

Abstract
Chronic ethanol consumption adversely affects the respiratory activity of rat liver mitochondria. It causes increased cellular production of oxygen radical species and selectively decreases mitochondrial glutathione (GSH) levels. Here we show, using Southern hybridization techniques on total rat genomic DNA, that long-term (11-13 months) ethanol feeding, using the Lieber-DeCarli diet, results in a 36% (P < .05; n = 4) decrease in hepatic mitochondrial DNA (mtDNA) levels when compared with paired controls. UV quantitation of mtDNA isolated from hepatic mitochondria showed that chronic ethanol intake (11-13 months) causes a 44% (P < .01; n = 6) decrease in the amount of mtDNA per milligram of mitochondrial protein. No significant decline in mtDNA levels was seen in ethanol-fed animals maintained on the diet for 1 to 5 months. Ethanol feeding caused a 42% (P < .01; n = 4) and a 132% (P < .05; n = 3) increase in 8-hydroxydeoxyguanosine (8-OHdG) formation in mtDNA in animals maintained on the diet for 3 to 6 months and 10 to 11 months, respectively. In addition, agarose gel electrophoresis revealed a 49% increase (P < .05; n = 3) in mtDNA single-strand breaks (SSB) in animals fed ethanol for more than 1 year. These findings suggest that chronic ethanol consumption causes enhanced oxidative damage to mtDNA in older animals along with increased strand breakage, and that this results in its selective removal/degradation by mtDNA repair enzymes.
Chronic ethanol consumption has been shown to result in multiple structural and functional alterations in hepatic mitochondria. In experimental animals, ethanol treatment decreases levels of mitochondrially encoded components of the electron transport chain within 3 to 5 weeks,1-3 and selectively depletes hepatic mitochondrial glutathione (GSH) levels over a period of 16 weeks.4-6 In addition, chronic ethanol treatment enhances oxidative stress7 and increases mitochondrial reactive oxygen species (ROS) production.8 In previous studies, we reported that chronic ethanol treatment of rats by a pair-feeding protocol is associated with oxidative damage to liver mitochondrial DNA (mtDNA) as reflected in increased levels of 8-hydroxydeoxyguanosine (8-OHdG) adducts.9 This increase in oxidative damage combined with the lower levels of antioxidants (GSH is required for glutathione peroxidase activity within the mitochondria) suggests that ROS may play a role in the actions of ethanol on the mitochondria and, more particularly, on mtDNA.
mtDNA exists in the rat as a circular 16,300–base pair plasmid-like structure encoding 2 rRNAs, 22 tRNAs, and 13 subunits of the electron transport chain. It is significantly more susceptible to oxidative damage than its nuclear counterpart.10,11 This is largely the result of its lack of protective histones and its location adjacent to the inner mitochondrial membrane,12 which is a major source of ROS production in the form of superoxide and hydrogen peroxide.13 During the aging process, mtDNA accumulates increased levels of deletions and oxidative damage.9,10,14-17 These deleterious events have been suggested to play a role in the senescence of an organism, although, at present, there is a paucity of data available to causally link these phenomena. If damage to mtDNA contributes to the mechanisms underlying aging, then any mitochondrial disease state or xenobiotic metabolism that enhances these events may be expected to have a premature aging effect.
Some of our earlier studies9 addressed the question of ethanol-elicited increases in oxidative damage to mtDNA in rat liver over a period of 6 months. It was not clear, however, whether this represented a new steady-state level of oxidative damage or whether the damage would continue to increase the longer the animals were fed ethanol. In addition, it was uncertain what the consequences of the damage would be with regards to the progression of alcoholic liver disease. It is not known whether or not 8-OHdG formation in mtDNA represents a lethal event and, if it does, whether repair enzymes are capable of removing the adduct before the damage becomes lethal.
In vitro studies have shown a linear correlation between the production of 8-OHdG and the presence of single-strand breaks (SSB).18 It may be that the increased oxidative damage detected during chronic ethanol intake is an indicator of more serious damage occurring to the mitochondrial genome. We have therefore conducted studies to quantitate mtDNA levels and to assay for mtDNA SSB. Results show that ethanol causes selective depletion of hepatic mtDNA in older animals along with a higher level of SSB. In addition, we have extended our analyses of ethanol-elicited oxidative damage to mtDNA to animals maintained on the diet for 1 year or more. Our findings suggest that ethanol consumption elevates oxidative damage to a new steady-state level in hepatic mitochondria, possibly by increasing production of ROS or by causing a decline in mtDNA repair mechanisms. These data are consistent with the hypothesis that chronic ethanol intake leads to alterations in the structural integrity of mtDNA and that damaged mtDNA is selectively degraded.
MATERIALS AND METHODS
Chemicals
All chemicals and reagents were purchased from Sigma (St. Louis, MO) unless otherwise stated. Sst I, double-stranded DNA ladders, and primers for polymerase chain reaction (PCR) were obtained from Life Technologies (Grand Island, NY). DNase I was purchased from ICN Biomedicals Inc. (Aurora, OH). 8-OHdG for use as a high-performance liquid chromatography standard was purchased from Wako Pure Chemical Industries (Richmond, VA). Amplitaq Gold was obtained from Perkin Elmer (Roche Molecular Systems Inc., Branchburg, NJ).
Animals
Male Sprague-Dawley rats obtained from Harland were fed a liquid diet19 with ethanol constituting 36% of the calories for a minimum of 41 days.20 Control animals were pair-fed the same diet but with maltose-dextrin isocalorically substituted for ethanol. All animals received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH publication 86-23, revised 1985).
Isolation of mtDNA
Mitochondria were isolated from rat livers by differential centrifugation using the method of Cohen et al.21 mtDNA was isolated using a Qiagen Plasmid Midi kit as previously described9 and dissolved in either TE (10 mmol/L Tris-HCl [pH 8.0], 1 mmol/L ethylenediaminetetraacetic acid) buffer or autoclaved water, depending on the subsequent application. Samples were stored at −20°C until use.
Analysis of K+–Sodium Dodecyl Sulfate Pellets for mtDNA-Protein Cross-link Formation
K+–sodium dodecyl sulfate (SDS) pellets, obtained during the mtDNA isolation process,9 from animals maintained on the diet for 11 to 13 months were resuspended in 50 mmol/L Tris-HCl (pH 8.0) and 10 mmol/L ethylenediaminetetraacetic acid and treated with 0.5 mg/mL proteinase K for 1 hour at 55°C. The K+-SDS pellets were then repelleted and any mtDNA in the supernatant extracted using phenol/chloroform/isoamyl alcohol, followed by ethanol precipitation.
