

Abstract
The nuclear factor-κB (NF-κB) family of transcription factors plays a central role in numerous physiological processes including development, cell survival, immunity and inflammation. We generated a series of stable clonal lines in mouse embryonic fibroblasts carrying NF-κB-GFP plasmid as a reporter. These cell lines were selected by flow cytometry for their high responsiveness to tumor necrosis factor (TNFα) or lipopolysaccharide (LPS), two classic NF-κB inducing stimuli. Although all clones were generated from the same parental cell line, they each had a distinctive pattern of response to NF-κB stimuli. While exhibiting distinct profiles with regard to the GFP reporter, analysis of endogenous NF-κB downstream targets did not always show the same variability. This suggests that in the absence of confirmation of the signaling outcomes using endogenous outputs, considerable caution must be exercised in the interpretation of data using stable reporter systems.
Keywords: NF-κB, GFP, reporter systems, Cell signaling, Toxoplasma
Introduction
NF-κB belongs to a family of transcription factors formed by dimerization between members of the Rel containing proteins (RelA, c-Rel, RelB, p105/p50, p100/p52) and bind to a common DNA sequence known as κB sites [15, 26]. NF-κB plays a crucial role in immune response, apoptosis and cell proliferation [4, 25]. Activation of the NF-κB pathway, triggered by microbial or viral infections, exposure to mitogens, or proinflammatory cytokines, results in the activation of the transcription of a number of key cytokines and cytokine receptors [5].
Inactive forms of NF-κB (the canonical p65/p50 and other heterodimers of the Rel family) are held captive in the cytoplasm by binding to the inhibitors of NF-κB (IκBα, IκBβ, IκBε) [15]. The basic activation pathway of NF-κB is achieved when, in response to various stimuli, the IkappaB kinase (IKK) signaling complex is activated by phosphorylation. Activated IKK in turn phosphorylates IκBα (on Ser 32/36 for IκBα) resulting in its subsequent degradation and the release of NF-κB. Freed NF-κB translocates to the nucleus, where it binds to κB sequences and activates the transcription of its target genes (Fig. 4A) [3, 15]. Two classic activators of NF-κB pathway are TNFα, a potent pro-inflammatory cytokine and LPS, a cell wall component of gram negative bacteria. Their signals are transmitted to IKK through a series of adaptor molecules by binding to TNFα receptor [11] and toll like receptor 4 (TLR4) respectively [23].
Figure 4.

Detection of NF-κB activation using the transient luciferase reporter system: Unmodified MEF and reporter clones representing the groups responding by flow cytometry were transfected for 40 hours with the NF-κB-luc reporter system plasmid and β-galactosidase expression plasmid as transfection control. The cells were unstimulated (UN) or stimulated 2 hours with TNFα (20ng/ml) or 5 hours with LPS (100ng/ml) and whole-cell lysates were subjected to luciferase assays to measure luciferase expression levels under NF-κB control. Luciferase activity off the NF-κB-luc plasmid was normalized to the corresponding β-galactosidase activity off the co-transfected plasmid. The mean luminescence from three independent experiments while revealing differential responses, exhibit virtually identical response patterns when comparing TNFα and LPS stimulated cells. This is in marked contrast to the GFP expression profiles in response to these stimuli off the NF-κB-GFP reporter.
Considerable work has been done using endogenous pathway analysis and reporter systems to elucidate signaling of the NF-κB pathway and finding targets that allows manipulation of the pathway to control cell proliferation and tumorgenesis (reviewed in [28]). The most common of these are reporter plasmids, in which a reporter gene is expressed under the control of a promoter containing multiple NF-κB binding sites as surrogate for transcriptional output [13, 14]. Stable cell lines expressing NF-κB GFP reporter plasmids have become commercially available (eg from System Biosciences, Mountain View, CA). These can potentially be used in screening for different inhibitors or activators of NF-κB as stable reporter systems are amenable to high throughput screens [28]. We developed such “in house” cell lines with the aim of using them in a high throughput screen to identify genes in the protozoan parasite Toxoplasma gondii responsible for NF-κB activation [20]. T.gondii is an obligate intracellular parasite that can infect any warm blood animal [27] and is known to manipulate multiple intracellular signaling pathways of the host. One of those is the NF-κB pathway, whose activation is required for inhibition of host apoptosis [24]. We have previously shown that activation of NF-κB is partially achieved by phosphorylation of a subset of host IκBα [21]. However, it soon became clear that manipulation of this pathway is a complex process involving multiple elements[22], which lead us to develop the reporter cell line system to screen for NF-κB defective mutants of the parasite.
