

Abstract
Objective
Coagulation factor VIIa (VIIa) binding to its cellular receptor, tissue factor (TF), not only initiates the coagulation cascade but also induces cell signaling by activating G-protein coupled protease-activated receptors. The objective of the present study is to investigate the role of lipid rafts and caveolae in modulating TF-VIIa signaling and coagulant functions.
Methods and Results
TF-VIIa coagulant function was measured in factor X activation assay and the signaling function was evaluated in phosphoinositide hydrolysis and IL-8 gene induction. Buoyant density gradient centrifugation and immunofluorescence confocal microscopy were used to determine cellular localization of TF and protease-activated receptor 2. The data show that a substantial fraction of TF and protease-activated receptor 2 resides in lipid rafts/caveolae, and disruption of lipid rafts by cholesterol depletion or modification reduced TF-VIIa—induced cell signaling. Disruption of caveolae with caveolin-1 silencing had no effect on the TF-VIIa coagulant activity but inhibited the TF-VIIa-induced cell signaling.
Conclusion
Overall our data show that lipid raft/caveolae play a selective role in modulating the TF-VIIa signaling function without affecting the TF-VIIa coagulant activity.
Keywords: caveolae, factor VIIa, lipid rafts, signaling, tissue factor, protease-activated receptors
Tissue factor (TF) is a cellular receptor for clotting factor VIIa, and the formation of TF-VIIa complexes on cell surfaces not only triggers the coagulation cascade but also transduces cell signaling via activation of protease-activated receptors (PARs), particularly PAR2.1,2 Although a number of recent studies provide valuable information on intracellular signaling pathways that are activated by TF-VIIa,1–3 the role of various cell surface components in mediating the interaction of TF-VIIa with PARs, and the subsequent signal transmittance are unknown. Unlike thrombin and trypsin, VIIa has to bind to TF to activate PARs. Although there is no quantitative data available on kinetics of PAR2 cleavage by TF-VIIa versus trypsin, a potent activator of PAR2, inability of TF-VIIa to trigger Ca2+ signaling and failure to desensitize signaling of subsequently added trypsin suggest that the TF-VIIa is a poor activator of PAR2.4–6 Despite this, a number of studies have shown that VIIa is as effective as trypsin or PAR2 agonist peptide in activating intracellular signaling pathways and gene expression.4–7 Although the potential mechanism for this phenomenon is unknown, compartmentalization of TF, PAR2, and G-proteins in the plasma membrane microdomains could facilitate a robust TF-VIIa—induced PAR2-mediated cell signaling.
Cholesterol plays a critical role in differentiating and maintaining cell surface microdomains of differing lipid composition. Cholesterol association with glycosphingolipids and other saturated long-chained lipids in biomembranes causes a phase transition in the membrane leading to segregated specialized microdomains, lipid rafts.8,9 Cholesteroland sphingolipid-rich rafts in association with structural protein caveolin-1 form caveolae, flask-shaped invaginations in the plasma membrane.10,11 Various receptors and signaling molecules are shown to be concentrated in lipid rafts and caveolae. For example, when compared with the rest of plasma membrane, lipid rafts/caveolae are enriched in Src-family kinases, epidermal growth factor receptor, platelet-derived growth factor receptor, various G-protein-coupled receptors and their down-stream signaling molecules, and a number of other signaling molecules.10–13 Further, cholesterol in plasma membrane can interact directly with membrane receptors and thus can have a strong influence on the affinity state, binding capacity, and signal transduction properties of receptors.14 Our recent studies with fibroblasts show that TF is localized in both lipid rafts and caveolae and disruption of these microdomains alters the coagulant activity of TF.15
Although certain G-protein-coupled receptors and G-proteins are known to be segregated into lipid rafts and caveolae,16–18 little is known whether G-protein-coupled PARs are segregated into lipid rafts and caveolae, and how such segregation might influence their activation by TF-VIIa and the subsequent cell signaling by coupling to G-proteins. To obtain answers to some of these questions, in the present study, we investigated the role of membrane cholesterol and lipid raft/caveolae in modulating the TF-VIIa signaling and coagulant functions in tumor cells.
Materials and Methods
Human breast carcinoma cell line MDA-MB-231 and COS-7 cells were obtained from ATCC (Rockville, Md). Quiescent monolayers were treated with methyl-β-cyclodextrin (mβCD) or filipin to disrupt lipid rafts/caveolae. Cell surface TF-VIIa activity was determined in factor X activation assay. TF antigen levels at the cell surface were determined by 125I-FVIIa or 125I-TF mAb binding assays. Phosphoinositide hydrolysis was determined by loading the cells with myo-[2-3H] inositol and monitoring the release of total 3H-inositol phosphates. Fluorescence microscopy was used for the measurement of calcium influxes in cells loaded with Fluo- 4. Lipid rafts were separated by buoyant density gradient centrifugation. Total RNA was extracted with Trizol reagent (Invitrogen, Carlsbad, Calif) and RNA was subjected to size fractionation on 1.0% agarose gel in formaldehyde buffer system. Northern blot analysis was performed using 32P-labeled IL-8 cDNA. The fixed cells, either nonpermeabilized or permeabilized with 0.1% Triton X-100, were stained with antibodies against TF, PAR2, or caveloin-1, followed by Oregon Green or Rhodamine Red-conjugated secondary antibodies. Cleavage of PAR2 was evaluated in cells transfected transiently with alkaline phosphatase (AP)-PAR2 expression vector and measuring the activity of AP that was released in to conditioned media following experimental treatments. Cells were transfected with control siRNA or caveolin specific siRNAs using SiPort amine.