Yields of mtDNA and Protein Determination
mtDNA yields were calculated from A260nm values or, for more accurate quantitation, by using the Picogreen assay kit (Molecular Probes, Inc., Eugene, OR). Protein measurements were performed using the BIORAD-Detergent-Compatible Protein Assay kit (BioRad Laboratories, Hercules, CA) with bovine serum albumin as the protein standard.
Enzymatic Digestion of mtDNA and High-Performance Liquid Chromatography Analysis of 8-OhdG
mtDNA was digested using nuclease P1 and Escherichia coli alkaline phosphatase as described previously.9 Nucleosides were injected onto a Supelcosil LC-18-DB deactivated reversed-phase column (Supelco Inc., Bellefonte, PA), and 8-OHdG and deoxyguanine were measured by electrochemical detection and UV detection, respectively.9
Extraction of Total Genomic DNA
Total genomic DNA was extracted using the QIAamp Tissue kit (QIAGEN Inc., Valencia, CA). Briefly, small pieces of rat liver (25-50 mg) were solubilized in the presence of proteinase K (360 μg/25 mg liver) at 55°C. Samples were loaded onto a QIAamp spin column and DNA eluted with autoclaved water or TE buffer preheated to 70°C. Samples were stored at −20°C until use.
PCR Analyses
PCR analysis was employed to prepare a mtDNA probe for subsequent Southern hybridizations. Primers 15805F (-gtccatacgttccccttaaa-) and 303R (-aaatttaccaaccctgagag-) were used to amplify a 778–base pair region extending from the cytochrome b region of the genome into the start of the D-loop. Conditions used for the PCR were: 95°C hot start for 10 minutes, followed by 35 cycles of 94°C for 30 seconds, 50°C for 30 seconds, and 72°C for 10 seconds. This was followed by a 10-minute extension at 72°C. Reaction products were examined by electrophoresis through 1% agarose gels and were confirmed by restriction digest.
Southern Hybridization
mtDNA or total genomic DNA was electrophoresed through 0.7% agarose gels. Gels were then soaked in 0.25N HCl for 15 minutes, denaturation solution (1.5 mol/L NaCl, 0.5N NaOH) for 30 minutes, and neutralization solution (100 mmol/L Tris-HCl [pH 8.0], 1.5 mol/L NaCl) for a further 30 minutes before being transferred overnight by upward capillary action in 10× SSC buffer (1.5 mol/L NaCl, 150 mmol/L trisodium citrate [pH 7.0]) onto Biodyne A Membranes (Gibco BRL, Grand Island, NY). mtDNA was cross-linked to the membranes by baking at 80°C for 2 hours and detected using the PCR product from the cytochrome b region (see Polymerase Chain Reaction), which was labeled with Fluor-12-dUTP using the Prime-It Fluor Fluorescence Labeling kit (Stratagene, La Jolla, CA). Membranes were prehybridized in QuickHyb (Stratagene) for 15 minutes at 68°C and hybridized with the fluorescent probe along with 1 mg sonicated salmon sperm DNA for 2 hours at 68°C in a hybridization incubator (Fisher Scientific, Pittsburgh, PA). Membranes were washed for 15 minutes at room temperature in 0.1× SSC/0.1% SDS and then twice with 0.1× SSC/0.1% SDS for 15 minutes at 60°C. Blocking was performed using 2% bovine serum albumin in TBS (50 mmol/L Tris-HCl [pH 8.0], 150 mmol/L NaCl) for 30 minutes at 60°C. All subsequent steps were performed at room temperature. Membranes were washed twice in TBST (TBS + 0.05% Tween) before being incubated in TBST with an antifluoroscein-antibody-alkaline phosphatase conjugate for 1 hour. Membranes were then washed three times with TBST before being incubated in an assay buffer (0.96% diethanolamine, 1 mmol/L MgCl2) for 5 minutes. Bands were detected by adding a dioxetane substrate and exposing the resulting chemiluminescence to x-ray film.
Restriction Digests
Mitochondrial or genomic DNA was restricted using Sst I or Bam HI (2U/1 μg DNA) in the appropriate buffer.
Analysis of mtDNA SSB
Isolated mtDNA was electrophoresed through 0.5% agarose gels prepared in TBE buffer. The number of SSB was calculated using the equation:
Statistical Analysis
Values reported in the Tables and Figures represent means ± SE of 3 or more independent samples. Statistical analyses were performed using the paired and unpaired t test employing the Microsoft Excel program.
RESULTS
Long-Term Ethanol Consumption Decreases mtDNA Yields
Total genomic DNA was isolated from the livers of animals maintained on an ethanol-containing diet for approximately 2 months and 1 year, respectively, along with their paired controls. The DNA was then accurately quantitated using the Picogreen assay (Molecular Probes Inc., Eugene, OR) and restricted using Sst I. This endonuclease cuts rat mtDNA at one site within the NADH subunit 1 region of the mitochondrial genome, and linearizes high-molecular-weight mtDNA, i.e., complex, open circular, and supercoiled conformations. These then electrophorese as a single band within an agarose gel, migrating to the position normally occupied by the linear form. Equal amounts of restricted, genomic DNA from control and ethanol-fed animals were subjected to agarose gel electrophoresis and Southern hybridization (see Materials and Methods), and mtDNA levels were analyzed using a probe to the cytochrome b and D-loop regions of the mitochondrial genome. Figure 1 shows autoradiograms obtained from 2-month-old animals maintained on an ethanol-containing diet for 2 to 3 months (Fig. 1A) and for 11 to 13 months (Fig. 1B) along with their paired controls. In the case of the chronic, short-term feeding regimen, no significant alteration in high-molecular-weight mtDNA was detected. In contrast, however, a consistent decrease in mtDNA content was seen in the animals fed ethanol for longer periods (Fig. 1B).
Fig. 1.