The most widely used reporters in live cells for such applications, are fluorescent proteins (e.g. GFP, YFP etc) and firefly Luciferase. Green fluorescent protein (GFP) from the jellyfish Aequorea victoria and its various variants [18] now form a cornerstone of both in vivo imaging of cells and tissues and in vitro fluorescence labeling. GFP-fusion proteins can be used to analyze the expression, localization, movement, interaction of proteins in addition to studying enhancers and promoters [7, 9]. GFP is unique in that the GFP fluorophore spontaneously forms intracellularly [12]. Therefore, the emitted fluorescence intensity provides a direct readout of GFP protein levels [6] that can be measured at the single-cell level without any processing steps. This makes GFP reporting a very convenient method for monitoring expression of inducible reporters using flow cytometry [2]. In contrast, bioluminescent reporters, based on Photinus pyralis firefly luciferase [8] rely on their enzymatic activity. The level of luciferase in a cell lysate is quantitated using a luminometer that measures the light produced when luciferase catalyzes its substrate luciferin [1]. Luciferase based system, which are highly sensitive, are frequently used in transient transfection studies to analysis promoters. An important assumption in these studies is that the levels of the enzymatic activity of luciferase protein are directly related to the transcriptional activity of the promoter under study. This is not always the case but can serve as a good first approximation.
In this study, we developed several stable cell lines to study NF-κB activation pathway using a GFP reporter system. These clonal cell lines showed distinct patterns of GFP expression in response to NF-κB stimulation by two classic stimuli despite being developed from the same parental cell population. To address this variability, and validate the reporter system, we examined both endogenous markers and alternative transient (NF-κB-luc) reporter systems. Surprisingly the variability observed by the NF-κB-GFP reporter was not observed with either the endogenous markers nor NF-κB-luc, suggesting that stably-integrated NF-κB-GFP may be less than an honest reporter in individual clonal lines. This provides a cautionary note in the interpretation of reporter systems data.
Material and Methods
Cell lines, plasmids, antibodies and chemical reagents
Wild type mouse embryonic fibroblast (WT MEF) were maintained in alpha minimum essential medium (αMEM) (Gibco-Invitrogen, Rockville, MD) supplemented with 14% fetal bovine serum (FBS), (Gemini Bio-Products, Woodland, CA), 100 U ml 1 penicillin, 100 μg ml 1 streptomycin and 2 mM L-glutamine (Gibco BRL, Rockville, MD). OptiMEM medium for transfection was from Gibc NF-κB-hrGFP reporter plasmid was purchased from Stratagene (La Jolla, CA). pSVβgal was from Promega (Madison, WI). NF-κB-Luc reporter plasmid (Mercury Pathway) was from Clontech Laboratory (Palo Alto, CA). The polyclonal antibodies to phospho-IκBα (sc-21869-R), IκBα (sc-371) and IKKα/β (sc-7607) were from Santa Cruz Biotechnology (Santa Cruz, CA). The goat anti-rabbit-HRP secondary antibody was from Jackson Laboratories (Bar Harbor, ME).
Murine recombinant TNFα was from R&D Systems (Minneapolis, MN). E. coli Lipopolysaccharide (LPS) (# L4391) was from Sigma-Aldrich (St. Louis, MO).
