

Abstract
Sarcophine-diol (SD), a structural modifications of sarcophine, has shown chemopreventive effects on 7,12-dimethylbenz(a)anthracene-initiated and 12-O-tetradecanoylphorbol-13-acetate-promoted skin tumor developments in mice. Tumorigenesis is associated with uncontrolled cell growth and loss of apoptosis. In the present study, the effects of SD on cell growth and apoptosis in human epidermoid carcinoma A431 cells were determined to assess whether SD could inhibit cell growth and/or induce apoptosis, thus elucidating possible mechanism of action. MTT assay was used for cell viability; bromodeoxyuridine incorporation assay was used for cell proliferation; fluorescence-activated cell sorting analysis of annexin V/propidium iodide staining and TUNEL assay were used for determining apoptotic cells; Western blot analysis was used for determining the expression of caspase-3 and colorimetric caspase activity assays were used for determination of caspase-3, -8, and -9 activity. The results showed that SD treatment at concentration of 200 to 600 µM resulted in a concentration-dependent decrease in cell viability and cell proliferation in A431 cells, which largely inhibited cell growth. Sarcophine-diol treatment induced a strong apoptosis and significantly (P < .05) increased DNA fragmentation in A431 cells. Furthermore, SD treatment significantly (P < .05) increased the activity and expression of caspase-3 through activation of upstream caspase-8 in A431 cells rather than the activation of caspase 9. Sarcophine-diol treatment is relatively much less cytotoxic in monkey kidney normal CV-1 cells. These results suggest that SD decreases cell growth and induces apoptosis through caspase-dependent extrinsic pathway in A431 cells, and this may contribute to its overall chemopreventive effects in mouse skin cancer models.
Introduction
Cancer is the second most common cause of death in the US accounting for one of every four deaths. Among all the cancers, the incidence of nonmelanoma skin cancer, including basal cell carcinoma and squamous cell carcinoma are the most common malignant neoplasms in humans [1]. It has been estimated that more than 1 million cases of BCC and squamous cell carcinoma are diagnosed each year in the US alone [2], which is equivalent to the incidence of malignancies in all other organs combined [3]. Therefore, the development of effective chemopreventive or chemotherapeutic agents is useful to address the risk of cutaneous malignancies.
Chemoprevention refers to the administration of naturally occurring and/or synthetic compounds to prevent the initiation and/or promotion and/or progression associated with carcinogenesis. This approach is promising because chemotherapy and surgery have not been fully effective against the high incidence of most of the cancers [4].
Recently, there has been a considerable interest in the use of marine natural products for the chemopreventive activity against skin tumor development [5–10]. Sarcophytol A, which is a cembranoid isolated from the Okinawan soft coral Sarcophyton glaucum, was reported for anticancer activity, particularly for skin cancer [11,12] and was studied by the National Cancer Institute at a preclinical trial level [6]. However, the major limitation with sarcophytol A is its supply because it is available only in minute quantities in the soft coral [13].
Sarcophine is one of the most abundant cembranolide also isolated from the Red Sea S. glaucum with yields up to 3% of animal dry weight [14]. It has been reported that semisynthesis of sarcophine derivatives such as sarcophine-diol (SD) and sarcophine-triol showed high chemopreventive effects against skin carcinogenesis [7–10]. In our previous work, topical application of SD (30 µg/100 µl in acetone per application), one of the structural modifications of sarcophine, has showed chemopreventive effects on 7,12-dimethylbenz(a)anthracene (DMBA)-initiated and 12-O-tetradecanoylphorbol-13-acetate (TPA)-promoted skin tumor development in female CD-1 mice [9]. However, the mechanism(s) of its efficacy is/are not fully investigated. In the mechanism(s) of chemopreventive agents, studies focused on blocking the activation of carcinogens and induction of detoxification pathway in the 1980s [4,15]. However, studies in recent years are more focused on the modulation of cell survival pathways such as cell cycle arrest and induction of apoptosis [16]. Tumorigenesis is associated with uncontrolled cell replication and loss of apoptotic death of cells [17]. Accordingly, in the present study, the effects of SD on cell proliferation and apoptosis in human epidermoid carcinoma A431 cells in in vitro system were determined to assess whether SD could inhibit cell growth and/or induce cell apoptosis.
Materials and Methods
Materials and Reagents
The soft coral S. glaucum was collected from several locations of the Red Sea in Egypt. Thiazolyl blue tetrazolium bromide (MTT), N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), EDTA disodium salt, sodium chloride, sodium dodecyl sulfate (SDS), Triton, sucrose, and phenylmethanesulphonylfluoride (PMSF) were purchased from Sigma Chemical Co. (St. Louis, MO). Dulbecco's modified Eagle's medium (DMEM), Roswell Park Memorial Institute medium (RPMI), fetal bovine serum (FBS), trypsin EDTA and phosphate buffered saline (PBS) were from Mediatech, Inc. (Herndon, VA). Dimethyl sulfoxide (DMSO) was obtained from Fisher Scientific (Fair Lawn, NJ). Cell proliferation ELISA kit, leupeptin, and pepstatin were from Roche Diagnostics GmbH (Mannheim, Germany). Vybrant Apoptosis Kit 2 and APO-BrdU TUNEL assay kit were purchased from Molecular Probes (Eugene, OR). Primary antibody against caspase-3 was from Cell Signaling Technology (Beverly, MA). Horseradish peroxidase-conjugated goat antirabbit and antimouse secondary antibodies were purchased from BD Biosciences (Rockville, MD). ECL Kit was bought from Amersham Biosciences (Piscataway, NJ). Caspase-3, -8, and -9 colorimetric protease assay kits were from BioSource (Camarillo, CA). Other reagents were obtained in their highest purity grade available commercially.
