

Abstract
A close structural analogue of bryostatin 1, which differs from bryostatin 1 only by the absence of the C30 carbomethoxy group (on the C13 enoate of the B-ring), has been prepared by total synthesis. Biological assays reveal a crucial role for substitution in the bryostatin 1 A-ring in conferring those responses which are characteristic of bryostatin 1 and distinct from those observed with PMA.
The bryostatins are a family of highly complex marine natural products originally isolated by Pettit using activity against P-388 leukemia cells to guide the fractionation of the crude extracts.1 Of the twenty known members, by far the most extensively studied is bryostatin 1 (1).2 Bryostatin 1 has been shown to exhibit a remarkable profile of biological effects, including potent activity against a number of cancers. Bryostatin 1 has also been shown to stimulate the immune system,3 which stands in contrast to many established oncolytic agents and which may play a role in the observed antitumor effects. Recently, bryostatin 1 has been shown to exhibit profound effects on memory in animal models4 and to have significant activity against Alzheimer's disease in transgenic mouse models.5 An even more recent exciting finding on the neurological effects of this agent is its ability to reverse damage from stroke and to effect neural growth and repair.6
Bryostatin 1 has also shown remarkable synergistic effects with a number of established oncolytic agents, including vincristine, paclitaxel, gemcitibine, and flavopyridol.7 These intriguing synergies have been shown to be quite complex in that the results depend heavily upon dosing schedules (including which agent is administered first) and methods of administration. In some cases, studies focusing on the underlying mechanisms for the synergies have been performed.8
Although the precise mechanisms by which bryostatin 1 leads to these observed biological results have not been rigorously established, it is thought that they are a consequence of binding to the C1 domains of PKC isozymes,9 and to other C1 domain containing proteins such as the RasGRPs, the chimerins, and the Munc13 proteins.10 Thus bryostatin 1 is well known to have very high binding affinity for the PKC's (Ki = 1.35 nM with PKCα) and these proteins play critical roles in cell signaling processes relevant to cellular events including proliferation, differentiation, motility, adhesion, and apoptosis.11 In addition to the natural activators of PKC (diacylglycerols, DAG's) numerous other activators with high affinity for PKC are known; the most thoroughly studied of these are the phorbol esters. However, whereas bryostatin 1 binds PKC (Ki with PKCα = 1.35 nM) with similar affinity to that of phorbol-12-myristate-13-acetate (PMA, Ki = 1.17 nM), the events induced subsequent to binding are quite different for the two agents.12 In particular, bryostatin 1 is not tumor promoting and functionally antagonizes many of the responses induced by the phorbol esters.
In an effort to identify the mechanisms responsible for the unique activity of bryostatin 1, we are attempting to determine, through chemical synthesis, the structural features of bryostatin that are responsible for its unique biological profile. Toward this end, we have developed and reported on powerful enabling methodology for the construction of pyran rings bearing flexible and malleable substitution precisely where it is needed for the preparation of a variety of bryostatin analogues13 and applied this to the synthesis of the bryopyran core structure.14
In an earlier report, we described the synthesis of three agents (2-4, Figure 2) based on the bryostatin tris-pyran core structure.15 These close mimics of the bryostatin 1 structure were found to have very high affinity for PKC (Ki with PKCα = 0.70-1.05 nM) but to be similar to PMA in terms of the results of both proliferation and attachment assays with U937 leukemia cells, a system where exposure to bryostatin 1 and PMA has been established to give very different biological endpoints and where bryostatin 1 antagonizes the PMA response.16 Characterization in multiple additional assays where bryostatin 1 is distinguished from phorbol esters in terms of biological response further supports the initial finding that these bryostatin analogues largely function as phorbol ester mimics (unpublished observations). No dependence on the nature of the C20 substituent was seen. Thus, although these compounds are close structural analogues of bryostatin 1, they are close functional analogues of PMA.
Figure 2.

Structures of Previous Bryopyran Analogues
In this paper, we report studies which reveal that the A-ring functionality of bryostatin 1 is critical in conferring bryostatin-like biological responses as opposed to those characteristic of the tumor-promoting phorbol esters. This was accomplished through total synthesis and biological evaluation of the “almost bryostatin” analogue 12, which lacks only the C30 carbomethoxy group of bryostatin 1 itself.
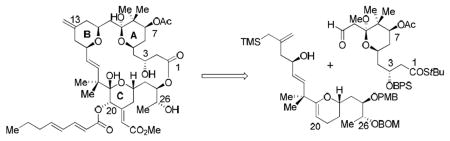
The synthetic plan uses our pyran annulation methodology13 to join preformed A-ring and C-ring subunits with concomitant formation of the B-ring, according to an overall strategy which has been suggested previously.14, 17 Although superficially similar to the route used previously to prepare 2-4, this synthesis reverses the roles of the coupling partners for the critical pyran annulation. The resulting differences then track back to a totally different synthetic route for the A-ring component than that utilized previously, as well as significant differences in the functionality present after pyran annulation.