A detailed Methods section is available in the online supplement at http://atvb.ahajournals.org.
Results
Localization of TF and PAR2 in Lipid Rafts and Caveolae
To investigate whether TF and PAR2 are colocalized in cholesterol-rich sphingolipid rafts and/or caveolae in breast carcinoma cells, we isolated lipid rafts from MDA-MB-231 cell extracts based on their insolubility in Triton X-100, followed by their buoyant density in sucrose gradient. The gradient fractions were analyzed by immunoblotting for the presence of TF, caveolin-1, GM1, and PAR2. The lipid raft markers, GM1 and caveolin-1, were fractionated mostly into detergent-insoluble low-buoyant density fractions, fractions 3 to 7. A substantial amount of both TF and PAR2 are also distributed into these fractions (Figure 1A). When antigen levels were normalized to total cellular protein content, ≈32% of TF and PAR2 are localized in the low-density fractions (n=3). We further evaluated the localization of TF and PAR2 on the tumor cell surface by dual immunofluorescence confocal microscopy using specific antibodies against TF, PAR2, and caveolin-1. Caveolin-1 antibodies revealed punctate staining pattern marking the caveolae on the cell surface. Likewise, TF also displayed a significant punctate cell surface staining, indicating their elevated concentration within the discrete plasma membrane microdomains (Figure 1B). Overlap of TF and caveolin-1 fluorescence showed a high degree of colocalization of TF and caveolin-1 (0.68±0.05), suggesting TF localization in caveolae. Similar to TF, PAR2 also displayed significant punctuate staining and colocalization with caveloin-1 (0.49±0.01). A substantial amount of PAR2 at the cell surface is colocalized with TF (0.56±0.02; Figure 1B).
Figure 1.

Localization of tissue factor and PAR2 in lipid rafts. A, Presence of TF and PAR2 antigens in low-density, detergent-insoluble microdomains. MDA-MB-231 cells were lysed with 1% Triton X-100 and fractionated on sucrose gradient centrifugation. Equal amounts of protein from each fraction were separated on SDS-PAGE and transferred for immunoblotting with polyclonal antibodies to TF and PAR2, mAB to caveolin-1, and HRP-conjugated cholera toxin. Fractions are labeled from the top to the bottom of the sucrose gradient. B, Immunofluorescence microscopy of TF, PAR2, and caveolin-1. MDA-MB-231 cells were fixed, and intact (for TF and PAR2 colocalization) or permeabilized cells were stained with polyclonal or monoclonal antibodies to TF, PAR2, and caveolin-1. Right panels depict overlay of images, and yellow in the panel represents colocalization of TF and PAR2 with caveolin-1. The images shown are montages of z-stacks.
Effects of mβCD and Filipin Treatments on Membrane Cholesterol, Caveolae, and TF Activity
mβCD is a membrane-impermeable agent that binds to cholesterol with high specificity and depletes membrane cholesterol.19 Filipin does not deplete membrane cholesterol but alters the physical distribution of the cholesterol in the membrane by forming filipin— cholesterol complexes.20 Treatment of MDA-MB-231 cells with increasing concentrations of mβCD (1 to 10 mmol/L) reduced the cholesterol content in a dose-dependent manner (supplemental Figure IA, please see http://atvb.ahajournals.org); 10-mM mβCD treatment for 1 hour reduced the cholesterol content by ≈65%. In contrast, filipin treatment had no effect on the cholesterol content in the membrane. Examination of cells treated with mβCD and filipin under light microscopy revealed no gross differences in morphology between control and treated cells. Under our experimental conditions, these treatments had no significant effect on the cell viability or cell attachment to the culture dish (data not shown). Both mβCD and filipin treatment, as revealed by transmission electron microscopy, disrupted caveolar structures in MDA-MB-231 cells. While caveolae invaginations are clearly visible in control cells, there are very few morphologically recognizable caveolae in the cell membrane after cholesterol depletion/modification (supplemental Figure IB). Immunoblot analysis of detergent-resistant membrane domain fractions of cells treated with mβCD and filipin showed a reduced TF and PAR2 antigen in low-density fractions (Figure 2A). Determination of TF and PAR2 content by densitometry of immunoblots of sucrose gradient fractions revealed that TF and PAR2 content was deceased to 12% in mβCD-treated cells and to 17% in filipin-treated cells (32% of TF and PAR2 was localized in low-density fractions in control cells). Consistent with these data, immunofluorescence microscopy studies revealed that removal of plasma membrane cholesterol by mβCD treatment reduced TF and PAR2 association with the caveolae (Figure 2B). Colocalization analysis of TF or PAR2 with caveolin-1 at the plasma membrane from ≈80 to 100 randomly selected regions gave the following correlation coefficients: for TF and caveolin-1, control 0.68±0.05 and mβCD-treated 0.49±0.01 (P<0.001); and for PAR2 and caveolin-1, control 0.49±0.014 and mβCD-treated 0.41±0.012 P<0.001). Filipin treatment reduced the TF colocalization with caveolin-1 by 18%.
Figure 2.