Synthesis of SD
Sarcophine was isolated from the soft coral S. glaucum by multiple extractions with petroleum ether at room temperature following the reported procedure [6,12] at the laboratories of Faculty of Pharmacy, Misr International University, Cairo, Egypt. The dried extract was evaporated under reduced pressure and chromatographed on silica gel column using hexane-ethyl acetate (1:2) as eluent. Pure sarcophine was obtained by crystallization from ethanol. Sarcophine-diol was synthesized according to the following procedure: sarcophine was reduced to its lactone opened ring analog (50 mg, 0.16 mmol) to which selenium dioxide (35.5 mg, 0.32 mmol) in dry 1,4-dioxane (15 ml) was added, and the reaction mixture was stirred at room temperature for 4 hours.Water was then added to the reaction mixture, and the product was extracted with CH2Cl2. Saturated NaHCO3 solution was used to wash the CH2Cl2 layer, which was dried over anhydrous Na2SO4. The solvent was evaporated and the residue was chromatographed on silica gel using hexane-acetone (1:1) as an eluent to obtain SD (7 mg, 13%) [6].
The structure of SD was fully characterized by spectroscopic methods as shown in Figure 1 and was identical to the analytical sample prepared according to previously reported method of synthesis [6,12]. Purity is confirmed by HPLC.
Figure 1.

The structure of SD.
Cell Culture
Human epidermoid carcinoma A431 cell line and monkey kidney CV-1 cell line were purchased from American Type Culture Collection (Manassas, VA). A431 cells were grown in DMEM, and CV-1 cells were grown in RPMI, both of which were supplemented with 10% heat-inactivated FBS, 100 U/ml penicillin and 100 µg/ml streptomycin in a humidified atmosphere containing 5% CO2 and 95% air at 37°C.
SD Solution
Sarcophine-diol was dissolved in DMSO to make 0.3-M stock solution, and the stock solution of SD was diluted in DMEM at different concentrations and was immediately used. In all the assays, the final concentrations of DMSO in DMEM were 0.333%.
Analysis of Cell Viability
Cell viability was determined by MTT assay as described by Dariusz et al. [18]. A431 cells were plated at a density of 7500 cells per well, and CV-1 cells were plated at a density of 5000 cells per well in a 96-well plate. After 24 hours, cells were treated as either DMEM with 0.333% DMSO alone as control or various concentrations of SD (50–600 µM) for 24, 48, and 72 hours. At the end of each treatment, MTT stock solution (5 mg/ml) was diluted to 0.5 mg/ml by DMEM and immediately used. The medium covering the cells was aspirated off, and then cells were incubated with 50 µl of 0.5 mg/ml MTT solution for 4 hours at 37°C. Thereafter, 150 µl of DMSO was added to each well to dissolve the dye crystal formazan, and the plate was allowed to stand for 1 hour at 37°C and then mixed with the microplate shaker for 5 minutes to make sure that all purple crystals were dissolved. Absorbance was measured by SpectraMax M2 microplate reader (Molecular Devices, Sunnyvale, CA) at 570 nm, with the absorbance at 650 nm to correct for background in the presence of an appropriate blank (without cells). The blank reading was subtracted from experimental readings, and cell viability was expressed as the percentage of the absorbance values of SD-treated groups to untreated controls.
Analysis of Cell Proliferation
Cell proliferation was assessed by bromodeoxyuridine (BrdU) incorporation using colorimetric ELISA Kit (Roche Diagnostics, GmbH, Mannheim, Germany). Cells were plated and treated as described for the MTTassay. At the end of each treatment period, cells were labeled with BrdU by incubating for 3 hours at 37°C under 5% CO2 to allow BrdU to incorporate in place of thymidine into the DNA of proliferating cells. Then, the labeling medium was removed by tapping off the plate. This was followed by adding FixDenat solution (provided in the kit) and incubating the plate for 30 minutes at room temperature according to the protocol provided by manufacturer. Subsequently, the FixDenat solution was removed by tapping off, and cells were incubated with anti-BrdU-POD solution for approximately 90 minutes at room temperature. Then, the antibody solution was removed and wells were rinsed with a washing solution. The substrate solution was added to each well, and the plate was incubated for 20 minutes at room temperature. Thereafter, 1 M H2SO4 was added to each well to stop the reaction and mixed thoroughly with the shaker for 1 minute. The absorbance of the samples was measured using a microplate reader (Molecular Devices) at 450 nm with the absorbance at 690 nm as reference. The blank (without cells) was performed in each experimental setup. The absorbance value of blank was subtracted from other experimental values, and cell proliferation was expressed as the percentage of the absorbance values of SD-treated groups to untreated controls.
Quantitation of Apoptosis by Annexin V/PI Staining
Vybrant Apoptosis Kit 2 (Molecular Probes) was used to quantitate apoptosis. The kit contains annexin V labeled with a fluorophore that can identify apoptotic cells by binding phosphatidylserine (PS) exposed on the cytoplasmic surface of the cell membrane of apoptotic cells. In addition, the kit includes a red fluorescent propidium iodide (PI) nucleic acid binding dye that stains dead cells. In brief, 2 x 105 cells in 2-ml medium were plated to each well of the six-well plate. After 24 hours, cells were treated with either DMEM with 0.333% DMSO alone as control or with various concentrations of SD (50, 100, and 400 µM) for 48 hours. At the end of the treatment, adherent and nonadherent cells were harvested and washed twice with ice-cold PBS and then resuspended cells with annexin-binding buffer to get a final concentration of 1 x 106 cells/ml. Approximately 1 x 105 cells in 100-µl buffer were incubated in the dark with 5 µl of annexin V and 1 µl of the 100 µg/ml PI solution for 15 minutes at room temperature. After incubation, 400 µl of annexin-binding buffer was added to each sample, and the sample was analyzed with BD FACScan flow cytometry (BD Biosciences, San Jose, CA) using CellQuest Software (BD Biosciences), which quantitated the percentage of apoptotic cells in the cell samples.