Stable NF-κB reporter cell lines construction
Reporter cell lines were generated by stably transfecting mouse fibroblast with the pNF-κB-hrGFP plasmid containing the hygromycin resistance gene. After hygromycin selection (150μg/ml), surviving cells where treated with TNFα (5ng/ml) overnight then sorted using Fluorescence Activated Cell Sorting (FACS) for high GFP expressing. Sorted cells were allowed to recover then individual clones were isolated by cloning ring technique [10].
Flow cytometry and cell sorting
Cells grown overnight were left untreated or treated with TNFα (5ng/ml) or LPS (100ng/ml) or infected with the T. gondii RH strain (MOI 5:1) overnight. Cells were then harvested, following trypsinization, in 1X PBS for flow cytometry analysis or in 1X PBS supplemented with 2% FBS for cell sorting. Cells were analyzed using FACSCalibur (Becton Dickinson) and sorted using MoFlo Cell Sorter (Dako Cytomation). Sorted cells were collected in 50% FBS and plated overnight in 15 % FBS containing αMEM medium then plated at low density to isolate individual colonies, which were ring cloned.
Luciferase reporter assay
Cells were plated in 6 well plates (Falcon) then transiently transfected at 80% confluency, with 0.5μg NF-κB-luc reporter plasmid along with 0.3μg SV40-β-galactosidase expression construct (pSVβgal), for an internal control, and 1.5μg of the empty vector, pCR3.1 as a filler (from Invitrogen Life Technologies) using Lipofectamine LTX and Plus Reagent (from Invitrogen Life Technologies) following the manufacturer’s protocol. Briefly, DNA was mixed with 200 μl of serum free OptiMEM and 2.5 μl of Plus Reagent followed by 5 minutes incubation. Lipofectamine LTX was added and complexes allowed for form for 20 minutes before adding to cells covered with 800 μl of OptiMEM. After four hours, transfection media was replaced by 10% FBS containing αMEM medium. Following overnight incubation, cells were transferred into two 96 well plates and selected wells were left untreated or stimulated with LPS (100ng/ml) for 5h or with TNFα (20ng/ml) for 2 hours before lysis. Cell lysates were examined for luciferase activity using LucLite kit from PerkinElmer or for β-galactosidase activity using 2-NitrophenylB-D-Galactopyranoside (ONPG) (# 73660) from Sigma. The luciferase activity in each reaction was normalized with respect to the corresponding β-galactosidase activity and expressed as relative luc activity.
Western blot analysis
Cells were plated into 6 well plates and left untreated or stimulated with LPS(100ng/ml) for 24h or with TNFα (20ng/ml) for 6 minutes before harvesting in 2x SDS-PAGE sample buffer [17] and subjected to polyacrylamide gel electrophoresis on 10% gels using Bio-Rad Mini-PROTEAN II apparatus. The gels were transferred onto nitrocellulose (Pall, East Hill) using a semi-dry blotting apparatus (BioRad) as recommended by the manufacturer. Transferred proteins were visualized using Ponceau S (Sigma) and blocked for 1 hour using TBS-Tween (TBS with 0.1% Tween 20) containing 5% dry milk. Incubations with the P-IκB primary antibody (1:500) was conducted overnight at 4°C in TBS-Tween containing 5%BSA. Blots were washed three times in TBS-Tween-BSA for 15 minutes each prior to the addition of secondary antisera (1:2000) conjugated to horseradish peroxidase (HRP) for 2 hours. Following three washes as above, the immunoreactive bands were visualized using enhanced chemiluminescence [19]. IKK was used for normalization of loading. The antibodies against IκB and IKK were diluted 1:1000 in PBS-0.2% Tween with 5% Milk and incubated for 1hour.
RT-PCR
Cells were grown overnight in 10cm dishes then left untreated or stimulated with TNFα (5ng/ml) or LPS (100ng/ml) overnight. Total RNA was isolated using mini RNeazy kit from Qiagen Inc. cDNA was prepared from 1 μg of total RNA using reverse transcription system from Promega. 1.5 μl of the resulting cDNA was used to perform PCR using the Reaction Ready Hot Start “sweet” PCR (PA-007) and primers for IL-6, CXCL-1 and β-actin (#PPM03015A-22, PPM03058A-200 and PPM02945A-200 respectively) from Superarray. The PCR program used was: 95°C for 15 minutes followed by 24 cycles (β-actin) or 27 cycles (IL-6 and CXCL-1) of 95°C for 15 seconds, 55°C for 30 seconds and 72°C for 30 seconds. β-actin -specific primers was used for loading control.
