- 27.Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G. EMBO J. 1991;10:4025–4031. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li P, Schwarz EM, O'Keefe RJ, Ma L, Looney RJ, Ritchlin CT, Boyce BF, Xing L. Arthritis Rheum. 2004;50:265–276. doi: 10.1002/art.11419. [DOI] [PubMed] [Google Scholar]
- 29.Habelhah H, Frew IJ, Laine A, Janes PW, Relaix F, Sassoon D, Bowtell DD, Ronai Z. EMBO J. 2002;21:5756–5765. doi: 10.1093/emboj/cdf576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li YP, Chen Y, John J, Moylan J, Jin B, Mann DL, Reid MB. FASEB J. 2005;19:362–370. doi: 10.1096/fj.04-2364com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 32.Dehoux MJM, van Beneden RP, Fernandez-Celemin L, Lause PL, This-sen JM. FEBS Lett. 2003;544:214–217. doi: 10.1016/s0014-5793(03)00505-2. [DOI] [PubMed] [Google Scholar]
- 33.Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, Takaoka A, Yokochi T, Oda H, Tanaka K, Nakamura K, Taniguchi T. Nature. 2000;408:600–605. doi: 10.1038/35046102. [DOI] [PubMed] [Google Scholar]
- 34.Pray TR, Parlati F, Huang J, Wong BR, Payan DG, Bennett MK, Issakani SD, Molineaux S, Demo SD. Drug Resist. Update. 2002;5:249–258. doi: 10.1016/s1368-7646(02)00121-8. [DOI] [PubMed] [Google Scholar]
- 35.Lam J, Ross FP, Teitelbaum SL. J. Bone Miner. Res. 2001;16(Suppl 1):S150. [Google Scholar]
- 36.Tsuboi M, Kawakami A, Nakashima T, Matsuoka N, Urayama S, Kawabe Y, Fujiyama K, Kiriyama T, Aoyagi T, Maeda K, Eguchi K. J. Lab. Clin. Med. 1999;134:222–231. doi: 10.1016/s0022-2143(99)90201-9. [DOI] [PubMed] [Google Scholar]
- 37.Kitajima I, Nakajima T, Imamura T, Takasaki I, Kawahara K, Okano T, Tokioka T, Soejima Y, Abeyama K, Maruyama I. J. Bone Miner. Res. 1996;11:200–210. doi: 10.1002/jbmr.5650110209. [DOI] [PubMed] [Google Scholar]
- 38.Xing L, Venegas AM, Chen A, Garrett-Beal L, Boyce BF, Varmus HE, Schwartzberg PL. Genes Dev. 2001;15:241–253. doi: 10.1101/gad.840301. [DOI] [PMC free article] [PubMed] [Google Scholar]