As shown in Scheme 1, reaction of the A-ring aldehyde 5 with hydroxy allylsilane 6,17 using TMSOTf in ether, gave the desired tricyclic adduct 7.18 Oxidative functionalization of the C-ring glycal afforded ketone 8, which was converted to enoate 9 by an aldol-elimination sequence.19 Luche reduction and acylation of the resulting alcohol provided 10 as essentially a single diastereomer. The correct C20 and C34 stereochemistry was confirmed by the observation of a strong nOe between the respective protons at these positions.
Scheme 1.

Synthesis of the “Almost Bryostatin” Bryopyran 12.
This intermediate was readied for macrolactonization by a sequence designed to avoid conducting this reaction on a dihydroxy acid, which has proven problematic. Removal of the BPS group at C3 proved necessary to allow for hydrolysis of the thiolester using LiOH and H2O2. Attempted hydrolysis with the BPS protected thiolester was slow and resulted in the hydrolysis of the C7 acetate as well. The resulting hydroxy acid was reprotected at C3 using TESCl. Following removal of the PMB group at C25, macrolactonization using the Yamaguchi20 conditions afforded the desired macrolactone in 62% yield. Global deprotection (hydrolysis of two mixed ketals, C3TES, and BOM ether) using the Lipshutz21 method (LiBF4 in aqueous acetonitrile) then gave the desired “almost bryostatin” analogue 12.
Initial assay of this analogue for PKC binding showed, as expected, high affinity for PKCα like that shown by 1 (Ki = 0.52 ± 0.06 nM, average of three experiments). Initial assays for function used the U937 leukemia proliferation and attachment assays previously mentioned in connection with analogues 2-4.16 The results for these assays are shown in Figures 3 and 4.
Figure 3.
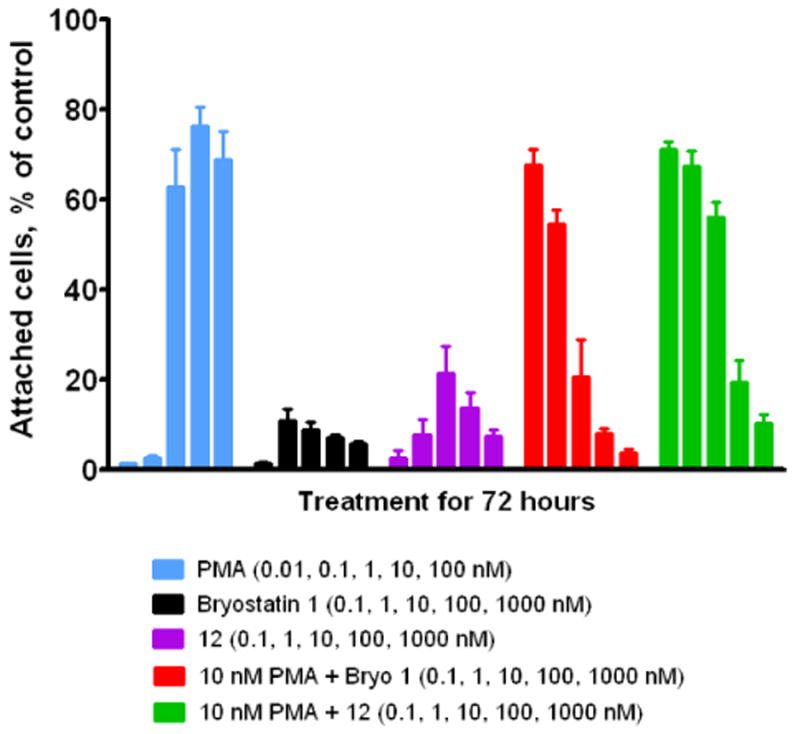
Effect of Analogue 12 on U937 Cell Attachment
Figure 4.

Inhibition of Proliferation Assay for Analogue 12
In the attachment assay (Figure 3), PMA induces attachment while bryostatin 1 shows a much diminished effect. Moreover, when both agents are administered together, bryostatin 1 blocks the effect of the phorbol ester in a dose-dependent manner. In marked contrast to analogue 4, the response of agent 12 in the attachment assay approaches that of bryostatin 1 itself. Analogue 12 can be seen even to display the dose-dependent biphasic response characteristic of exposure to bryostatin 1. In addition, analogue 12, like bryostatin 1, is a functional antagonist of PMA and blocks the effect of the phorbol ester when the two agents are administered together.
The results with analogue 12 in the proliferation assay (Figure 4) are likewise very similar to those for bryostatin 1. In this assay, bryostatin 1 is not strongly antiproliferative, whereas the phorbol ester PMA is. Moreover, bryostatin 1 is able to block the effect of PMA in a dose dependent manner. Both of these aspects of bryostatin 1 induced biological response are captured by 12.