Effects of membrane cholesterol depletion/modification on TF and PAR2 association with lipid rafts and caveolae. MDA-MB-231 cells cultured in T-75 flask (A) or chambered-glass slides (B) were treated with a control vehicle or mβCD (10 mmol/L) for 1 hour, or filipin (5 μg/mL) for 15 minutes. A, Cells were lysed in 1% Triton X-100, subjected sucrose gradient centrifugation; 10 μg of protein from each fraction were separated on SDS-PAGE for immunoblotting with polyclonal antibodies to TF and PAR2, and HRP-conjugated cholera toxin (as a raft marker). Fractions are labeled from the top to the bottom of the sucrose gradient. GM1 and PAR2 panels for control vehicle-treated cells in this figure and Figure 1A were the same. B, Immunofluorescence microscopy of TF, PAR2, and caveolin-1. MDA-MB-231 cells were fixed; nonpermeabilized (top panel) or permeabilized (middle and bottom panels) cells were stained with polyclonal or monoclonal antibodies to TF, PAR2, and caveolin-1. Right panels depict overlay of images, and yellow in the panel represents colocalization of TF and PAR2 with caveolin-1. The images represent single X-Y-plane of a mid-section from z-stacks.
Depletion of cholesterol from the plasma membrane by mβCD treatment reduced the cell surface TF-VIIa activity significantly, and the extent of decrease in the activity is correlated with the extent of cholesterol depletion from the plasma membrane (supplemental Figure IIA). In contrast, filipin treatment increased the cell surface TF-VIIa activity significantly. The opposing effects of mβCD and filipin on the TF-VIIa activity is consistent with our earlier observations made with fibroblasts.15 To address whether the cholesterol depletion/modification might have altered the availability of negatively charged phospholipids at the cell surface, which could influence the TF-VIIa activity, we examined the effect of mβCD (10 mmol/L) and filipin (5 μg/mL) on the ability of MDA-MB-231 cells to support factor Xa/factor Va-catalyzed activation of prothrombin, which also depends on negatively charged phospholipids. We found no differences in rates of thrombin generation in control cells and cells treated with either mβCD or filipin (supplemental Figure IIB). Analysis of annexin V binding to control, mβCD-treated, and filipin-treated cell by flow cytometry gave identical and overlapping fluorescence histograms (data not shown). These data rule out the possibility of potential alterations in negatively charged phospholipids as a reason for the altered TF-VIIa coagulant activity in mβCD-treated or filipin-treated cells.
Modulation of TF-VIIa—Induced Signaling
To investigate the role of lipid rafts and caveolae in TF-VIIa—induced cell signaling, we studied the effects of cholesterol depletion and lipid raft/caveolae disruption on TF-VIIa—induced phosphoinositide (PI) hydrolysis and IL-8 gene expression. VIIa treatment increased the rate of PI hydrolysis in MDA-MB-231 cells by >4-fold compared with untreated cells. Treatment of carcinoma cells with 10 mmol/L mβCD before their exposure to VIIa fully attenuated the TF-VIIa—induced increase in PI hydrolysis (Figure 3A). mβCD treatment had no significant effect on the basal PI hydrolysis measured in untreated cells. Restoration of membrane cholesterol to the cholesterol-depleted cells by cholesterol loading restored the TF-VIIa ability to induce PI hydrolysis (Figure 3B). This observation also rules out a potential nonspecific effect of mβCD because the cells were exposed to a same concentration of mβCD to load the cholesterol as that was used to deplete the cholesterol. Similar to mβCD, filipin treatment also markedly reduced the VIIa-induced PI hydrolysis in MDA-MB-231 cells. Consistent with these data, both mβCD and filipin treatments reduced the TF-VIIa—induced IL-8 gene expression (Figure 4). These data suggest that the cholesterol depletion or modification of membrane cholesterol, which disrupt lipid rafts and caveolae, impair the TF-VIIa—induced cell signaling.
Figure 3.
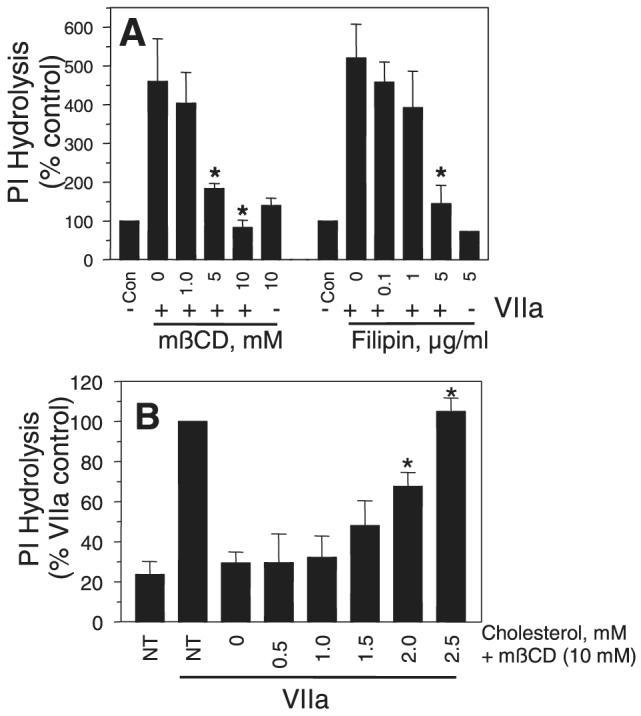
Effects of mβCD and filipin treatments on TF-VIIa—induced PI hydrolysis. A, MDA-MB-231 cells loaded with myo-[2-3H] inositol were treated with either varying concentrations of mβCD or filipin, for 1 hour and 15 minutes, respectively. Thereafter, the cells were exposed to VIIa (10 nM) for 2 hours and the total 3H-inositol phosphates released were quantified (n=4 to 6 ±SEM). B, Myo-[2-3H] inositol-loaded MDA-MB-231 cells were first treated for 30 minutes at 37°C with mβCD (10 mmol/L) to deplete cholesterol. After washing the cells, cholesterol was reintroduced to the cells by incubating the cholesterol-depleted cells with varying concentrations of cholesterol in mβCD (10 mmol/L) for 30 minutes. After cholesterol loading, the cells were washed and exposed to VIIa (10 nM) for 2 hours and the total 3H-inositol phosphates released were quantified (n=3 ±SEM). *Significantly (P<0.05) differs from the control cells that are exposed to VIIa (A) or cholesterol-depleted cells (mβCD +0 cholesterol; B). NT, not treated with either mβCD or cholesterol.