Results
GFP reporter responds differently to different NF-κB stimuli
Low efficiency of transfection usually hinders use of reporter plasmids in some cell types (e.g. MEF) especially when combined with a reporter with low sensitivity like GFP. To overcome this problem we generated stable reporter cell lines to study mechanisms of regulation of NF-κB pathway. These reporter cell lines were established to allow for a rapid screening of mutants of the Toxoplasma gondii parasites defective in activating NF-κB in the host cells. We have previously shown that T. gondii inhibits apoptosis in an NF-κB dependent manner in host fibroblasts [20, 24]. A stable NF-κB-GFP reporter cell line (PHS2) previously developed in our lab for screening of mutants of the T. gondii, lost its response to NF-κB activation over time, despite repeated selection of positive clones (data not shown). To generate new reporter cell lines we stably transfected mouse fibroblast cells (MEF) with the reporter plasmid pNF-κB-hrGFP which has a GFP reporter gene under the control of 4 κB binding sites and a hygromycin resistance gene. Hygromycin selected cells were stimulated with TNFα [9] (R3 in Fig. 1C) or LPS (LN#) and the high responders were sorted by FACS. Eighty individual clones were isolated by ring cloning and out of those; thirty clones were tested for response to TNFα and LPS by flow cytometry analysis. Interestingly, a panel of phenotypes, represented by clones in Figure 2A, were isolated. Each clone had distinct intensity and patterns of GFP expression (Fig. 2), ranging from high response to TNFα activation with very low to LPS (e.g. TNh1) to almost equal expression to both stimuli (e.g. LN9), regardless of the initial stimulus used for sorting. In addition, several non-responsive clones were isolated (e.g. LN16). Accordingly, the responsive cell lines were divided into three groups as described in Figure 2B: Group I (TNF+++/LPS-): High TNFα responders with low LPS response, Group II (TNF+++/LPS++): High TNFα response with intermediate LPS response (with regard to intensity, number of cells or both) and finally Group III ( TNF+++/LPS+++): those that responded equally to both stimuli. We picked a representative clone from each group for further analysis: TNh1, selectively TNFα responsive but LPS non-responsive (group I), LN4 responds better to TNFα than LPS (group II) and LN9 with a comparable response to both TNFα and LPS (group III).
Figure 1.

Sorting of NF-κB-hrGFP stable cell lines: Parental MEFs (A), untreated NF-κB-hrGFP stably transfected cells (B) or TNFα (5ng/ml overnight) treated cells (C) were analyzed by FACS. Cells that responded to TNFα stimulation by increased GFP expression (R3) were selected and plated for subsequent selection of individual clones. Inset is a histogram representation of the scatter plot results.
Figure 2. Flow cytometric analysis of NF-κB-GFP reporter cell clones.

A. Stable clonal cell lines selected following the initial screen for GFP expression using either TNFα (pink trace, TN# or TNh# series) or LPS (orange trace, LN# series) as the stimulating agent were tested for background GFP fluorescence (black trace, unstimulated-UN) or following overnight stimulation with TNFα (5ng/ml) or LPS (100ng/ml). The percentage of cells expressing GFP under each condition is indicated as an inset for each clone.
B. The distribution of GFP expression in individual clones was grouped according to the response to TNFα or LPS. The number of clones characterized in each group are indicated.
C. A single representative of each responding group was selected for further analysis. These clonal cell lines TNh1 (Group I), LN9 (Group II) and LN4 (Group III) were subjected to TNFα or LPS treatment or left untreated. The mean response, presented as percentage of cells expressing GFP, across three experiments. Error bars represent standard deviation from the mean.