Quantitation of DNA Fragmentation by TUNEL Assay
The amount of DNA fragmentation in A431 cells induced by SD treatment was detected by APO-BrdU TUNEL assay kit (Molecular Probes) as described by the manufacturer. Briefly, cells were plated and treated as described for annexin V/PI staining. After collecting cells by trypsinization and washing cells by ice-cold PBS two times, cells were fixed by 1% (w/v) paraformaldehyde in PBS followed by 70% (v/v) ethanol. The fixed cells were incubated with BrdU-labeled nucleotide solution followed by incubation with Alexa Fluor 488 dye-labeled anti-BrdU antibody for staining fragmented DNA in apoptotic cells. The cells were then analyzed by flow cytometry. Positive and negative control cells provided in the kit were run with each assay to ensure the proper performance of the assay.
Determination of Cleaved Caspase-3 Expression by Western Blot Analysis
Cells (2 x 106) in 20-ml medium were plated to each 75-cm2 flask before drug treatment. After 24 hours of cell attachment, cells were treated with either DMEM with 0.333% DMSO alone as control or with SD (100 and 400 µM) for 48 hours. At the end of the treatment, adherent and nonadherent cells were harvested and washed twice with ice-cold PBS and lysed with 100 µl of lysis buffer containing 10 mM HEPES, 1% Triton X, 5 mM EDTA, 350 mM sucrose, 1 µg/ml leupeptin, 1 µg/ml pepstatin, and 1 mM PMSF. Samples then were then passed through the 25 G needle 30 times and were kept in -20°C freezer for 30 minutes. Samples were processed again with 25 G needle 30 times and centrifuged at 10,000g for 5 minutes to remove nucleus and nonprotein material. Finally, proteins for the assay were obtained by collecting the supernatant and were used for the determination of cleaved caspase-3 by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot analysis as described by Zhang et al. [9].
Protein concentration in the supernatant was determined by BCA protein assay kit (Pierce, Rockford, IL) with albumin as a standard.
Equal amounts of proteins (60 µg) from control, 100-µM SD, and 400-µM SD were loaded to 12.5% SDS-PAGE, and electrophoresis was run to separate proteins. The proteins in gels were transferred to nitrocellulose membranes. Membranes were blocked with 5% nonfat milk in TBS (10 mM Tris and 100 mM NaCl) for 1 hour and probed with primary antibodies against cleaved caspase-3 and β-actin followed by appropriate horseradish peroxidase-conjugated secondary antibody and ECL detection. Western blots were quantitated by using a UVP Biochem Gel Documentation system (UVP, Inc., Upland, CA).
Colorimetric Caspase-3, -8, and -9 Activity Assays
Activities of caspase-3, -8, and -9 were measured by using ApoTarget Kits (BioSource International, Camarillo, CA), which were used to determine the caspase proteolytic activities in lysates of mammalian cells. The protocol provided by the manufacturer was used. In brief, cells were plated and treated as described for the determination of cleaved caspase-3 expression. At the end of the treatment, cells were harvested, washed twice with ice-cold, PBS and lysed for 30 minutes on ice in the lysis buffer provided by the kit. Proteins were collected by centrifuging at 10,000g for 1 minute. Protein concentration in the supernatant was determined by BCA protein assay kit, and samples were diluted to a concentration of 1 mg/ml using lysis buffer. Samples containing 50 µg of proteins in 50-µl lysis buffer were added to the reaction buffer and different substrates to test the different caspase activities [DEVD (Asp-Glu-Val-Asp)-pNA substrate for caspase-3, IETD (Ile-Glu-Thr-Asp)-pNA substrate for caspase-8 and DEVD (Asp-Glue-Val-Asp)-pNA substrate for caspase-9]. The samples were incubated overnight at 37°C. On cleavage of respective colorimetric substrate peptides conjugated to p-nitroanilide (pNA) to free pNA by various caspases, absorption of light by free pNA can be quantitated using a spectrophometer at 400 nm. Comparison of the absorbance of free pNA from SD-treated samples with controls allows determination of the fold increase in caspase-3, -8, and -9 activities, respectively.
Statistical Analysis
Data were analyzed with INSTAT software (Graph Pad, San Diego, CA). Analysis of variance followed by the Tukey test was applied to compare the statistical difference of different SD treatment groups with untreated controls. Significance in all the experiment was considered at P < .05. Values were expressed as mean ± SEM.
Results
SD Reduced Cell Viability of A431 Cells
Our aim was to investigate whether SD treatment could inhibit the growth of skin cancer cells. Therefore, using human epidermoid carcinoma A431 cells, the effects of SD on cell viability were assessed by using MTT assay. Cells were treated with various concentrations of SD (50–600 µM) for 24, 48, and 72 hours. As shown in Figure 2A, SD (200–600 µM) treatment for 24, 48, and 72 hours resulted in 10% to 55%, 16% to 76%, and 7% to 90% (P < .05 to .0001), respectively, decreases in cell viability. Interestingly, SD treatment for 48 hours at low concentrations (100–200 µM) provided a greater degree of inhibition of cell viability compared with 72 hours treatment. However, SD treatment at high concentration (300–600 µM) for 72 hours provided a greater decrease in cell viability of A431 cells than that of 48 hours treatment.
Figure 2.
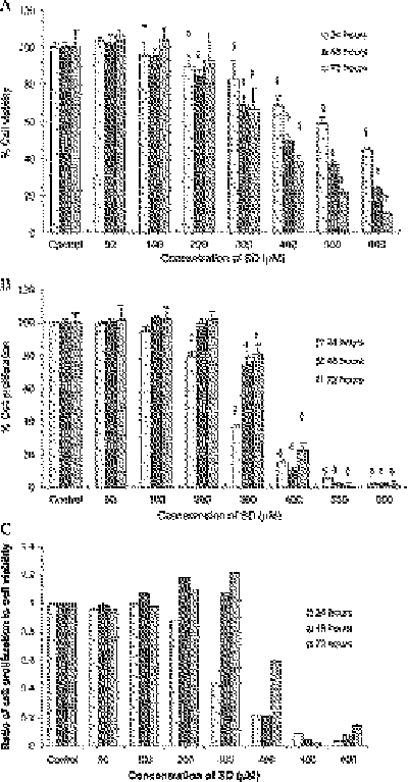
Effects of SD on cell viability and cell proliferation in A431 cells. Cells were treated with SD (0–600 µM) for 24, 48, and 72 hours, respectively. At the end of respective treatment, MTT assay (A) and BrdU incorporation assays (B) were performed as detailed in the Materials and Methods section. Values of MTT assay (A) are mean ± SE of eight replicates in each treatment, whereas values of BrdU incorporation assay (B) are mean ± SE of four samples in each treatment. (C) Ratio of proliferating cells to viable cells. *P < .05; †P < .01; ‡P < .001; §P < .0001 indicate statistical significance in SD-treated groups compared with the control.