Figure 4.

Membrane cholesterol depletion/modification impairs TF-VIIa—induced IL-8 gene expression. Monolayers of MDA-MB-231 cells were treated with varying concentrations of mβCD (A and B) or filipin (C and D), and then stimulated with VIIa (10 nM) for 90 minutes. Total RNA was extracted and subjected to Northern blot analysis to probe IL-8 gene expression. Data shown in (A) and (C) represent mean±SEM from 4 to 6 experiments, and representative Northern blots were shown in (B) and (D). *Significantly (P<0.05) differs form the value obtained with control (not treated with mβCD or filipin) cells exposed to VIIa.
In additional studies, we evaluated the effect of lipid raft/caveolae disruption by mβCD and filipin treatments on trypsin and PAR2 agonist peptide-induced PI hydrolysis and IL-8 gene expression. The data showed both mβCD and filipin treatments also impaired trypsin and PAR2 agonist peptide-induced PI hydrolysis and IL-8 gene expression (data not shown). These data are not completely unexpected because lipid raft/caveolae disruption is shown to impair coupling of some heterotrimeric G-proteins to their activated receptors,11,21 a step that follows after the activation of PAR2 at the cell surface. However, these data raise a question on whether lipid raft disruption impaired the TF-VIIa signaling because it disrupted TF-VIIa activation of PAR2 at the cell surface or impaired the interaction of G-proteins with their activated receptors.
To address this question, we investigated the effect of mβCD and filipin treatment on PAR2 agonist peptide-induced Ca2+ signaling. Exposure of MDA231 cells to PAR2 agonist peptide rapidly increased the concentration of intracellular Ca2+, and pretreatment of MDA231 cells with mβCD (10 mmol/L) or filipin (5 μg/mL) had no effect on PAR2 AP-induced increase in intracellular Ca2+ (Figure 5A). Similar data were obtained with trypsin (because VIIa does to induce Ca2+ signaling, we cannot use this assay to test the effect of VIIa). Next, we investigated the effect of lipid raft disruption on the cleavage of PAR2 by TF-VIIa and trypsin. For these studies, COS-7 cells were transiently transfected with a PAR2 cleavage reporter construct (AP-PAR2). Extent of AP release into the conditioned medium correlates the extent of PAR2 cleavage. As shown in Figure 5B, both mβCD and filipin treatments markedly reduced TF-VIIa cleavage of PAR2. In contrast, they have no effect or slightly increased trypsin cleavage of PAR2. These data suggest that lipid raft disruption specifically impaired TF-VIIa cleavage of PAR2.
Figure 5.

Membrane cholesterol depletion/modification specifically impairs TF-VIIa activation of PAR2. A, MDA231 cells cultured in glass-chambered slides were loaded with Fluo-4 and treated with mβCD (10 mmol/L for 1 hour) or filipin (5 μg/mL for 15 minutes). After mounting the slide on a microscope stage, live fluorescence images were obtained for 30 seconds, and then control vehicle or PAR2 agonist peptide (SLIGKV, 25 μmol/L) was added to the cells, and the imaging was continued for 3 minutes. Increase in intracellular Ca2+ levels were shown as increase in fluorescence at 530 nm emission wavelength. B, COS-7 cells transiently transfected with TF plus AP-PAR2 expression vectors were treated with a control vehicle, mβCD (10 mmol/L for 1 hour), or filipin (5 μg/mL for 15 minutes), and thereafter exposed to VIIa or trypsin (10 nM) for 1 hour, then soluble AP activity in the conditioned medium was measured. Results are expressed as AP activity in cells treated with VIIa or trypsin minus that in cells not treated with proteases, but subjected to mβCD and filipin treatments (n=3 ±SEM). *Significantly (P<0.05) differs from the control cells exposed to the same protease.
Effect of Caveolin-1 Silencing on TF-VIIa Coagulant and Signaling Functions
Although both mβCD and filipin are widely used and became standard tools to disrupt caveolae, these reagents could potentially exert broad ranging effects unrelated to caveolae. To overcome this possibility, we selectively disrupted caveolae by inhibiting caveolin-1, an integral protein required for caveolae formation using siRNA technology. Transfection of MDA-MB-231 cells with caveolin siRNA markedly reduced the caveolin-1 protein expression and had no effect on either TF antigen or activity levels at the cell surface (supplemental Figure IIIA). Further, caveolin-1 silencing had no effect on the surface expression of TF as evidenced by similar TF-specific VIIa binding to control, negative siRNA, and caveolin-1 siRNA transfected cells (supplemental Figure IIIB). Consistent with these data, caveolin-1 silencing did not decrease the TF coagulant activity (supplemental Figure IIIC). Colocalization studies employing immunofluorescence confocal microscopy revealed a significant decrease in TF and PAR2 colocalization in caveloin-1 silenced cells compared with control cells. Caveloin-1 silencing, as expected, also had no effect on the expression of PAR2 as measured in Ca2+ signaling assay using PAR2 agonist peptide as the activator (data not shown). Caveolin-1 silencing reduced the TF-VIIa—mediated cleavage of PAR2 (Figure 6A) and IL-8 gene expression (Figure 6B). It is important to note that caveloin-1 silencing had no effect on trypsin cleavage of PAR2, which establishes that caveolin-1 silencing specifically affects TF-VIIa cleavage of PAR2. Caveolin-1 silencing significantly impaired TF-VIIa—induced expression of IL-8 (Figure 6B). In contrast to its marked effect on VIIa signaling, caveolin-1 silencing had a minimal effect on PAR2 agonist peptide-induced IL-8 expression (Figure 6B) and partial inhibitory effect on trypsin-induced IL-8 expression (data not shown).