Effect of chronic ethanol consumption on hepatic mtDNA levels in aged rats as assessed by Southern hybridization of genomic DNA. Genomic DNA was isolated from control and ethanol-fed animals as described in Materials and Methods and 1 μg electrophoresed through 0.7% agarose gel in TBE buffer. mtDNA was detected using a fluorescently labeled probe to the cytochrome b region of the mitochondrial genome. (A) Chronic short-term feeding. (B) Chronic long-term feeding.
Densitometric analyses of the Southern blots shown in Fig. 1 reveal a 15% (not significant; n = 4) and 37% (P < .05; n = 4) decrease in mtDNA content in those animals fed ethanol for 2 to 3 and 11 to 13 months, respectively. In addition to the predominant bands seen in Fig. 1B, 2 further bands of low intensity can be detected in the animal fed an ethanol-containing diet for 382 days. These bands are likely to represent mtDNA deletion products and were included in the densitometric calculations of total mtDNA in the sample.
Fig. 2.
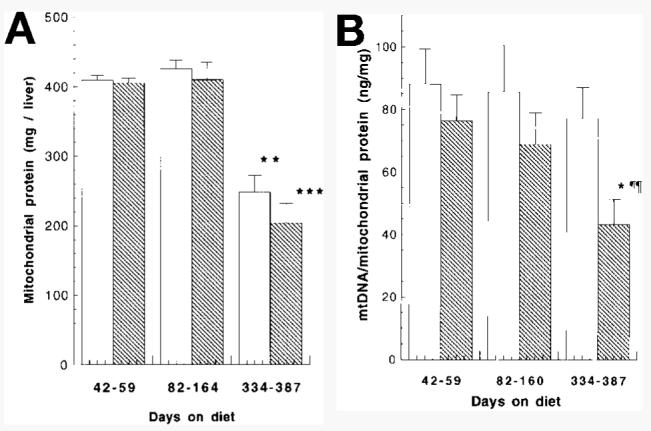
Effect of chronic ethanol consumption on (A) mitochondrial protein and (B) mtDNA content in rat liver. Mitochondria and mtDNA were isolated as described in Materials and Methods. Results represent means ± SE of at least 5 animals per group. Statistical significance of difference of age groups compared with the 42-to-59-day group (*P < .05, **P < .01, ***P < .001) using the Student t test. Statistical significance of difference between ethanol-fed animals and pair-fed controls (¶¶P < .01) using the paired t test. (□), Control; ( ), ethanol.
), ethanol.
The lower levels of mtDNA seen in the Southern analyses of genomic DNA may arise from an ethanol-elicited depression in the number of hepatic mitochondria or, alternatively, from decreased levels of mtDNA per mitochondrion. To clarify which of these phenomena were occurring, mitochondria were obtained from animals maintained on an ethanol-containing diet for up to 13 months along with their paired controls. mtDNA was isolated as described in Materials and Methods and the yield measured by UV absorbance at A260nm. Results were grouped into those from animals maintained on the diet for approximately 1 to 2 months (42-59 days), 2 to 6 months (82-160 days), and 11 to 13 months (334-387 days). Aging was associated with a significant decrease in the amount of mitochondrial protein isolated from both control (39% decrease compared with the 42-to-59–day group; P < .01) and ethanol-fed (50% decrease; P < .001) animals (Fig. 2A) maintained on the diet for 334 to 387 days. Ethanol had no effect on the yield of mitochondrial protein. By contrast, ethanol feeding caused a significant (44%; P < .05) decrease in the level of mtDNA per milligram of mitochondrial protein (Fig. 2B) in those animals maintained on an ethanol-containing diet for 334 days or longer when compared with paired controls. Aging had no effect on the levels of mtDNA isolated from pair-fed control animals at any of the time periods studied. There was, however, a significant age-related decrease (43%; P < .05) in mtDNA yields from the ethanol-fed animals after 334 days on the diet (Fig. 2B) when compared with the group on the diet for 42 to 59 days. These results suggest that, while decreased yields of mitochondria are obtained from aged animals, the lower levels of mtDNA seen in ethanol-fed animals (Fig. 1B) are the result of decreases in the levels of mtDNA per mitochondrion. Comparison of the two methodologies used to measure mtDNA levels in the same animals show good agreement and are summarized in Table 1.
Table 1.
Comparison of Methodologies Used to Assess Ethanol-Elicited Decreases in Hepatic mtDNA Content
|
Method of Analysis |
||
|---|---|---|
| Months on Diet |
Southern Analyses Followed by Densitometry |
A260nm Yields |
| 2-3 | 16% (NS, n = 4) | 13% (NS, n = 4) |
| 11-13 | 36% (P < .05, n = 4) | 44% (P < .01, n = 6) |
In an attempt to determine the origin of the ethanol-elicited depletion in hepatic mtDNA seen in aged animals, the K+-SDS pellets produced during the isolation procedure (see Materials and Methods) were extracted and assayed for mtDNA. Conceivably, increases in oxidative damage seen during chronic ethanol exposure9 may result in increased formation of mtDNA-protein cross-links,23-25 leading to selective removal of the mtDNA in ethanol-fed animals during isolation. However, treatment of the K+-SDS pellets from aged animals (more than 334 days on the diet) with proteinase K, followed by phenol/chloroform extraction, failed to recover greater amounts of mtDNA in the samples from ethanol-fed animals (Fig. 3).
Fig. 3.
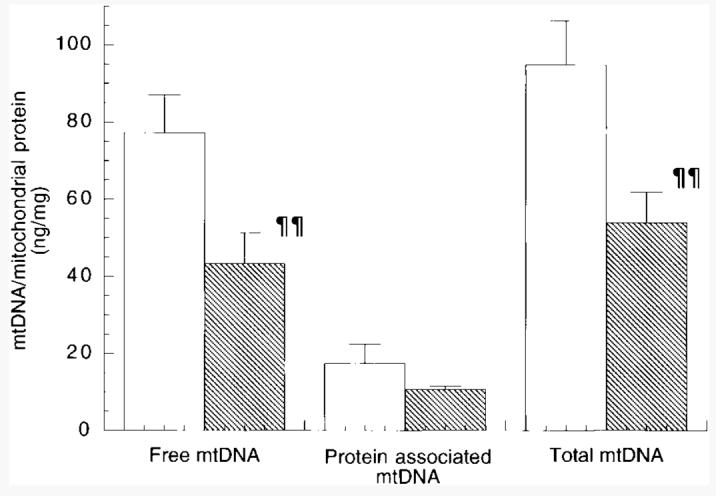
Effect of chronic long-term ethanol feeding on formation of mtDNA-protein cross-links. Both free and protein associated mtDNA was isolated from control and ethanol-fed animals maintained on the LieberDeCarli for 334 to 387 days as described in Materials and Methods. Results represent means ± SE of 6 pairs of animals. Statistical significance of difference between ethanol-fed group and pair-fed control group (¶¶P < .01) using the paired t test. (□), Control; ( ), ethanol.