SD Inhibited Cell Proliferation of A431 Cells
In similar treatments as above, the effects of SD on cell proliferation in A431 cells were investigated by BrdU incorporation assay. Sarcophine-diol (300–600 µM) treatment for 24, 48, and 72 hours resulted in 63% to 98%, 26% to 98%, and 19% to 99% (P < .05 to .0001) decreases in cell proliferation, respectively. Sarcophine-diol treatment at 200 µM for 24 hours also significantly (P < .001) inhibited cell proliferation as shown in Figure 2B. Overall results suggest that SD treatment (100–300 µM) for 24 hours could lead to a greater inhibition of cell proliferation in A431 cells compared with those of 48 and 72 hours of treatment.
Furthermore, the ratio of BrdU incorporation to viable cells for each treatment was calculated to investigate the role of either inhibition of cell viability or inhibition of cell proliferation caused by SD on the decrease of cell growth in A431 cells. As shown in Figure 2C, inhibitory effects of SD treatment at an early period (24 hours) and at higher concentrations (400–600 µM) were mainly due to the inhibition of cell proliferation because the ratio of proliferative cells to viable cells was less than 1, whereas inhibitory effects caused by lower concentrations of SD (100–300 µM) for higher treatment periods (48 and 72 hours) were largely due to the decrease in cell viability.
SD Induced a Concentration-Dependent Apoptotic Cell Death
We next assessed whether the apoptotic cell death in A431 cells was observed in response to the treatment of SD. Both control and SD-treated cells were stained using Vybrant Apoptosis Kit 2. The apoptotic cells stained with annexin V labeled with Alexa Fluor 488 (green fluorescence) and necrotic cells stained with PI (red fluorescence) were analyzed by flow cytometry. As shown in Figure 3A, a significant number of A431 cells shifted from viable state (lower left quadrant) to apoptotic state (lower right quadrant) and showed higher staining for annexin V (FL1-H) after treatment with different concentrations of SD for 48 hours. Figure 3, B and C, summarized the percentages of apoptotic cells and necrotic cells in control and SD-treated cell populations obtained after analysis of data from three independent experiments using CellQuest Software. Sarcophine-diol treatment for 48 hours at 50-, 100-, and 400-µM concentrations resulted in 19.1%, 41.2%, and 48.6% of apoptotic cell death compared with 8.8% of apoptotic cells in the DMSO-treated control A431 cell population. However, as shown in Figure 3C, SD treatment for 48 hours at 50-, 100-, and 400-µM concentrations could not significantly (P < .05) induce necrotic cell death. These results suggest that SD treatment (50–400 µM) significantly (P < .05) induced apoptotic cell death but not necrotic cell death in A431 cells in a concentration-dependent manner.
Figure 3.

Effects of SD on apoptosis in A431 cells as judged by annexin V/PI staining. Cells were treated with SD (0–400 µM) for 48 hours, and then cells were collected by brief trypsinization. (A) Dot plot of annexin V (FL1-H)/PI (FL2-H) staining of A431 cells by flow cytometry. The lower left quadrant contains viable cells, which exclude PI and are negative for annexin V staining. The lower right quadrant shows apoptotic cells, which still exclude PI but bind to green fluorescence labeled annexin V through PS exposed on the cell surface of apoptotic cells. The upper quadrants represent necrotic cells that do not exclude PI and display red fluorescence. Percentages of apoptotic cells (B) and necrotic cells (C) of each treatment after analysis of the FACS data using CellQuest software. In each case, data represent mean ± SE of three observations. *P < .05 indicates statistical significance in SD-treated groups compared with the control.
SD Induced DNA Fragmentation in A431 Cells
Annexin V/PI staining represents an early stage of apoptosis, whereas DNA fragmentation is the biochemical hallmark of apoptosis, an irreversible event that commits the cell to die. Using TUNEL assay, we therefore sought to determine whether SD treatment could increase DNA fragmentation, a late event in the apoptotic cascade, in A431 cells [19]. As shown in Figure 4A, after treating A431 cells with 400-µM SD for 48 hours, the marker (M1) in SD-treated sample contained more number of cells that were DNA-fragmented as indicated by a higher number of green fluorescent cells in SD-treated sample compared with control cultures. Figure 4B represented the mean percentages of cells with DNA fragmentation from three independent experiments. Data were analyzed using CellQuest Software. Sarcophine-diol at 400 µM induced a significant (P < .05) amount of DNA fragmentation in 11.0% of cells, accounting for three-fold of DNA fragmentation induction over the DMSO-treated control A431 cells. However, SD at lower concentrations (50–100 µM) did not induce a significant amount of DNA fragmentation in A431 cells. These observations suggest that SD could cause DNA fragmentation in A431 only at a higher concentration.
Figure 4.

Effects of SD on DNA fragmentation in A431 cells. Cells were treated with SD (0–400 µM) for 48 hours, and the cells were collected by brief trypsinization. (A) Histograms show the data obtained by TUNEL assay. The marker (M1) includes the apoptotic cells with fragmented DNA, which were positive for green fluorescence. (B) Percentages of apoptotic cells with fragmented DNA in the SD-treated and untreated cell populations. In each case, data represent mean ± SE of three observations. *P < .05 indicates statistical significance in SD-treated groups compared with the control.