Taken together with the previous results for analogue 4, these results clearly show that: (1) the C30 carbomethoxy group is not essential to obtain bryostatin-like biological responses with these analogues, and (2) the structure in the C7-C9 region of the A-ring is critical in conferring bryostatin-like biological responses as opposed to those characteristic of the tumor-promoting phorbol esters. Practical implications are that bryostatin analogues simplified in the B ring may provide effective biological surrogates of bryostatin 1 for therapy, whereas bryologues lacking appropriate substitution in the A ring instead fall within the broad family of potent PKC activators but do not capture the special behavior of bryostatin 1.
For a long time, most other work on assessment of synthetic bryostatin analogues has focused on binding and simple measures of biological potency, largely skipping over the nature of the biological responses induced by the analogues.22 This has led to the view that the A- and B-rings of the structure serve merely as “spacer domains” which simply hold other groups in the molecule in the proper positions.23 Clearly this is not the case when function is considered. Our results show that analogues such as 2-4, although based on the bryostatin platform, appear to living cells to be much like phorbol esters, and that bryostatin-like function can be restored upon inclusion of appropriate A-ring functionality.
The critical structural features of bryostatin 1 primarily responsible for its unique behavior are now reduced to substitution at just three carbons. Efforts to elucidate the role of substituents at these positions on function are in progress.
Supplementary Material
Experimental procedures, assay results, and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
Figure 1.

Structure of Bryostatin 1
Acknowledgments
Financial support was provided by the NIH through grant GM28961 and through the Intramural Research Program, CCR, NCI, NIH.
Contributor Information
Gary E. Keck, University of Utah, Department of Chemistry, 315 South 1400 East, RM 2020, Salt Lake City, Utah 84112.
Yam B. Poudel, University of Utah, Department of Chemistry, 315 South 1400 East, RM 2020, Salt Lake City, Utah 84112
Dennie S. Welch, University of Utah, Department of Chemistry, 315 South 1400 East, RM 2020, Salt Lake City, Utah 84112.
Matthew B. Kraft, University of Utah, Department of Chemistry, 315 South 1400 East, RM 2020, Salt Lake City, Utah 84112
Anh P. Truong, University of Utah, Department of Chemistry, 315 South 1400 East, RM 2020, Salt Lake City, Utah 84112.
Jeffrey C. Stephens, University of Utah, Department of Chemistry, 315 South 1400 East, RM 2020, Salt Lake City, Utah 84112
Noemi Kedei, LCBG, Center for Cancer Research, NCI, NIH, Bethesda, MD 2082.
Nancy E. Lewin, LCBG, Center for Cancer Research, NCI, NIH, Bethesda, MD 2082
Peter M. Blumberg, LCBG, Center for Cancer Research, NCI, NIH, Bethesda, MD 2082
References
- 1.Pettit GR, Herald CL, Douobek DL, Herald DL. J Am Chem Soc. 1982;104:6846–6848. [Google Scholar]
- 2.Hale KJ, Hummersome MG, Manaviazar S, Frigerio M. Nat Prod Rep. 2002;19:413–453. doi: 10.1039/b009211h. [DOI] [PubMed] [Google Scholar]
- 3.Koutcher JA, Motwani M, Zakian KL, Li XK, Matei C, Dyke JP, Ballon D, Yoo HH, Schwartz GK. Clin Cancer Res. 2000;6:1498–1507. [PubMed] [Google Scholar]
- 4.(a) Alkon DL, Epstein H, Kuzirian A, Bennett MC, Nelson TJ. Proc Natl Acad Sci U S A. 2005;102:16432–16437. doi: 10.1073/pnas.0508001102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sun MK, Alkon DL. European Journal of Pharmacology. 2005;512:43–51. doi: 10.1016/j.ejphar.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 5.(a) Etcheberrigaray R, Tan M, Dewachter I, Kuiperi C, Van der Auwera I, Wera S, Qiao L, Bank B, Nelson TJ, Kozikowski AP, Van Leuven F, Alkon DL. Proc Natl Acad Sci U S A. 2004;101:11141–11146. doi: 10.1073/pnas.0403921101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Alkon DL, Sun MK, Nelson TJ. Trends Pharmacol Sci. 2007;28:51–60. doi: 10.1016/j.tips.