), ethanol.
Experiments were also conducted to label mtDNA by monitoring the uptake of [methyl-3H]thymidine into coupled, actively respiring mitochondria isolated from control and ethanol-fed animals that had been maintained on a liquid diet for 1 year. After a rapid initial uptake of [methyl-3H]thymidine into mitochondria, mtDNA continued to incorporate radiolabel at a steady rate over a 2-hour period. Analysis of the K+-SDS pellets from these mitochondria after 1 hour revealed no significant radiolabeled thymidine association. mtDNA isolated from 1 mg of mitochondrial protein incorporated 48 fmol dTTP in ethanol preparations and 58 fmol dTTP in paired controls (means of 2 experiments). A further 0.11 and 0.09 fmol dTTP were found associated with ethanol and control pellets, respectively (means of 2 experiments). This amount represents a minor fraction of the total radioactivity incorporated into mtDNA, suggesting that only a minimal level of replicating mtDNA is associated with the pellets. No difference in the rate of radiolabel incorporation into mtDNA was seen in hepatic mitochondria isolated from control or ethanol-fed animals (data not shown). In addition, experiments investigating the effect of proteinase K, used during extraction of total genomic DNA (see Materials and Methods) on mtDNA showed that the enzyme did not selectively degrade mtDNA from ethanol-fed animals (data not shown).
In summary, all the further experiments performed failed to support the hypothesis that mtDNA was missing because of its selective removal during the isolation procedure. We conclude that mtDNA levels are decreased in aging animals maintained on an ethanol-containing diet.
Ethanol Consumption Enhances Age-Dependent Increase in Oxidative Damage to mtDNA
Previously, we reported increased levels of 8-OHdG in mtDNA isolated from ethanol-fed animals.9 In those studies, however, animals were only fed ethanol for a period of up to 6 months. During the same period, it has been reported that mitochondrial GSH levels decrease by 85%6 in ethanol-fed animals. Our results pointed toward an association between mitochondrial GSH levels and oxidative damage to mtDNA. The findings of decreased mtDNA levels in aged, ethanol-consuming animals prompted us to extend our studies of 8-OHdG levels to animals maintained on an ethanol diet for 1 year. mtDNA was isolated from male Sprague-Dawley rats maintained on an ethanol-containing liquid diet for varying time periods up to 11 months and assayed for 8-OHdG (a standard marker for oxidative DNA damage) formation. The results, which lie within the ranges previously reported,10 were then grouped into those maintained on the diet for 1 to 2 months (42-59 days), 3 to 6 months (105-164 days), and 11 months (334-339 days). 8-OHdG formation increased in ethanol-fed animals by 4-fold over the course of 11 months (P < .05) (Table 2). Pair-fed control animals also showed increases in oxidative damage to mtDNA, but to a lesser extent than that seen in the ethanol-fed animals (2-fold over 11 months; P < .01). Comparison of data from control and ethanol-fed animals showed that ethanol caused a significant enhancement in 8-OHdG formation in those animals maintained on the diet for more than 105 days (Table 2).
Table 2.
Effect of Ethanol and Aging on 8-OHdG Formation in Rat Hepatic mtDNA
| Days on Diet | 42-59 | 105-164 | 334-339 |
|---|---|---|---|
| Control | 3.98 ± 0.50 (5) | 6.95 ± 1.01* (4) | 8.33 ± 1.02† (3) |
| Ethanol | 4.76 ± 0.93 (5) | 9.91 ± 1.13†§ (4) | 19.34 ± 6.28*‡ (3) |
NOTE. Data expressed as 8-OHdG/100,000dG.
Results represent means ± SE of (n) animals. Statistical significance of age groups compared with the 42-59–day group
P < .05
P < .01
Statistical significance of difference between ethanol-fed group and pair-fed control group
P < .05
P < .01
Effect of Aging and Ethanol Consumption on mtDNA Strand Breakage
Figure 4A shows an agarose gel analysis of hepatic mtDNA isolated from animals maintained on an ethanol-containing diet for 13 months, along with their paired controls. The gel clearly shows a decrease in the levels of supercoiled mtDNA in the ethanol-fed animals and indicates increased SSB formation. The number of SSB were calculated according to the formula described in Materials and Methods, and the results are represented graphically in Fig. 4B. Ethanol consumption caused a 49% (P < .05; n = 3) increase in the number of SSB occurrences.
Fig. 4.

Effect of chronic long-term ethanol feeding on formation of mtDNA SSB. (A) mtDNA was isolated from control and ethanol-fed animals as described in Materials and Methods and 200 ng electrophoresed through a 0.7% agarose gel in TBE buffer. The gel was stained with ethidium bromide. Lanes 1-6, unrestricted mtDNA; lanes 7 and 8, mtDNA restricted with Sst I; lanes 9 and 10, mtDNA restricted with Bam HI; lane 11, 1-kb DNA ladder; lane 12, supercoiled DNA ladder. (B) Results from (A) represented graphically. SSB =−ln{(1.4 × supercoiled)/[(1.4 × supercoiled) + open circular]}. (□), Control; ( ), ethanol.
), ethanol.
DISCUSSION
The results presented in this article show that ethanol consumption results in structural modification of mtDNA in aging animals. The data suggest that enhanced oxidative stress associated with ethanol intake may be a cause of these modifications. While aging resulted in elevated levels of 8-OHdG in control and chronic ethanol-fed animals, the increase was significantly more pronounced in those maintained on the ethanol-containing diet (Table 2). Mitochondria are known to consume the vast majority of the oxygen (>90%) used in a cell via oxidative phosphorylation. Of this, a small percentage (1%-5%) is converted by the electron transport chain to superoxide13 and hydrogen peroxide.26,27 It is possible that this hydrogen peroxide may be cleaved inside the mitochondria by heavy metals such as copper to yield hydroxyl radicals, which would then be capable of attacking the mtDNA.28 mtDNA is especially susceptible to free radical attack because of its close proximity to the ROS-producing inner mitochondrial membrane12 and because of the fact that, unlike its nuclear counterpart, it does not contain protective histones. The continued production of ROS by the mitochondria over the lifetime of an organism is believed to result in a gradual increase in oxidative modification of mtDNA, which has been hypothesized to contribute to the aging process.29,30 Although this may explain the association of age with increased levels of 8-OHdG shown in Table 2, it does not explain the enhanced oxidative damage seen during ethanol consumption.