The fact that a single transfection gave rise to several lines with differential responses suggested either that the NF-κB activation pathway has been altered by the random insertion of the reporter plasmid or that the individual clones possess a normal activation profile that is not honestly reported by the GFP reporter system.
The insertion site of the plasmid is an important factor that is reflected in the high background (high GFP expression in untreated cells) in LN4 and LN9 (Fig. 2). This may result from the insertion of the plasmid in a transcriptionally active part of the genome. These clones are still useful since they respond nicely to the used stimuli despite a high background.
At this point, we felt that this apparent differential response to TNFα and LPS could be used to identify both common and unique components of signaling pathways and stimuli resulting in NF-κB activation. To test this possibility, we used the three selected clones to monitor NF-κB activation by the protozoan Toxoplasma gondii (strain RH). As seen in Figure 3, each clone showed a different pattern of response to Toxoplasma infection. TNh1 did not respond to the NF-κB activation by the parasite, while LN9 showed a response similar to that of LPS and LN4 response was less than that seem with TNFα and LPS. We know from earlier studies in our lab and others [16, 20] that T. gondii activates NF-κB. The results obtained in Figure 3 suggested that using reporter cells lines to screen for parasite mutants defective in NF-κB activation depends primarily on the clone used for this purpose.
Figure 3.

Effect of Toxoplasma gondii (Strain RH) infection of reporter cell lines. Reporter clones TNh1, LN9 and LN4, representing Groups I, II and III respectively were left unstimulated (black trace) or stimulated overnight with TNFα (5ng/ml) (pink trace), LPS (100ng/ml) (orange trace) or infected with T. gondii strain RH (blue trace) at a multiplicity of infection of 5:1 and analyzed for GFP expression using flow cytometry. Toxoplasma infection yielded differential responses depending on the reporter cell line used.
In order to evaluate the utility of these reporter cells, we examined NF-κB activation in these lines, using alternative readouts, to validate stable NF-κB-GFP cell lines as reporter systems.
Analysis of the reporter cell lines using the Luciferse transient reporter system
To confirm the results obtained by the flow cytometry using the GFP reporter lines, we decided to use a transient NF-κB reporter system. Despite the low efficiency of the transfection known for primary mouse embryonic fibroblast, we were able to get a robust readout because of the higher sensitivity of the luciferase reporter gene. NF-κB-luc was transiently transfected into the different clones followed by TNFα (20 ng/ml for 2 hours) or LPS (100 ng/ml for 5 hours) stimulation. Wild type immortalized mouse fibroblasts (WT MEF) were used as a control. Interestingly, the luciferase reporter system did not reproduce the results seen with the GFP reporter system. In all the tested cell lines, including the control WT MEF, LPS induced high activation the NF-κB pathway using NF-κB-luc as the reporter (Fig. 4). Group I (TNh1 cells), characterized by being selectively responsive to TNFα and non-responsive to LPS with the flow cytometry, showed excellent response to LPS with the luciferase reporter. This implies that the LPS pathway is intact in the TNh1 cell line and the results seen with the GFP reporter may be due to the site of integration of the NF-κB-hr-GFP into the genome. In addition, the generally higher response to LPS in all cell lines may reflect the high sensitivity of the luciferase reporter system. These results suggest that the process of generating the stable reporter cell lines altered the signaling pathways in these cells and that the insertion site of the pNF-κB-hrGFP plasmid into the genome affected its ability to respond to LPS activation even though the signaling pathway is intact.
Phosphorylation of IκB confirms the GFP-reporter results
The reporter plasmid experiments suggested that the activation of NF-κB in response to TNFα and LPS is different in each of the examined clones. In order to understand the effects of generating the stable cell line on the endogenous NF-κB activation pathways we first had to examine intrinsic events of the pathway in the normal (WT MEF) and reporter cell lines. Phosphorylation of IκB is the common point where the NF-κB activation pathways converge (Fig. 5A). Phospho-IκB (P-IκB) is subjected to ubiquitination followed by degradation thus allowing the free NF-κB to translocate to the nucleus and to initiate gene expression of target genes.