Figure 6.
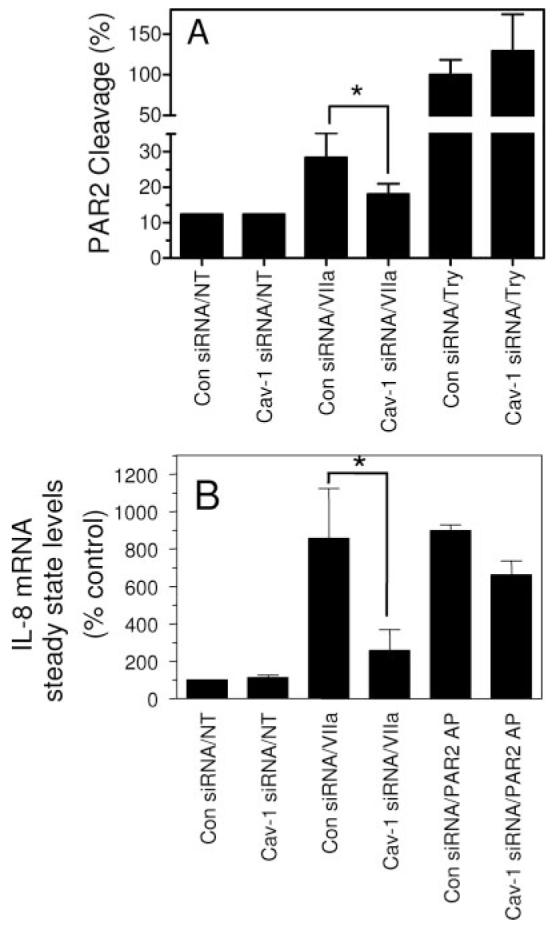
Effect of caveolin-1 silencing on TF-VIIa cleavage of PAR2 and PAR2-mediated IL-8 gene expression. A, MDA-MB-231 cells were transfected with caveolin-1 siRNA or control siRNA. After 48 hours, the cells were infected with adenovirus expressing AP-PAR2. Sixteen hours later, monolayers of MDA 231 were treated with control vehicle (NT), VIIa, or trypsin (10 nM) for 1 hour. The conditioned medium collected and AP activity was determined. Alkaline phosphatase activity released with trypsin (10 nM) in control siRNA transfected cells was arbitrarily taken as 100% of PAR2 cleavage (n=6, mean ±SD). B, MDA-MB-231 cells were transfected with control siRNA or caveolin siRNA were treated with control vehicle (NT), VIIa (10 nM), or PAR2 peptide agonist (50 μmol/L) for 90 minutes. Total RNA was extracted and subjected to Northern blot analysis for IL-8 mRNA expression, and hybridization signal intensities were quantified using phosphor imaging (n=3). *Significantly (P<0.05) differs from the corresponding control.
Discussion
Unlike thrombin and trypsin, VIIa has to bind its cellular receptor, TF, to activate PARs. Therefore, TF-VIIa could only activate PARs that are in the close vicinity of TF. Unless TF and PAR2 are present in high density, it is unlikely that TF-VIIa encounters PAR2 on the cell surface. This raises an important question on how TF-VIIa could activate PAR2-mediated cell signaling. In this study, we show that a fraction of both TF and PAR2 are compartmentalized in cholesterol-rich specialized plasma membrane domains, lipid rafts/caveolae, in breast carcinoma cells (MDA-MB-231), a cell system in which TF-VIIa was shown to induce robust PAR2-mediated cell signaling.6 Depletion or sequestration of plasma membrane cholesterol impaired the TF-VIIa—induced cell signaling in these cells. Silencing of caveloin-1, an integral protein of caveolae, diminished the TF-VIIa—induced cell signaling without affecting the TF-VIIa coagulant activity. Overall, the data presented in this report suggest that sequestration of TF and PAR2 in lipid rafts/caveolae, probably coupled with differential segregation of heterotrimeric G-proteins into these microdomains, facilitates an effective environment for TF-VIIa—induced cell signaling.