One possible mechanism could revolve around the decreased levels of respiratory chain components known to occur during chronic ethanol feeding.1-3 Subunits of respiratory complexes encoded for by the mitochondrial genome are known to be selectively depressed during ethanol consumption, possibly as a result of an ethanol-elicited impairment of mitochondrial ribosomal subunit association.31 It may be that this alteration in inner mitochondrial membrane structure results in elevated production of ROS. Increased levels of mitochondrially produced hydrogen peroxide have been shown to occur in hepatocytes isolated from ethanol-fed rats.8 Another possibility is that the increased production of hydrogen peroxide or other ROS, caused by the ethanol-elicited induction of cytochrome P450 IIEI in the endoplasmic reticulum32 and the rotenone-insensitive NADH-cytochrome c reductase in the outer mitochondrial membrane,33 may lead to increased diffusion of hydrogen peroxide into the mitochondria, thus providing the potential for greater intra-mitochondrial levels of ROS. In addition, chronic ethanol feeding has been reported to deplete mitochondrial GSH levels up to 85% by impairing its import into the organelle.5,6,34 As reduced GSH is a substrate for GSH peroxidase within the mitochondria, its depletion may result in a diminished capacity to remove hydrogen peroxide. Ethanol feeding may also have an effect on mtDNA repair mechanisms.
Recently, a number of mitochondrial enzymes involved in mtDNA repair have been identified including a mitochondrial oxidative damage endonuclease, which recognizes and excises 7,8-dihydro-8-oxoguanine.35 An ethanol-elicited decline in the activity or content of enzymes such as this may result in increased 8-OHdG adduct formation. Any one or all of the potential mechanisms alluded to above may result in elevated levels of oxidative damage to mtDNA.
The elevated levels of 8-OHdG seen in ethanol-fed animals may be paralleled by increased mtDNA strand breaks and lead to accumulation of mtDNA mutations. With regard to the former, 8-OHdG formation can be correlated with production of SSB.36 Both are known to arise from DNA attack by hydroxyl radicals.37 Recent studies by Suter and Richter have shown that fragmented mtDNA contains a high level of 8-OHdG residues.38 It has also been demonstrated in vitro that mitochondria isolated from ethanol-fed animals, in the presence of NADH, are capable of causing nicks in exogenously added plasmid DNA.33 In the case of mtDNA mutations, in vitro studies have shown that the mtDNA polymerase g misreads 8-OHdG bases and pairs a deoxyadenosine base opposite.39 This results in the production of G-to-T transversions. At present, such transversions have not been detected in vivo.
Our studies support the idea that there is a correlation between oxidative damage to mtDNA and strand breakage. In animals maintained on an ethanol-containing diet for more than 1 year, a significant increase in SSB occurrence was observed (Fig. 4A and 4B). The consequences of this may be a selective removal of damaged mtDNA by mitochondrial endonucleases, such as mitochondrial endonuclease G (Endo G). It has been shown that oxygen radical–elicited SSB formation increases the susceptibility of DNA to attack by endonuclease G.40 This model could provide an explanation for the selective removal of high-molecular-weight mtDNA in ethanol-fed animals. While aging had no effect on the yields of mtDNA from control animals, there was a significant decrease in the yields from aged animals maintained on an ethanol-containing diet (Fig. 2B). Our results also show that ethanol consumption caused a very significant decrease in mtDNA yields when compared with animals maintained on the control diet, but only in those animals fed the diet for ≥11 months (Figs. 1 and 2B, Table 1). In these animals, ethanol had no effect on the amount of mitochondrial protein recovered from the liver (Fig. 2A). This indicates that the mtDNA depletion in the liver arises not from decreased numbers of mitochondria, but from diminished levels of mtDNA per mitochondrion. Furthermore, analyses of K+-SDS precipitates, formed during the isolation of mtDNA (Fig. 3), in conjunction with studies incorporating radiolabeled [3H-methyl]thymidine into replicating mtDNA (see Results), suggests that the loss of mtDNA is not the result of ethanol-induced DNA-protein cross-link formation. Animals fed ethanol for ≥11 months also exhibited the greatest levels of oxidative damage to mtDNA (Table 2) and showed increased strand breaks (Fig. 4A and 4B). These observations are consistent with the selective removal of damaged mtDNA by the organelle. Further studies from our laboratory also suggest a link between acute oxidative stress and mtDNA depletion in mitochondria from ethanol-fed rats. Mitochondria isolated from control and ethanol-fed animals maintained on a liquid diet for 2 months were exposed to an oxygen radical–generating system (hydrogen peroxide, Cu2+, and ascorbic acid) and the mtDNA was isolated. While no depletion was seen in control animals, significant decreases in mtDNA yields were found in ethanol-fed animals (manuscript in preparation), suggesting that ethanol consumption enhances the sensitivity of mtDNA to oxidative stress, resulting in its selective depletion.
While the increase in SSB formation seen in hepatic mtDNA isolated from ethanol-fed animals (Fig. 4A and 4B) is believed to be occurring in vivo, the possibility cannot be excluded that chronic ethanol consumption results in structural modifications of mtDNA that render it more susceptible to damage during the isolation procedure. The mechanism(s) behind oxidative stress–induced SSB formation in cells is not clear. Recent evidence suggests that the accessible surface areas of the hydrogen atoms of the DNA backbone play a significant role in DNA strand breakage mediated by the hydroxyl radical.37 In our studies, extreme care is taken to minimize shearing of the mtDNA during its extraction, phenol is not used, and isolated mtDNA is aliquoted before storage at −20°C to prevent damage during freeze-thawing.