Apoptosis Induced by SD Involves Caspase-3 Cleavage
Because caspase-3 cleavage is the key event in the process of apoptosis, it is used as a marker of apoptosis induction [20]. Next, the effects of SD treatment on the expression of cleaved caspase-3 were examined. As shown in Figure 5, SD treatment at 400 µM for 48 hours exhibited an intense band at 17 kDa and a light band at 19 kDa, both of which were bands for cleaved caspase-3. The caspase-3 bands for control and 100-µM SD treatment were very faint, and moreover, only cleaved caspase-3 at 17 kDa was observed for control and 100-µM SD. These results suggest that SD treatment at 400 µM induce apoptosis involving the cleavage of caspase-3.
Figure 5.

Effects of SD on expression of cleaved caspase-3 in A431 cells. Cells were treated with SD (0–400 µM) for 48 hours, and then cells were collected by brief trypsinization. Total cell lysates were prepared and loaded to SDS-PAGE and Western blot analysis. Membranes were then probed with cleaved caspase-3 and β-actin antibodies followed by appropriate secondary antibody and ECL detection.
Activation of Caspase-3 by SD Is Caspase-8 Dependent But Caspase-9 Independent
On the basis of results that SD treatment at 400 µM for 48 hours could cause a strong expression of cleaved caspase-3 in A431 cells, we also assessed whether SD could increase the activity of caspase-3. Results in Figure 6A showed that SD treatment at 400 µM for 48 hours increased 1.7-fold the induction of the activity of caspase-3 over DMSO-treated control.
Figure 6.

Effects of SD on activities of caspase-3, -8, and -9 in A431 cells. Cells were treated with SD (0–400 µM) for 48 hours, and then enzymatic activity in cell lysates was quantified by measuring chromophores obtained from cleaved substrates. (A) Caspase-3 activity was assessed as the cleavage of DEVE-pNA to synthetic tetrapeptide DEVD and chromophore pNA. (B) Caspase-8 activity was determined by the cleavage of IETD-pNA to synthetic tetrapeptide IETD and chromophore pNA. (C) Caspase-9 activity was investigated by the cleavage of LEHD-pNA to synthetic tetrapeptide LEHD and chromophore pNA as detailed in the Materials and Methods section. Data are shown as the percentage of the absorbance values to controls and represent mean ± SE of three independent samples. *P < .05 indicates statistical significance in SD-treated groups compared with the control.
In a classic apoptotic cascade, activation of an executioner caspase such as caspase-3 is mediated through two pathways: an extrinsic pathway that is mediated by caspase-8 and an intrinsic pathway that is regulated by caspase-9 [21]. Because SD could induce the activity of caspase-3, the activities of caspase-8 and -9 were also investigated as upstream effectors leading to caspase-3 activation. Figure 6B showed that SD treatment at 400 µM for 48 hours significantly (P < .05) induced a 1.9-fold increase of the caspase-8 activity over DMSO-treated control, whereas SD treatment at 400 µM for 48 hours cause comparable caspase-9 activity as control (Figure 6C). However, SD at lower concentration (50 and 100 µM) could not induce the activity of caspase-3, -8, and -9 (data not shown). These results indicated that SD treatment at 400 µM for 48 hours caused the activation of caspase-3, which was caspase-8 dependent but caspase-9 independent.
SD Did Not Increase Caspase-3 Activity in CV-1 Cells
Next, we wanted to investigate the effects of SD on the normal cell line. Therefore, using monkey kidney CV-1 cells, the effects of SD on cell viability and apoptosis were assessed. Results showed that SD treatment for 72 hours at 800 µM inhibited 44% of cell viability in CV-1 cells, and SD treatment at 400 µM for 48 hours did not significantly induce the activity of caspase-3 (data not presented).
Discussion
In recent years, marine natural products have gained considerable attention as cancer chemopreventive agents [5–10], such as sarcophytol A, a cembranoid isolated from the Okinawan soft coral Sarcophyton. Sarcophytol A was studied by the National Cancer Institute at a preclinical trial level for skin cancer [6]. Sarcophine is one of the most abundant cembranolide isolated from the Red Sea Sarcophyton. It has been reported that semisynthesis of sarcophine derivatives such as SD and sarcophine-triol showed high chemopreventive effects against skin carcinogenesis in mouse models [7–10]. In our pervious studies, SD showed chemopreventive effects on DMBA-initiated and TPA-promoted skin tumor developments in female CD-1 mice by enhancing the expressions of caspase-3 and -8 and decreasing COX-2 levels [9]. But the detailed mechanism(s) of its efficacy were not well studied. The present study was designed to elucidate the mechanism(s) of action of SD because it may result in the design of novel approaches by which cancer chemopreventive agents could target. Inhibition of cell growth and induction of apoptosis are two key events that many chemopreventive agents target [16,22–24]. Accordingly, in this study, the effects of SD on cell growth and apoptosis in skin cancer-derived human epidermoid carcinoma cells were investigated. The hypothesis that was tested in this study was whether SD could inhibit cell growth and/or induce apoptosis.
Our data demonstrated that SDtreatment resulted in a concentration-dependent loss of cell viability as judged by testing cell's mitochondrial metabolic activity usingMTTassay. Sarcophine-diol treatment also inhibited A431 cell proliferation as measured by testing the amount of BrdU incorporated to DNA during DNA synthesis. Both inhibitions of cell viability and cell proliferation contributed to the overall inhibition of cell growth by SD treatment in A431 cells.