Cholesterol plays a critical role in differentiating and maintaining cell surface microdomains of differing lipid composition, particularly sphingolipid rafts. Cholesterol- and sphingolipid-rich rafts in association with a structural protein, caveolin-1, form caveolae, flask-shaped invaginations in the plasma membrane. Because these microdomains have been shown to be enriched with a variety of signaling molecules,13,16,22–24 it is believed that they play a role in compartmentalizing signaling molecules at the cell surface and modulating signaling functions.9,10,18,21,25 Although certain G-protein-coupled receptors and G-proteins are known to be segregated into lipid rafts and caveolae,16–18 we are not aware of any report on whether PARs are segregated into lipid rafts. Consistent with our recent observations in fibroblasts,15 TF in MDA-MB-231 tumor cells is also localized in lipid rafts/caveolae. Further, our studies also show that a fraction of PAR2 is colocalized with TF presumably in lipid rafts/caveolae. Segregation of a fraction of PAR2 with TF into the lipid rafts may facilitate PAR2 access to VIIa bound to the TF. TF-VIIa cleavage of PAR2 in these microdomains could activate the signaling pathways as these microdomains were also shown to be enriched with certain heterotrimeric G-proteins16,21 and a number of other signaling molecules.10–13
The integrity of cholesterol-rich membrane microdomains appears to be critical for the TF-VIIa—induced PAR2-mediated cell signaling. Depletion of membrane cholesterol, which leads to the loss of the caveolar structure as observed by transmission electron microscopy, reduced TF and PAR2 association with low-density membrane microdomains/caveolae, and impaired the TF-VIIa—induced PI hydrolysis and IL-8 gene expression. Although the depletion of membrane cholesterol also impairs the assembly of TF-VIIa complex at the cell surface,15 this alone cannot explain the complete loss of TF-VIIa—induced cell signaling in the cholesterol-depleted cells. In this context, it is pertinent to point out that cholesterol depletion by 10 mmol/L mβCD treatment reduced the TF-VIIa activation of factor X by ≈50%, whereas it completely attenuated the TF-VIIa—induced cell signaling. The importance of the integrity of cholesterol-rich membrane microdomains in the TF-VIIa—induced cell signaling is better illustrated in cells where membrane cholesterol is sequestered rather than depleted. Filipin, which sequesters the membrane cholesterol by forming cholesterol-filipin complexes,20 impaired the signaling function of TF-VIIa despite the increased TF-VIIa coagulant activity in these cells. The increased TF-VIIa coagulant activity observed in filipin-treated cells could have been the result of increased concentration of cholesterol in membranes patches because filipin treatment is shown to result in cholesterol aggregation in the membrane20 or movement of TF from inactive glycosphingolipid-rich microdomains to active anionic phospholipid region of the membrane.
Although exact underlying mechanisms are not fully known, it is well-established that most of the TF activity at the cell surface is encrypted.26,27 Ultrastructural localization of TF in smooth muscle cells,28 activated endothelial cells,29 and fibroblasts15 showed that a fraction of TF in these cells is associated with caveolae. Based on the increased TF activity and the enlargement of caveolar structures in smooth muscle cells after their detachment, Mulder et al28 speculated that caveolae-associated TF might function as a latent pool of procoagulant activity, which can rapidly be activated at sites in which vessel wall integrity is lost. Recently, Lupu et al30 reported that caveolae may regulate TF activity indirectly through regulation of tissue factor pathway inhibitor activity. These investigators showed caveolin-1 silencing decreased the tissue factor pathway inhibitor activity on activated endothelial cells and thereby increases TF activity by several fold. The data presented in this report show that caveloin-1 silencing had no significant effect on TF coagulant activity at the surface of tumor cells. Similar results were also obtained in lung fibroblasts (data not shown). These data strongly suggest that the caveolae are not negative regulators of TF procoagulant activity, as previously thought. In this context, it may be pertinent to note that, unlike endothelial cells, other cell types synthesize little or no tissue factor pathway inhibitor and most of it was secreted and not associated with the cell surface.31,32 Similarly, MDA-MB-231 tumor cells, the cell model system used in the present study, do not produce tissue factor pathway inhibitor (unpublished data of the authors). Thus, it is unlikely that tissue factor pathway inhibitor plays a role in localizing TF into caveolae or modulating TF-VIIa activity or signaling in our model system.
In contrast to its lack of effect on TF coagulant activity, caveloin-1 silencing significantly reduced the TF-VIIa—induced cell signaling in tumor cells. It is likely that the loss of the structural integrity of caveolae might have caused the relocalization of TF and PAR2 at the cell surface without affecting their functions per se. Such reorganization could have placed the PAR2 beyond the physical reach of TF-VIIa, thus impairing the ability of TF-VIIa to activate PAR2. This hypothesis is supported by our observation that showed lipid raft/caveolar disruption by cholesterol binding drugs or caveolin-1 silencing impaired TF-VIIa cleavage of PAR2 but not trypsin cleavage of PAR2.
A number of G-protein-coupled receptors and their interacting proteins are known to be localized within lipid rafts and caveolin-enriched microdomains.16,21,33,34 Further, caveolins may act as scaffolding proteins to cluster and regulate signaling molecules targeted to the caveolae, such as Src-family tyrosine kinase, epidermal growth factor receptor, and G-protein α subunits.12 Therefore, disruption of lipid rafts or caveolae may also cause uncoupling of G-proteins and other signaling proteins with their membrane receptor. This could explain why lipid raft disruption by cholesterol binding drugs had no effect on trypsin activation of PAR2 at the cell surface but impaired trypsin-induced PAR2-mediated cell signaling.