It has been suggested by Pessayre et al.41 that alcoholic patients may suffer from premature aging. The free radical theory of aging states that the continuous exposure of mtDNA to ROS attack over the course of an organism's lifetime eventually leads to impaired mitochondrial function and decreased energy production to a level below the threshold of that necessary for cell survival, resulting in senescence.29,30 There is, however, no direct evidence linking oxidative damage and mtDNA mutation to the death of an organism other than the timing of their rapid increase during the later years of an organism's lifetime.16 If oxidative damage, strand breaks, and mtDNA mutations are causal mechanisms of senescence, then ethanol consumption might indeed be considered a contributing factor to premature death. However, there is no evidence that chronic ethanol intake causes premature death in experimental animals such as the rat in the feeding protocol used in these studies.
The ramifications of our findings in relation to the progression of alcoholic liver disease remain to be seen. Analysis of liver pathology revealed no signs of increased microvesicular steatosis during long-term ethanol feeding (data not shown). Many of the major effects of chronic ethanol consumption (Lieber-DeCarli diet) on liver mitochondria, e.g., decreased levels of mitochondrially encoded electron transport chain components and depletion of mitochondrial GSH,4-6 are seen within the first 4 months of ethanol feeding. Elevated cytochrome P450 IIEI levels, which are likely to cause increased oxidative stress within the cell, are seen within the first 10 days.32 The animals, however, continue to survive for a regular lifespan, indicating that the mitochondria are capable of withstanding many of the apparently deleterious effects induced by ethanol. It may be that these phenomena are not harmful as long as the energy requirements of the liver remain below a metabolically stressful threshold level. If, however, the mitochondria are called upon to participate in cellular processes requiring increased energy demand, such as liver regeneration or defense against viral infection, then the ethanol-elicited impairments may be more damaging. Mitochondria are reported to possess between 2 to 10 genomes per mitochondrion, depending on the tissue type,42,43 and their replication is semiautonomous with regards to the cell cycle. It is not known what triggers mtDNA replication or, indeed, which genome(s) will replicate at any one time. The consequences of mtDNA depletion are therefore speculative. It may be that the remaining genomes replicate at greater rates, thereby providing the potential for an increased frequency of replication errors. Support for this comes from studies performed by Larrson et al.44 on heterozygous Tfam (mitochondrial transcription factor A) knockout mice. In these animals, the mtDNA copy number in all tissues investigated, including the liver, was decreased by 34%. At the same time, no change in hepatic mitochondrial transcripts was detected, indicating an increase in transcription of the remaining genomes. Alternatively, it may be that mtDNA replication cannot compensate for the depletion and the mtDNA levels continue to decrease until a threshold level is crossed at which insufficient transcripts are produced for regular mitochondrial functioning. The hepatic mitochondria of rats fed the Lieber-DeCarli diet may not reach this threshold level during their lifetime. Other alcohol-feeding protocols may achieve this condition at an earlier time, e.g., the Tsukamoto-French model.45,46 Experiments are currently underway to assess these phenomena in other animal models of ethanol feeding.
Finally, a comment should be made as to how our findings relate to the observation of decreased respiratory chain activity1-3,47 in ethanol-fed animals. Our results suggest that the decreased activity of electron transport chain complexes is not the result of ethanol-elicited damage to mtDNA. Respiratory chain deficiencies are known to occur within the first month of ethanol consumption,1,2,47,48 whereas damage to mtDNA is a much later process. If the two phenomena are related, it is more likely that depressions in electron transport chain activity give rise to mtDNA damage by promoting ROS formation through the respiratory chain. In other systems, respiratory defects appear to correlate with an enhanced frequency of mtDNA mutations. For instance, mutated mtDNA molecules have been shown to accumulate in respiratory-deficient cells in the skeletal muscle of patients with Kearns-Sayre syndrome.49,50
In conclusion, our studies suggest a mechanism for ethanol-elicited impairment of mitochondrial function in older animals. Increased oxidative damage to hepatic mtDNA arising from chronic ethanol consumption accompanies a greater number of mtDNA strand breaks. The damaged mtDNA may then be selectively removed by mitochondrial endonucleases, resulting in mtDNA depletion. When mtDNA depletion becomes severe and exceeds the threshold for maintaining energy supply in the liver, tissue damage may result.
Acknowledgments
Supported by Alcoholic Beverage Medical Research Foundation grant F41701 and by National Institutes of Health grants AA07186 and AA07215.
Abbreviations
- GSH
glutathione
- ROS
reactive oxygen species
- mtDNA
mitochondrial DNA
- 8-OHdG
8-hydroxydeoxyguanosine
- SSB
single-strand breaks
- PCR
polymerase chain reaction
- SDS
sodium dodecyl sulfate
REFERENCES
- 1.Coleman WB, Cunningham CC. Effects of chronic ethanol consumption on the synthesis of polypeptides encoded by the hepatic mitochondrial genome. Biochim Biophys Acta. 1990;1019:142–150. doi: 10.1016/0005-2728(90)90136-r. [DOI] [PubMed] [Google Scholar]