Apoptosis or programmed cell death is the physiological process by which unwanted or undesirable cells are eliminated during development and other normal biologic process without causing damage to surrounding tissues. An increasing number of chemopreventive agents have been shown to stimulate apoptosis in premalignant and malignant cells in vitro or in vivo [16]. Thus, the present study also investigated whether SD treatment could induce apoptosis in A431 malignant cancer cells in vitro. During apoptosis, many different cell changes occur such as loss of phospholipids asymmetry of the plasma membrane, cell shrinkage, activation of proteases, mitochondrial changes, and internucleosomal DNA fragmentation [25]. These apoptotic cell alterations can be detected by different flow cytometric techniques. In an early stage of apoptosis, PS is translocated from the inner side of the plasma membrane to the outer layer, and thus, PS becomes exposed at the external surface of the cell. Alexa Fluor 488-labeled annexin V, in the presence of calcium ions, immediately adheres to PS, which results in green fluorescence of the apoptotic cells [26–28]. However, the translocation of PS to the external surface of the cell not only is unique to apoptosis but also occurs during cell necrosis, which is an accidental cell death, a pathologic process occurring when cells are exposed to a serious physical or chemical insult [27,28]. The difference between these two forms of cell death is that during the initial stages of apoptosis, the cell membrane remains intact, whereas necrosis is accompanied by loss of cell membrane integrity and leakage of cellular constituents into the environment [27]. Therefore, another red fluorescent PI nucleic acid binding staining is performed in conjunction with annexin V to distinguish apoptotic and necrotic cells. Necrotic cells do not exclude PI and show red fluorescence, whereas apoptotic cells exclude PI with little or no red fluorescence.
In the later stage of apoptosis, the biochemical hallmark is the fragmentation of the genomic DNA, an irreversible event that commits the cell to die [28,29]. DNA fragmentation can be detected by incorporating labeled nucleotides such as 5-bromo-2′-deoxyuridine 5′-triphosphate (BrdUTP) onto the free 3′-OH ends of the fragments using terminal deoxynucleotidyl transferase (TdT) enzyme followed by detection of the labeled molecule with fluorescein isothiocyanate-labeled antibody. This type of assay, often referred to as the TUNEL (TdT dUTP Nick End Labeling) assay, allows monitoring the percentage of DNA-fragmented cells with green fluorescence in the cell samples [26].
Our data showed that SD treatment (50–400 µM) for 48 hours could significantly (P < .05) induce apoptotic cell death but not necrotic cell death as judged by annexin V/PI staining. Sarcophine-diol treatment at 400 µM also caused a significant (P < .05) amount of DNA fragmentation as judged by TUNEL assay in A431 cells. These results suggest that SD at low concentrations (50 and 100 µM) could cause apoptotic cell death by loss of phospholipids asymmetry of the plasma membrane but not DNA fragmentation in A431 cells. However, a high concentration of SD (400 µM) could induce apoptosis in A431 by both loss of the plasma membrane's phospholipids asymmetry and DNA fragmentation.
In a classic apoptosis, apoptotic cell death can be initiated by either death receptor pathway (extrinsic pathway) or mitochondrial pathway (intrinsic pathways), both of which can activate caspase-3. Activation of caspase-3 results in the cleavage of the inhibitor of the caspase-activated deoxyribonuclease, and the caspase-activated deoxyribonuclease becomes active leading to DNA fragmentation and apoptotic cell death [30]. For the death receptor pathway, activation of death receptors using ligands leads to caspase-8 activity. Two routes have been identified to activate caspase-3 by caspase-8. In one route, caspase-8 directly processes pro-caspase-3 in the downstream. In another route, caspase-8 cleaves Bid, a member of the Bcl-2 family. The truncated Bid then translocates to mitochondria and stimulates the release of cytochrome c, which activates caspase-9 together with Apaf-1. The activated caspase-9 causes processing of pro-caspase-3 to the activated form. For the mitochondrial pathway, the balance of proapoptotic and antiapoptotic members of the Bcl-2 family is thought to regulate the permeability of mitochondria and the release of cytochrome c from the mitochondria to cytosol. The cytochrome c then activates caspase-9 together with Apaf-1, and caspase-9 in turn activates caspase-3 [21,30]. Accordingly, in our study, first the expression of cleaved caspase-3 and the activity of caspase-3 were investigated to assess whether SD causes apoptosis through a classic pathway. The results showed that SD treatment at 400 µM for 48 hours led to a strong cleaved caspase-3 band compared with untreated control, and similarly, SD treatment at 400 µM for 48 hours increased 1.7-fold the induction of the activity of caspase-3 over untreated control. These results suggest that SD treatment at a higher concentration (400 µM) induces apoptosis through a caspase-dependent pathway. Furthermore, extrinsic pathway and/or intrinsic pathway triggered by SD in A431 cells were determined. Results showed that SD treatment at 400 µM for 48 hours significantly (P < .05) induced the activity of caspase-8 but not caspase-9, indicating that SD induced the activity of caspase-3 through extrinsic pathway, which was mediated by the activation of caspase-8. However, SD at lower concentrations (50 and 100 µM) could not induce the cleavage of caspases, although SD at those concentrations could significantly (P < .05) cause loss of the plasma membrane's phospholipids asymmetry in A431 cells. These observations suggested that SD might induce apoptosis through other mechanism(s) at lower concentrations, such as through caspase-independent pathway [31,32].
Results also showed that SD treatment for 72 hours at 800 µM inhibited 44% of cell viability in monkey kidney CV-1 cells compared with SD treatment at 400 µM that inhibited 62% of cell viability in A431-1 cells. Moreover, SD treatment at 400 µM for 48 hours increased 1.7-fold the induction of the activity of caspase-3 in A431 cells, whereas in monkey kidney normal cells, SD treatment at 400 µM for 48 hours did not significantly increase the activity of caspase-3. Those results suggest that SD exhibits much less cytotoxicity in normal cells than that in skin tumor cells.