In summary, the data presented herein demonstrate that TF localization at the cell membrane could influence different functions of TF differently. While caveolar localization of TF had no influence in propagating the procoagulant activity of TF, it is essential in supporting the TF-VIIa—induced cell signaling. The cholesterol content in the plasma membrane and not the structural integrity of the rafts/caveolae appear to influence the TF procoagulant activity, whereas the structural integrity of cholesterol-rich, caveolin-1— enriched microdomains plays a greater role in regulating the TF-VIIa signaling. Our data also suggest that localization of TF and PAR2 in membrane microdomains is critical for TF-VIIa to trigger PAR2-mediated cell signaling, whereas PAR2 localization is less critical for PAR2 peptide-mediated cell signaling. These findings may have several biological implications. This could explain why some cells that express both TF and functional PAR2 may fail to respond to FVIIa.35 Further, compartmentalization of TF-VIIa signaling to the microdomains may facilitate the cells to respond to other proteases in tandem or subsequent to FVIIa exposure because these proteases (or peptide agonists) can access the PAR2 that is not readily accessible to TF-VIIa. Because the cholesterol is one of the major components of the rafts/caveolae, their structure is sensitive to the amount of cholesterol in the membrane, and therefore the concentration of cholesterol in the membrane not only regulates the TF-VIIa coagulant function but also the TF-VIIa—induced cell signaling. Because recent studies suggest that TF-VIIa, in addition to triggering blood coagulation, plays a role in many pathophysiological processes,1,2 cholesterol lowering could provide additional health benefits, in addition to reducing atherosclerosis.
Supplementary Material
Acknowledgments
The authors thank Ki-Hyun Kim for the construction of AP-PAR2 expression vector.
Sources of Funding This work was supported by grants HL 65500 (to U.P.) and HL 58869 (to L.V.M.R.) from the National Institute of Health. S.M. is a recipient of postdoctoral fellowship award from the American Heart Association, Texas Affiliate.
Footnotes
Disclosure
None.
References
- 1.Rao LVM, Pendurthi UR. Tissue factor-factor VIIa signaling. Arterioscler Thromb Vasc Biol. 2005;25:47–56. doi: 10.1161/01.ATV.0000151624.45775.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belting M, Ahamed J, Ruf W. Signaling of the tissue factor coagulation pathway in angiogenesis and cancer. Arterioscler Thromb Vasc Biol. 2005;25:1545–1550. doi: 10.1161/01.ATV.0000171155.05809.bf. [DOI] [PubMed] [Google Scholar]
- 3.Versteeg HH, Spek CA, Peppelenbosch MP, Richel DJ. Tissue factor and cancer metastasis: the role of intracellular and extracellular signaling pathways. Mol Med. 2004;10:6–11. doi: 10.2119/2003-00047.versteeg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petersen LC, Thastrup O, Hagel G, Sorensen BB, Freskgard P-O, Rao LVM, Ezban M. Exclusion of known protease activated receptors in factor VIIa-induced signal transduction. Thromb Haemost. 2000;83:571–576. [PubMed] [Google Scholar]
- 5.Sorensen BB, Freskgard P-O, Nielsen LS, Rao LVM, Ezban M, Petersen LC. Factor VIIa-induced p44/42 mitogen-activated kinase activation requires the proteolytic activity of factor VIIa and is independent of the tissue factor cytoplasmic domain. J Biol Chem. 1999;274:21349–21354. doi: 10.1074/jbc.274.30.21349. [DOI] [PubMed] [Google Scholar]
- 6.Hjortoe GM, Petersen LC, Albrektsen T, Sorensen BB, Norby PL, Mandal SK, Pendurthi UR, Rao LV. Tissue factor-factor VIIa-specific up-regulation of IL-8 expression in MDA-MB-231 cells is mediated by PAR-2 and results in increased cell migration. Blood. 2004;103:3029–3037. doi: 10.1182/blood-2003-10-3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Camerer E, Huang W, Coughlin SR. Tissue factor- and factor X-dependent activation of protease-activated receptor-2 by factor VIIa. Proc Natl Acad Sci USA. 2000;97:5255–5260. doi: 10.1073/pnas.97.10.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bloch K. Cholesterol: Evolution of structure and function. In: Vance DE, Vance J, editors. Biochemistry of Lipids, Lipoproteins and Membranes. Elsevier Science; Amsterdam: 1991. pp. 363–381. [Google Scholar]
- 9.Simons K, Ehehalt R. Cholesterol, lipid rafts, and disease. J Clin Invest. 2002;110:597–603. doi: 10.1172/JCI16390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fielding CJ, Fielding PE. Cholesterol and caveolae: Structural and functional relationships. Biochim Biophys Acta. 2000;1529:210–222. doi: 10.1016/s1388-1981(00)00150-5. [DOI] [PubMed] [Google Scholar]
- 11.Schlegel A, Lisanti MP. The caveolin triad: caveolae biogenesis, cholesterol trafficking, and signal transduction. Cytokine Growth Factor Rev. 2001;12:41–51. doi: 10.1016/s1359-6101(00)00022-8. [DOI] [PubMed] [Google Scholar]
- 12.Okamoto T, Schlegel A, Scherer PE, Lisanti MP. Caveolins a family of scaffolding proteins for organizing preassembled signaling complexes at the plasma membrane. J Biol Chem. 1998;273:5419–5422. doi: 10.1074/jbc.273.10.5419. [DOI] [PubMed] [Google Scholar]