Figure 5. Phosphorylation of IκB in the reporter cells lines.

A. Schematic representation of NF-κB activation: Various stimuli, like TNFα and LPS, bind to their cell surface receptors resulting in activation of a signaling cascade leading to phosphorylation and activation of IKK complex and the subsequent phosphorylation of IκB. Degradation of IκB releases NF-κB (p65–p50 dimer) and allows it to transclocate to the nucleus where it initiates transcription of its target genes.
B. Immunoblot analysis of NF-κB activation monitoring IκBα phosphorylation and degradation in reporter cell lines: Cell monolayers were left unstimulated (U) or stimulated for 6 minutes with TNFα (20ng/ml) (T) or for 2 hours with LPS (100ng/ml) (L). The phosphorylation of IκBα (P-IκB), detected with a Phospho-IκBα antibody, exhibits a stimulus dependent accumulation of the phospho-protein. Total IκB shows degradation in response to its phosphorylation, following addition of TNFα or LPS indicating activation of the NF-κB pathway. The absence if P-IκBα signal in TNh1(L) coincident with markedly reduced total IκBα suggests that the degradation of the phospho-IκBα is responsible for its absence following LPS stimulation. Anti-I-kappa kinase (IKK) levels were used as the loading control.
To assess the possibility of a defect in the NF-κB activation pathways we looked at the common point of IκB phosphorylation. We then examined the total levels of IκBα to confirm that it got degraded in response to phosphorylation by the IKK complex. As a loading control, we examined the levels of IKK, which is a component of the NF-κB pathway whose level is not changed in response to different stimuli. Westerns were performed at different time points ( 6, 10, 15 30 minutes and 2, 4 and 18 hours) to determine the optimum time point for analyzing p-IκBα, since it is know to be a short-lived molecule (data not shown). TNFα results in a rapid degradation of P-IκBα with the best result seen with 6 minutes TNFα treatment. P-IκBα, induced by LPS is more stable, as a result we picked the 2 hours time point. Cells were treated for 6 min with TNFα or for 2 hours with LPS then harvested for western analysis. In the control parental cell lines (WT MEF) and group II (LN4 cell line), P-IκB showed no significant difference in response to either TNFα or LPS (Fig. 5B: lane T and L). Stimulation of group I cells (TNh1) with TNFα resulted in robust phosphorylation of IκB (Fig. 5B), but not with LPS. But it is important to note the decrease in total IκB compared to the U lane, which confirms that the LPS pathway is functional in this group. In group III (LN9 cell line) there was not much detectable increase of IκB phosphorylation with either TNFα or LPS compared to the unstimulated lane, which may be the result of the high background seem with the flow cytometry analysis in the unstimulated cells. On the other hand, western of total IκB shows that IκB is efficiently degraded in all groups and the control when stimulated with TNFα and not as well with LPS stimulation, which suggests that TNFα may be a better inducer of IκB phosphorylation and subsequent degradation.
NF-κB dependent gene expression
A defect in the activation of NF-κB should affect its ability to activate the downstream targets. Some of the previous experiments suggest that each of the three examined cell lines has changed to some degree by the process of their construction. To understand the effect of these changes on the endogenous NF-κB pathways, we looked at the expression of CXCL1/GRO1 and IL6 which are two cytokines similarly activated by TNFα and LPS through NF-κB [29]. If stably integrated NF-κB-GFP is an honest reporter then we would expect similar pattern for GFP and the expression of these endogenous genes. Expression of the two cytokines in group I (TNh1) cell line where consistent with the flow cytometry analysis and the phosphorylation of IκB, where TNFα stimulation resulted in higher activation of both IL-6 and CXCL1 genes compared to the unstimulated cells (Fig. 6). The pattern of the gene expression in TNh1 is very similar to that in the WT MEF control cells. It is important to note that LPS was still able to induce the expression of CXCL1, which again confirms that the LPS activation pathway is still intact. On the other hand, the expression of IL-6 and CXCL1 in groups II and III (LN4 and LN9 cells) was similar to each other (and different from the parental MEF cell line) in that LPS was able to induce higher expression of IL-6 compared to TNFα levels of induction (Fig. 6).