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 6.Sun MK, Hongpaisan J, Nelson TJ, Alkon DL. Proc Natl Acad Sci U S A. 2008;105:13620–13625. doi: 10.1073/pnas.0805952105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Mohammad RM, Wall NR, Dutcher Julie A, Al-Katib AM. Clin Cancer Res. 2000;6:4950–4956. [PubMed] [Google Scholar]; (b) Koutcher JA, Motwani M, Zakian KL, Li XK, Matei C, Dyke JP, Ballon D, Yoo HH, Schwartz GK. Clin Cancer Res. 2000;6:1498–1507. [PubMed] [Google Scholar]; (c) El-Rayes BF, Gadgeel S, Shields AF, Manza S, Lorusso P, Philip PA. Clin Cancer Res. 2006;12:7059–7062. doi: 10.1158/1078-0432.CCR-06-1419. [DOI] [PubMed] [Google Scholar]; (d) Roberts JD, Smith MR, Feldman EJ, Cragg L, Millenson MM, Roboz GJ, Honeycutt C, Thune R, Padavic-Shaller K, Carter WH, Ramakrishnan V, Murgo AJ, Grant S. Clin Cancer Res. 2006;12:5809–5816. doi: 10.1158/1078-0432.CCR-05-2730. [DOI] [PubMed] [Google Scholar]
- 8.(a) Wang S, Wang Z, Grant S. Mol Pharmacol. 2003;63:232–242. doi: 10.1124/mol.63.1.232. [DOI] [PubMed] [Google Scholar]; (b) Wang S, Wang Z, Dent P, Grant S. Blood. 2003;101:3648–3657. doi: 10.1182/blood-2002-09-2739. [DOI] [PubMed] [Google Scholar]
- 9.Reyland ME, Insel PA, Messing RO, Dempsey EC, Newton AC, Mochly-Rosen D, Fields AP. Am J Physiol Lung Cell Mol Physiol. 2000;279:429–438. doi: 10.1152/ajplung.2000.279.3.L429. [DOI] [PubMed] [Google Scholar]
- 10.Brose N, Rosenmund C. J Cell Sci. 2002;115:4399–4411. doi: 10.1242/jcs.00122. [DOI] [PubMed] [Google Scholar]
- 11.Parker PJ, Dekker LV, editors. Protein Kinase C. R G Landes; Austin: 1997. Molecular Biology Intellegence Unit. [Google Scholar]
- 12.Blumberg PM, Pettit GR. In: New Leads and Targets in Drug Research. Krosgaard–Larsen P, Christensen CB, Kodof H, editors. Munksgaard; Copenhagen: 1992. pp. 273–285. [Google Scholar]
- 13.Keck GE, Covel JA, Schiff T, Yu T. Org Lett. 2002;4:1189–1192. doi: 10.1021/ol025645d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Keck GE, Truong AP. Org Lett. 2005;7:2149–2152. doi: 10.1021/ol050511w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Keck GE, Truong AP. Org Lett. 2005;7:2153–2156. doi: 10.1021/ol050512o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keck GE, Kraft MB, Truong AP, Li W, Sanchez CC, Kedei N, Lewin N, Blumberg PM. J Am Chem Soc. 2008;130:6660–6661. doi: 10.1021/ja8022169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vrana JA, Saunders AM, Srikumar PC, Grant S. Differentiation. 1998;63:33–42. doi: 10.1046/j.1432-0436.1998.6310033.x. [DOI] [PubMed] [Google Scholar]
- 17.(a) Keck GE, Welch DS, Vivian PK. Org Lett. 2006;8:3667–3670. doi: 10.1021/ol061173h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Keck GE, Welch DS, Poudel YB. Tetrahedron Lett. 2006;47:8267–8270. doi: 10.1016/j.tetlet.2006.09.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Unreacted aldehyde 5 and the O-trimethylsilyl derivative of 6 were isolated in 35 % and 27 % yields respectively and subsequently recyled.
- 19.Use of 1.1 equiv of LDA gave incomplete conversion of ketone 8 to the desired aldol adduct. Attempts to increase conversion resulted in reaction also occurring at the C7 acetate.
- 20.Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull Chem Soc Jpn. 1979;52:1989–1993. [Google Scholar]
- 21.Lipschutz BH, Harvey DF. Synth Commun. 1982;14:267–277. [Google Scholar]
- 22.(a) Wender PA, Verma VA. Org Lett. 2008;10:3331–3334. doi: 10.1021/ol801235h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wender PA, DeChristopher BA, Schrier AJ. J Am Chem Soc. 2008;130:6658–6659. doi: 10.1021/ja8015632. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wender PA, Debrabander J, Harran PG, Jimenez JM, Koehler MFT, Lippa B, Park CM, Shiozaki M. J Am Chem Soc. 1998;120:4534–4535. [Google Scholar]; Note also references therein.
- 23.Wender PA, Baryza JL, Hilinski MK, Horan JC, Kan C, Verma VA. In: Drug Discovery Research: New Frontiers in the Post-Genomic Era. Huang Z, editor. Wiley-VCH; Hoboken, NJ: 2007. pp. 127–162. [Google Scholar]; Note also the references cited in reference 22 and references therein.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures, assay results, and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.