- 13.Liu J, Oh P, Horner T, Rogers RA, Schnitzer JE. Organized endothelial cell surface signal transduction in caveolae distinct from glycosylphosphatidylinositol-anchored. J Biol Chem. 1997;272:7211–7222. doi: 10.1074/jbc.272.11.7211. [DOI] [PubMed] [Google Scholar]
- 14.Burger K, Gimpl G, Fahrenholz F. Regulation of receptor function by cholesterol. Cell Mol Life Sci. 2000;57:1577–1592. doi: 10.1007/PL00000643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mandal SK, Iakhiaev A, Pendurthi UR, Rao LV. Acute cholesterol depletion impairs functional expression of tissue factor in fibroblasts: modulation of tissue factor activity by membrane cholesterol. Blood. 2005;105:153–160. doi: 10.1182/blood-2004-03-0990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chun M, Liyanage UK, Lisanti MP, Lodish H. Signal transduction of a g protein-coupled receptor in caveolae: colocalization of endothelin and its receptor with caveolin. Proc Natl Acad Sci USA. 1994;91:11728–11732. doi: 10.1073/pnas.91.24.11728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 18.Frank PG, Woodman SE, Park DS, Lisanti MP. Caveolin, caveolae, and endothelial cell function. Arterioscler Thromb Vas Biol. 2003;23:1161–1168. doi: 10.1161/01.ATV.0000070546.16946.3A. [DOI] [PubMed] [Google Scholar]
- 19.Klein U, Gimpl G, Fahrenholz F. Alteration of the myometrial plasma membrane cholesterol content with β-cyclodextrin modulates the binding affinity of the oxytocin receptor. Biochem. 1995;34:13784–13795. doi: 10.1021/bi00042a009. [DOI] [PubMed] [Google Scholar]
- 20.Orlandi PA, Fishman PH. Filipin-dependent inhibition of cholera toxin evidence for toxin internalization and activation through caveolae-like domains. J Cell Biol. 1998;141:905–915. doi: 10.1083/jcb.141.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oh P, Schnitzer JE. Segregation of heterotrimeric G proteins in cell surface microdomains. G (q) binds caveolin to concentrate in caveolae, whereas G (i) and G (s) target lipid rafts by default. Mol Biol Cell. 2001;12:685–698. doi: 10.1091/mbc.12.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sargiacomo M, Sudol M, Tang Z, Lisanti MP. Signal transducing molecules and glycosyl-phosphatidylinositol-linked proteins form a caveolin-rich insoluble complex in MDCK cells. J Cell Biol. 1993;122:789–807. doi: 10.1083/jcb.122.4.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song KS, Li S, Okamoto T, Quilliam LA, Sargiacomo M, Lisanti MP. Co-purification and direct interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains. Detergent-free purification of caveolae membranes. J Biol Chem. 1996;271:9690–9697. doi: 10.1074/jbc.271.16.9690. [DOI] [PubMed] [Google Scholar]
- 24.Huang C, Hepler JR, Chen LT, Gilman AG, Anderson RGW, Mumby SM. Organization of G proteins and adenylyl cyclase at the plasma membrane. Mol Biol Cell. 1997;8:2365–2378. doi: 10.1091/mbc.8.12.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson RGW. The caveolae membrane system. Annu Rev Biochem. 1998;67:199–225. doi: 10.1146/annurev.biochem.67.1.199. [DOI] [PubMed] [Google Scholar]
- 26.Rao LVM, Pendurthi UR. Tissue factor on cells. Blood Coag Fibrin. 1998;9(suppl 1):S27–S35. [PubMed] [Google Scholar]
- 27.Bach RR. Tissue factor encryption. Arterioscler Thromb Vasc Biol. 2006;26:456–461. doi: 10.1161/01.ATV.0000202656.53964.04. [DOI] [PubMed] [Google Scholar]
- 28.Mulder AB, Smit JW, Bom VJJ, Blom NR, Ruiters MHJ, Halie R, van der Meer J. Association of smooth muscle cell tissue factor with caveolae. Blood. 1996;88:1306–1313. [PubMed] [Google Scholar]
- 29.Mulder AB, Smit JW, Bom VJJ, Blom NR, Halie MR, van der Meer J. Association of endothelial tissue factor and thrombomodulin with caveolae. Blood. 1996;88:3667–3670M. [PubMed] [Google Scholar]
- 30.Lupu C, Hu X, Lupu F. Caveolin-1 enhances tissue factor pathway inhibitor exposure and function on the cell surface. J Biol Chem. 2005;280:22308–22317. doi: 10.1074/jbc.M503333200. [DOI] [PubMed] [Google Scholar]
- 31.Bajaj MS, Birktoft JJ, Steer SA, Bajaj SP. Structure and biology of tissue factor pathway inhibitor. Thromb Haemost. 2001;86:959–972. [PubMed] [Google Scholar]
- 32.Pendurthi UR, Rao LVM, Williams JT, Idell S. Regulation of tissue factor pathway inhibitor expression in smooth muscle cells. Blood. 1999;94:579–586. [PubMed] [Google Scholar]
- 33.de Weerd WF, Leeb-Lundberg LM. Bradykinin sequesters B2 bradykinin receptors and the receptor-coupled Galpha subunits Galphaq and Galphai in caveolae in DDT1 MF-2 smooth muscle cells. J Biol Chem. 1997;272:17858–17866. doi: 10.1074/jbc.272.28.17858. [DOI] [PubMed] [Google Scholar]
- 34.Feron O, Belhassen L, Kobzik L, Smith TW, Kelly RA, Michel T. Endothelial nitric oxide synthase targeting to caveolae. J Biol Chem. 1996;271:22810–22814. doi: 10.1074/jbc.271.37.22810. [DOI] [PubMed] [Google Scholar]
- 35.Camerer E, Rottingen J-A, Gjernes E, Larsen K, Skartlien AH, Iversen J-G, Prydz H. Coagulation factors VIIa and Xa induce cell signaling leading to up-regulation of the egr-1 gene. J Biol Chem. 1999;274:32225–32233. doi: 10.1074/jbc.274.45.32225. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.