Figure 6.
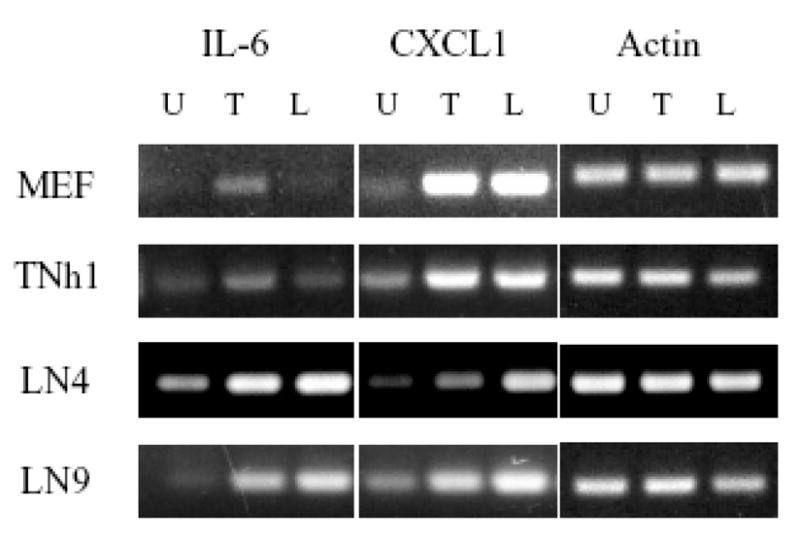
Expression of endogenous NF-κB target genes in unmodified MEF and reporter cell lines. The steady state levels of the NF-κB-regulated genes IL6 and CXCL1 were monitored by RT-PCR in untreated cells and cells stimulated overnight with TNFα (5ng/ml) or LPS (100ng/ml). Expression of the endogenous genes further reinforces the observation that the results from the NF-κB-GFP reporter transgene do not always accurately reflect the true expression patterns of endogenous genes. RT-PCR of β-actin message was used as a loading control
The results of the RT-PCR analysis were surprising because they did not follow the same response pattern seen in all previous experiments where TNFα activation typically gave a higher response. LPS was mostly able to induce higher or equal expression of CXCL1 compared to TNFα despite its absent or modest activation of the GFP reporter plasmid and P-IκB. This reflects the complexity of NF-κB signaling and the interplay of different pathways to fine-tune the expression of each individual target gene. Therefore, while the reporter system reflects the effects on NF-κB alone, the RT-PCR shows the cumulative effects of multiple integrated signaling pathways.
Discussion
In mammalian cells, transfection of plasmids in which a reporter gene is expressed under the control of an enhancer is widely used for studying transcription factors. Both GFP and luciferase are widely used reporter genes [30]. GFP has the advantage of requiring no processing as its expression can be monitored at single cell basis, which facilitates screening by FACS. On the other hand, the luciferase reporter gene offers a higher sensitivity but requires more processing of the samples and assessing of the activity results in the loss of cells.
The NF-κB pathway is an important drug target since it plays a central role in inflammation, immunity, cell survival and cancer. Several potential inhibitors of NF-κB are already being tested in preclinical and clinical trials [28]. More drugs are being sought and high throughput drug screens often rely on stable reporter systems.
We were interested in developing a stable NF-κB-GFP reporter system amenable to flow cytometry to screen for T. gondii mutants defective in activating NF-κB which is required to inhibit apoptosis in host cells [20]. Activation of NF-κB is partially achieved by phosphorylation of a subset of host IκBα [21]. However, it soon became clear that manipulation of the NF-κB pathway is more complex [22]. To address this issue, we developed a stable NF-κB-GFP reporter cell line (PSH2) for screening of mutants of the T. gondii that fail to activate NF-κB. This cell line was successfully used to identify several T. gondii mutants defective in activating NF-κB (data not shown). Unfortunately, PSH2 gradually lost its response to NF-κB activation, despite repeated selection of positive clones (data not shown). This loss is likely linked to the recombination at the railroaded identical NF-κB binding sites in the integrated reporter., This led us to develop additional cell lines and more extensively characterize their properties.