In conclusion, SD inhibits cell growth and induces apoptosis possibly through a caspase-dependent extrinsic pathway of A431 cells, which may contribute to the overall chemopreventive effects of SD observed in mouse skin cancer model [9]. Our previous investigation indicated that SD is a very potent chemopreventive agent in DMBA-TPA carcinogenesis protocol in CD-1 mice because it resulted in a significant decrease in skin tumor development at 30 µg per application compared with other chemopreventive agents that resulted in similar effects at milligram application [33–37]. For example, α-santalol inhibited DMBA-initiated and TPA-promoted and UVB-induced skin tumor development in mice at 5 mg per application [33,34]; curcumin at 1.1 to 3.7 mg per application inhibited DMBA-initiated and TPA-promoted skin tumor development in mice [35]; and topical application of (-)-epigallocatechin-3-gallate at 1-mg/cm2 skin area per application prevented photocarcinogenesis in wild type (C3H/HeN) mice [36] and silibinin at 9 mg per application prevented UV radiation-caused skin damages in SKH-1 hairless mice [37]. We anticipated that SD would be equally potent in in vitro assays as detailed in this study. However, SD was found to have significant effects on various parameters studied at relatively higher concentrations compared with other chemopreventive agents such as α-santalol [17], curcumin [38], (-)-epigallocatechin-3-gallate [39], and silibinin [39]. A possible explanation could be that SD is metabolized in vivo, and metabolite(s) is/are more potent than parent SD because chemopreventive agent(s) or SD is not as effective in human epidermoid carcinoma A431 malignant cancer cells as it is in preneoplastic cells under in vivo conditions. Further studies on the effects of metabolite(s) of SD on in vivo animal models and on in vitro cell culture studies are needed to elucidate the mechanism(s) of action of SD. Sarcophine-diol has an excellent potential to be a potent chemopreventive agent for the nonmelanoma skin cancer development.
Footnotes
This work was supported by Juhnke Funds from the South Dakota State University Foundation and by Governor's 2010 Research Initiative Individual Seed Grant (H.F.).
References
- 1.Mantena SK, Sharma SD, Katiyar SK. Berberine inhibits growth, induces G1 arrest and apoptosis in human epidermoid carcinoma A431 cells by regulating Cdki-Cdk-cyclin cascade, disruption of mitochondrial membrane potential and cleavage of caspase 3 and PARP. Carcinogenesis. 2006;27(10):2018–2027. doi: 10.1093/carcin/bgl043. [DOI] [PubMed] [Google Scholar]
- 2.American Cancer Society Publication, author. Cancer Facts & Figures. 2008 http://www.cancer.org.
- 3.Yusuf N, Irby C, Katiyar SK, Elmets CA. Photoprotective effects of green tea polyphenols. Photodermatol Photoimmunol Photomed. 2007;23:48–56. doi: 10.1111/j.1600-0781.2007.00262.x. [DOI] [PubMed] [Google Scholar]
- 4.Wattenberg LW. Chemoprevention of cancer. Cancer Res. 1985;45:1–8. [PubMed] [Google Scholar]
- 5.Haefner B. Drugs from the deep: marine natural products as drug candidates. Drug Discov Today. 2003;8:536–544. doi: 10.1016/s1359-6446(03)02713-2. [DOI] [PubMed] [Google Scholar]
- 6.Fahmy H, Khalifa S, Konoshima T, Zjawiony JK. An improved synthesis of 7, 8-epoxy-1, 3, 11-cembratriene-15R (α), 16-diol, a cembranoid of marine origin with a potent cancer chemopreventive activity. Mar Drugs. 2004;2:1–7. [Google Scholar]
- 7.Fahmy H, Zjawiony JK, Konoshima T, Tokuda H, Khan S, Khalifa S. Potent skin cancer chemopreventing activity of some novel semi-synthesis cembranoids from marine sources. Mar Drugs. 2006;4:1–9. [Google Scholar]
- 8.Kundoor V, Zhang X, Khalifa S, Fahmy H, Dwivedi C. A possible mechanism of action of the chemopreventive effects of sarcotriol on skin tumor development in CD-1 mice. Mar Drugs. 2006;4:274–285. doi: 10.3390/md504197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang X, Kundoor V, Khalifa S, Zeman D, Fahamy H, Dwivedi C. Chemopreventive effects of sarcophine-diol on skin tumor development in CD-1 mice. Cancer Lett. 2007;253:53–59. doi: 10.1016/j.canlet.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 10.Kundoor V, Zhang X, Bommareddy A, Khalifa S, Fahmy H, Dwivedi C. Chemopreventive effects of sarcotriol on ultraviolet B-induced skin tumor development in SKH-1 hairless mice. Mar Drugs. 2007;5:197–207. doi: 10.3390/md504197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fujiki H, Suganuma M, Takagi K. Sarcophytol A and its anologs. In: Huang MT, Ho CT, Lee CY, editors. Cancer Preventive Activity in Phenolic Compounds in Food and Their Effects on Health II, Antioxidants & Cancer Prevention. ACS Symp Ser. Washington, DC: American Chemical Society; 1992. pp. 380–387. [Google Scholar]
- 12.Katsuyama I, Fahmy H, Zjawiony JK, Khalifa S, Kilada RW, Konoshima T, Takasaki M, Tokuda H. Semisythesis of new sarcophine derivatives with chemopreventive activity. J Nat Prod. 2002;65:1809–1814. doi: 10.1021/np020221d. [DOI] [PubMed] [Google Scholar]
- 13.Ne'eman I, Fishelson L, Kashman Y. Sarcophine—a new toxin from the soft coral Sarcophyton glaucum (Alcyonaria) Toxicon. 1974;12:593–598. doi: 10.1016/0041-0101(74)90192-5. [DOI] [PubMed] [Google Scholar]
- 14.Sawant S, Youssef D, Mayer A, Sylvester P, Wali V, Arant M, Sayed KE. Anticancer and anti-inflammatory sulfur-containing semisynthetic derivatives of sarcophine. Chem Pharm Bull. 2006;54(8):1119–1123. doi: 10.1248/cpb.54.1119. [DOI] [PubMed] [Google Scholar]