- 2.Coleman WB, Cunningham CC. Effect of chronic ethanol consumption on hepatic mitochondrial transcription and translation. Biochim Biophys Acta. 1991;1058:178–186. doi: 10.1016/s0005-2728(05)80235-x. [DOI] [PubMed] [Google Scholar]
- 3.Coleman WB, Cahill A, Ivester P, Cunningham CC. Differential effects of ethanol consumption on synthesis of cytoplasmic and mitochondrial encoded subunits of the ATP synthase. Alcohol Clin Exp Res. 1994;18:947–950. doi: 10.1111/j.1530-0277.1994.tb00064.x. [DOI] [PubMed] [Google Scholar]
- 4.Fernandez-Checa JC, Garcia-Ruiz MC, Ookhtens M, Kaplowitz N. Impaired uptake of glutathione by hepatic mitochondria from chronic ethanol-fed rats. Tracer kinetic studies in vitro and in vivo and susceptibility to oxidant stress. J Clin Invest. 1991;87:397–405. doi: 10.1172/JCI115010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garcia-Ruiz C, Morales A, Ballesta A, Rhodes J, Kaplowitz N, Fernandez-Checa JC. Effect of chronic ethanol feeding on glutathione and functional integrity of mitochondria in periportal and perivenous rat hepatocytes. J Clin Invest. 1994;94:193–201. doi: 10.1172/JCI117306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirano T, Kaplowitz N, Tsukamoto H, Kamimura S, Fernandez-Checa JC. Hepatic mitochondrial glutathione depletion and progression of experimental alcoholic liver disease in rats. Hepatology. 1992;16:1423–1427. doi: 10.1002/hep.1840160619. [DOI] [PubMed] [Google Scholar]
- 7.Ishii H, Thurman RG, Ingelman-Sundberg M, Cederbaum A, Fernandez-Checa J, Kato S, et al. Oxidative stress in alcoholic liver injury. Alcohol Clin Exp Res. 1996;20:162A–167A. doi: 10.1111/j.1530-0277.1996.tb01768.x. [DOI] [PubMed] [Google Scholar]
- 8.Bailey SM, Cunningham CC. Acute and chronic ethanol increases reactive oxygen species generation and decreases viability in fresh, isolated rat hepatocytes. Hepatology. 1998;28:1318–1326. doi: 10.1002/hep.510280521. [DOI] [PubMed] [Google Scholar]
- 9.Cahill A, Wang X, Hoek JB. Increased oxidative damage to mitochondrial DNA following chronic ethanol consumption. Biochem Biophys Res Commun. 1997;235:286–290. doi: 10.1006/bbrc.1997.6774. [DOI] [PubMed] [Google Scholar]
- 10.Richter C, Park JW, Ames BN. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc Natl Acad Sci U S A. 1988;85:6465–6467. doi: 10.1073/pnas.85.17.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richter C. Oxidative damage to mitochondrial DNA and its relationship to ageing. Int J Biochem Cell Biol. 1995;27:647–653. doi: 10.1016/1357-2725(95)00025-k. [DOI] [PubMed] [Google Scholar]
- 12.Nass MMK. Mitochondrial DNA: I. Intramitochondrial distribution and structural relations of single- and double-length circular DNA. J Mol Biol. 1969;42:521–528. doi: 10.1016/0022-2836(69)90240-x. [DOI] [PubMed] [Google Scholar]
- 13.Richter C. Reactive oxygen and DNA damage in mitochondria. Mutat Res. 1992;275:249–255. doi: 10.1016/0921-8734(92)90029-o. [DOI] [PubMed] [Google Scholar]
- 14.Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci U S A. 1994;91:10771–10778. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mecocci P, MacGarvey U, Kaufman AE, Koontz D, Shoffner JM, Wallace DC, et al. Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann Neurol. 1993;34:609–616. doi: 10.1002/ana.410340416. [DOI] [PubMed] [Google Scholar]
- 16.Hayakawa M, Torii K, Sugiyama S, Tanaka M, Ozawa T. Age-associated accumulation of 8-hydroxydeoxyguanosine in mitochondrial DNA of human diaphragm. Biochem Biophys Res Commun. 1991;179:1023–1029. doi: 10.1016/0006-291x(91)91921-x. [DOI] [PubMed] [Google Scholar]
- 17.Hayakawa M, Hattori K, Sugiyama S, Ozawa T. Age-associated oxygen damage and mutations in mitochondrial DNA in human hearts. Biochem Biophys Res Commun. 1992;189:979–985. doi: 10.1016/0006-291x(92)92300-m. [DOI] [PubMed] [Google Scholar]
- 18.Toyokuni S, Sagripanti JL. Association between 8-hydroxy-2 -deoxyguanosine formation and DNA strand breaks mediated by copper and iron. Free Radic Biol Med. 1996;20:859–864. doi: 10.1016/0891-5849(95)02184-1. [DOI] [PubMed] [Google Scholar]
- 19.Lieber CS, DeCarli LM. Animal models of chronic ethanol toxicity. Methods Enzymol. 1994;233:585–594. doi: 10.1016/s0076-6879(94)33061-1. [DOI] [PubMed] [Google Scholar]
- 20.Ponnappa BC, Hoek JB, Jubinski L, Rubin E. Ethanol withdrawal stimulates protein synthesis in rat pancreatic lobules. Biochim Biophys Acta. 1990;1036:107–112. doi: 10.1016/0304-4165(90)90021-n. [DOI] [PubMed] [Google Scholar]
- 21.Cohen NS, Kyan FS, Kyan SS, Cheung C-W, Raijman L. The apparent Km of ammonia for carbamoyl phosphate synthase (ammonia) in situ. Biochem J. 1985;229(1):205–211. doi: 10.1042/bj2290205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richter C, Gogvadze V, Laffranchi R, Schlapbach R, Schweizer M, Suter M, et al. Oxidants in mitochondria: from physiology to diseases. Biochim Biophys Acta. 1995;1271(1):67–74. doi: 10.1016/0925-4439(95)00012-s. [DOI] [PubMed] [Google Scholar]
- 23.Dizdaroglu M, Gajewski E. Structure and mechanism of hydroxyl radical–induced formation of a DNA-protein cross-link involving thymine and lysine in nucleohistone. Cancer Res. 1989;49:3463–3467. [PubMed] [Google Scholar]
- 24.Dizdaroglu M, Gajewski E, Reddy P, Margolis SA. Structure of a hydroxyl radical induced DNA-protein cross-link involving thymine and tyrosine in nucleohistone. Biochemistry. 1989;28:3625–3628. doi: 10.1021/bi00434a071. [DOI] [PubMed] [Google Scholar]
- 25.Oleinick NL, Chiu S, Ramakrishnan N, Xue L. The formation, identification, and significance of DNA-protein cross-links in mammalian cells. Br J Cancer. 1987;55:133–140. [PMC free article] [PubMed] [Google Scholar]