- 15.Bertram JS, Kolonel LN, Meyskens FL. Rationale and strategies for chemoprevention of cancer in humans. Cancer Res. 1987;47:3012–3031. [PubMed] [Google Scholar]
- 16.Sun SY, Hail N, Jr, Lotan R. Apoptosis as a novel target for cancer chemoprevention. J Natl Cancer Inst. 2004;96:662–672. doi: 10.1093/jnci/djh123. [DOI] [PubMed] [Google Scholar]
- 17.Kaur M, Agarwal C, Singh RP, Guan X, Dwivedi C, Agarwal R. Skin cancer chemopreventive agent, α-santalol, induces apoptotic death of human epidermoid carcinoma A431 cells via caspase activation together with dissipation of mitochondrial membrane potential and cytochrome c release. Carcinogenesis. 2005;26(2):369–380. doi: 10.1093/carcin/bgh325. [DOI] [PubMed] [Google Scholar]
- 18.Dariusz S, Sarah JS, Richard HC, Conner H, Kohyama T, Wen FQ, Fang Q. An improved MTT assay. J Immunol Methods. 1993;157:203–207. doi: 10.1016/0022-1759(93)90088-o. [DOI] [PubMed] [Google Scholar]
- 19.Bentz BG, Hammer ND, Radosevich J, Haines GK. Nitrosative stress induces DNA stress breaks but not caspase mediated apoptosis in a lung cancer cell line. J Carcinog. 2004;3:16–31. doi: 10.1186/1477-3163-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nicholson DW, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci. 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- 21.Brady HJ. Apoptosis and leukaemia. Br J Haematol. 2003;123:577–585. doi: 10.1046/j.1365-2141.2003.04663.x. [DOI] [PubMed] [Google Scholar]
- 22.Kasibhatla S, Tseng B. Why target apoptosis in cancer treatment. Mol Cancer Ther. 2003;2:573–580. [PubMed] [Google Scholar]
- 23.Buolamwini JK. Cell cycle molecular target in novel anticancer drug discovery. Curr Pharm Des. 2000;6:379–392. doi: 10.2174/1381612003400948. [DOI] [PubMed] [Google Scholar]
- 24.Collins K, Jacks T, Pavletich NP. The cell cycle and cancer. Proc Natl Acad Sci USA. 1997;94:2776–2778. doi: 10.1073/pnas.94.7.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin SJ, Reutelingsperger C, McGahon AJ, Rader JA, Schie R, LaFace DM, Green DR. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Overbeeke R, Steffens-Nakken H, Vermes I, Reutelingsperger C, Haanen C. Early features of apoptosis detected by four different flow cytometry assays. Apoptosis. 1998;3:115–121. doi: 10.1023/a:1009649025439. [DOI] [PubMed] [Google Scholar]
- 27.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labeled annexin V. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- 28.Vermes I, Haanen C, Reutelingsperger C. Flow cytometry of apoptotic cell death. J Immunol Methods. 2000;243:167–190. doi: 10.1016/s0022-1759(00)00233-7. [DOI] [PubMed] [Google Scholar]
- 29.Schwartzman R, Cidlowski JA. Apoptosis: the biochemistry and molecular biology of programmed cell death. Endocr Rev. 1993;14(2):133–151. doi: 10.1210/edrv-14-2-133. [DOI] [PubMed] [Google Scholar]
- 30.Ghobrial IM, Witzig TE, Adjei AA. Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin. 2005;55(3):178–194. doi: 10.3322/canjclin.55.3.178. [DOI] [PubMed] [Google Scholar]
- 31.Lorenzo HK, Susin SA. Mitochondrial effectors in caspase-independent cell death. FEBS Lett. 2004;557:14–20. doi: 10.1016/s0014-5793(03)01464-9. [DOI] [PubMed] [Google Scholar]
- 32.Lockshin RA, Zakeri Z. Caspase-independent cell death? Oncogene. 2004;23:2766–2773. doi: 10.1038/sj.onc.1207514. [DOI] [PubMed] [Google Scholar]
- 33.Dwivedi C, Guan X, Harmsen WL, Voss AL, Goetz-Parten DE, Koopman EM, Johnson KM, Valluri HB, Matthees DP. Chemopreventive effects of α-santalol on skin tumor development in CD-1 and SENCAR mice. Cancer Epidemiol Biomarkers Prev. 2003;12:151–156. [PubMed] [Google Scholar]
- 34.Dwivedi C, Valluri HB, Guan X, Agarwal R. Chemopreventive effects of α-santalol on ultraviolet B radiation-induced skin tumor development in SKH-1 hairless mice. Carcinogenesis. 2006;27(9):1917–1922. doi: 10.1093/carcin/bgl058. [DOI] [PubMed] [Google Scholar]
- 35.Huang MT, Wang ZY, Georgiadis CA, Laskin JD, Conney AH. Inhibitory effects of curcumin on tumor initiation by benzo[a]pyrene and 7,12-dimethylbenz[a]anthracene. Carcinogenesis. 1992;13(11):2183–2186. doi: 10.1093/carcin/13.11.2183. [DOI] [PubMed] [Google Scholar]
- 36.Meeran SM, Mantena SK, Elmets CA, Katiyar SK. (-)-Epigallocatechin-3-gallate prevents photocarcinogenesis in mice through interleukin-12-dependent DNA repair. Cancer Res. 2006;66(10):5512–5520. doi: 10.1158/0008-5472.CAN-06-0218. [DOI] [PubMed] [Google Scholar]
- 37.Dhanalakshmi S, Mallikarjuna GU, Singh RP, Agarwal R. Silibinin prevents ultraviolet radiation-caused skin damages in SKH-1 hairless mice via a decrease in thymine dimer positive cells and an up-regulation of p53-p21/Cip1 in epidermis. Carcinogenesis. 2004;25(8):1459–1465. doi: 10.1093/carcin/bgh152. [DOI] [PubMed] [Google Scholar]
- 38.Bush JA, Cheung KJ, Li G. Curcumin induces apoptosis in human melanoma cells through a Fas receptor/caspase-8 pathway independent of p53. Exp Cell Res. 2001;271:305–314. doi: 10.1006/excr.2001.5381. [DOI] [PubMed] [Google Scholar]
- 39.Bhatia N, Agarwal C, Agarwal R. Differential responses of skin cancer-chemopreventive agents silibinin, quercetin, and epigallocatechin 3-gallate on mitogenic signaling and cell cycle regulators in human epidermoid carcinoma A431 cells. Nutr Cancer. 2001;39(2):292–299. doi: 10.1207/S15327914nc392_20. [DOI] [PubMed] [Google Scholar]