- 26.Boveris A, Oshino N, Chance B. The cellular production of hydrogen peroxide. Biochem J. 1972;128:617–630. doi: 10.1042/bj1280617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 28.Giulivi C, Boveris A, Cadenas E. Hydroxyl radical generation during mitochondrial electron transfer and the formation of 8-hydroxydesoxyguanosine in mitochondrial DNA. Arch Biochem Biophys. 1995;316:909–916. doi: 10.1006/abbi.1995.1122. [DOI] [PubMed] [Google Scholar]
- 29.Ames BN, Shigenaga MK, Hagen TM. Oxidants, antioxidants, and the degenerative diseases of aging. Proc Natl Acad Sci U S A. 1993;90:7915–7922. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wallace DC, Bohr VA, Cortopassi G, Kadenbach B, Linn S, Linnane AW, et al. Group report: The role of bioenergetics and mitochondrial DNA mutations in aging and age-related diseases. In: Esser K, Martin GM, editors. Molecular Aspects of Aging. Wiley; New York: 1995. pp. 199–225. [Google Scholar]
- 31.Cahill A, Baio DL, Ivester P, Cunningham CC. Differential effects of chronic ethanol consumption on hepatic mitochondrial and cytoplasmic ribosomes. Alcohol Clin Exp Res. 1996;20:1362. doi: 10.1111/j.1530-0277.1996.tb01135.x. [DOI] [PubMed] [Google Scholar]
- 32.Tsutsumi M, Lasker JM, Takahashi T, Lieber CS. In vivo induction of hepatic P4502E1 by ethanol: role of increased enzyme synthesis. Arch Biochem Biophys. 1993;304(1):209–218. doi: 10.1006/abbi.1993.1341. [DOI] [PubMed] [Google Scholar]
- 33.Kukielka E, Dicker E, Cederbaum AI. Increased production of reactive oxygen species by rat liver mitochondria after chronic ethanol treatment. Arch Biochem Biophys. 1994;309:377–386. doi: 10.1006/abbi.1994.1127. [DOI] [PubMed] [Google Scholar]
- 34.Garcia-Ruiz MC, Morales A, Colell A, Ballesta A, Rhodes J, Kaplowitz N, et al. Feeding S-adenosyl-l-methionine attenuates both ethanol-induced depletion of mitochondrial glutathione and mitochondrial dysfunction in periportal and perivenous rat hepatocytes. Hepatology. 1995;21:207–214. doi: 10.1002/hep.1840210133. [DOI] [PubMed] [Google Scholar]
- 35.Croteau DL, Rhys CMJ, Hudson EK, Dianov GL, Hansford RG, Bohr VA. An oxidative damage-specific endonuclease from rat liver mitochondria. J Biol Chem. 1997;272:27338–27344. doi: 10.1074/jbc.272.43.27338. [DOI] [PubMed] [Google Scholar]
- 36.Lloyd DR, Carmichael PL, Phillips DH. Comparison of the formation of 8-hydroxy-2 -deoxyguanosine and single- and double-strand breaks in DNA mediated by fenton reactions. Chem Res Toxicol. 1998;11:420–427. doi: 10.1021/tx970156l. [DOI] [PubMed] [Google Scholar]
- 37.Balasubramanian B, Pogozelski WK, Tullius TD. DNA strand breaking by the hydroxyl radical is governed by the accessible surface areas of the hydrogen atoms of the DNA backbone. Proc Natl Acad Sci U S A. 1998;95:9738–9743. doi: 10.1073/pnas.95.17.9738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suter M, Richter C. Fragmented mitochondrial DNA is the predominant carrier of oxidized DNA bases. Biochemistry. 1999;38(1):459–464. doi: 10.1021/bi9811922. [DOI] [PubMed] [Google Scholar]
- 39.Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349:431–443. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 40.Ikeda S, Ozaki K. Action of mitochondrial endonuclease G on DNA damage by l-ascorbic acid, peplomycin and cis-diamminedichloroplatinum (II) Biochem Biophys Res Commun. 1997;235:291–294. doi: 10.1006/bbrc.1997.6786. [DOI] [PubMed] [Google Scholar]
- 41.Mansouri A, Fromenty B, Berson A, Robin M-A, Grimbert S, Beaugrand M, et al. Multiple hepatic mitochondrial DNA deletions suggest premature oxidative aging in alcoholic patients. J Hepatol. 1997;27:96–102. doi: 10.1016/s0168-8278(97)80286-3. [DOI] [PubMed] [Google Scholar]
- 42.Borst P, Kroon AM. Mitochondrial DNA: physicochemical properties, replication, and genetic function. Int Rev Cytol. 1969;26:107–190. doi: 10.1016/s0074-7696(08)61635-6. [DOI] [PubMed] [Google Scholar]
- 43.Nass MM. Uptake of isolated chloroplasts by mammalian cells. Science. 1969;165:1128–1131. doi: 10.1126/science.165.3898.1128. [DOI] [PubMed] [Google Scholar]
- 44.Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, et al. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice [Comments] Nat Genet. 1998;18:231–236. doi: 10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- 45.Tsukamoto H, French SW, Benson N, Delgado G, Rao GA, Larkin EC, et al. Severe and progressive steatosis and focal necrosis in rat liver induced by continuous intragastric infusion of ethanol and low fat diet. Hepatology. 1985;5:224–232. doi: 10.1002/hep.1840050212. [DOI] [PubMed] [Google Scholar]
- 46.Tsukamoto H, French SW, Reidelberger RD, Largman C. Cyclical pattern of blood alcohol levels during continuous intragastric ethanol infusion in rats. Alcohol Clin Exp Res. 1985;9(1):31–37. doi: 10.1111/j.1530-0277.1985.tb05046.x. [DOI] [PubMed] [Google Scholar]
- 47.Thayer WS, Rubin E. Molecular alterations in the respiratory chain of rat liver after chronic ethanol consumption. J Biol Chem. 1981;256:6090–6097. [PubMed] [Google Scholar]
- 48.Thayer WS, Rubin E. Effects of chronic ethanol intoxication on oxidative phosphorylation in rat liver submitochondrial particles. J Biol Chem. 1979;254:7717–7723. [PubMed] [Google Scholar]
- 49.Muller-Hocker J, Seibel P, Schneiderbanger K, Zietz C, Obermaier-Kusser B, Gerbitz KD, et al. In situ hybridization of mitochondrial DNA in the heart of a patient with Kearns-Sayre syndrome and dilatative cardiomyopathy. Hum Pathol. 1992;23:1431–1437. doi: 10.1016/0046-8177(92)90065-b. [DOI] [PubMed] [Google Scholar]
- 50.Mita S, Schmidt B, Schon EA, DiMauro S, Bonilla E. Detection of “deleted” mitochondrial genomes in cytochrome-c oxidase-deficient muscle fibers of a patient with Kearns-Sayre syndrome. Proc Natl Acad Sci U S A. 1989;86:9509–9513. doi: 10.1073/pnas.86.23.9509. [DOI] [PMC free article] [PubMed] [Google Scholar]