Construction of a stable NF-κB reporter cell line resulted in the generation of cell lines with different phenotypes. Consequently, identification of factors affecting the NF-κB pathway was entirely dependent on the specific cell line chosen for the study. Our criterion for the initial screen of the transfected cell was a high response to TNFα, LPS, or both. We reasoned that the generation of pathway-selective reporters could be exploited to isolate specific components of the NF-κB activation cascades. After isolation of individual clones, each showed a different pattern and intensity of response to TNFα and LPS stimulation, regardless of the agent used in the initial screen. Accordingly, parasite infection results in differential induction of NF-κB in each clone, pointing to need for further validation of the reporter response.
Remarkably, despite widely different responses when assessed using the integrated stable NF-κB-GFP reporter we found that the examination of endogenous markers or transient NF-κB -luc expression that no significant differences were noted. These differences between the stable NF-κB-GFP reporter and endogenous outputs were not restricted to the stimulus used. Thus, while LPS did not induce a response in TNh1 using the GFP reporter plasmid, it did induce an excellent response (Fig. 4) using the NF-κB–luc reporter plasmid and induced a strong expression of CXCL1 cytokine. This suggests that the endogenous NF-κB signaling pathways were not altered during the course of constructing the stable cell lines and that the site of insertion of reporter plasmid affected the outcome specifically for the reporter. As a result, the GFP reporter system did not reflect accurately the NF-κB activity. Interpretation of RT-PCR results are further complicated by the possible involvement of multiple pathways in the activation of downstream targets but it is still useful in confirming the integrity of signaling through the receptors of the different stimuli used in our work. The use of transcriptional data therefore completes the tracing of the entire signaling cascade pointing to its integrity. It further reveals that a stable NF-κB-GFP reporter does not always accurately reflect the activation and activity of the pathway it is reporting on.
Our study further shows that use of one control stimulus (ie only TNFα) is not enough to validate the reporter system since each clone had a different response to other stimuli (ie LPS and T. gondii). If a larger number of stimuli are to be screened, a larger variation of responses can be seen for which reason validation using endogenous markers is essential.
Acknowledgments
We thank the staff of the Core Laboratory for Flow Cytometry at the University of Kentucky College of Medicine for their technical assistance. This work was supported by NIH grant RO-1 AI049367 (A.P.S.).
References
- 1.Alam J, Cook JL. Reporter genes: application to the study of mammalian gene transcription. Anal Biochem. 1990;188:245–254. doi: 10.1016/0003-2697(90)90601-5. [DOI] [PubMed] [Google Scholar]
- 2.Anderson MT, Tjioe IM, Lorincz MC, Parks DR, Herzenberg LA, Nolan GP, Herzenberg LA. Simultaneous fluorescence-activated cell sorter analysis of two distinct transcriptional elements within a single cell using engineered green fluorescent proteins. Proc Natl Acad Sci U S A. 1996;93:8508–8511. doi: 10.1073/pnas.93.16.8508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 4.Barkett M, Gilmore TD. Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6910–6924. doi: 10.1038/sj.onc.1203238. [DOI] [PubMed] [Google Scholar]
- 5.Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 6.Cheng L, Fu J, Tsukamoto A, Hawley RG. Use of green fluorescent protein variants to monitor gene transfer and expression in mammalian cells. Nat Biotechnol. 1996;14:606–609. doi: 10.1038/nbt0596-606. [DOI] [PubMed] [Google Scholar]
- 7.Chudakov DM, Lukyanov S, Lukyanov KA. Fluorescent proteins as a toolkit for in vivo imaging. Trends Biotechnol. 2005;23:605–613. doi: 10.1016/j.tibtech.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 8.de Wet JR, Wood KV, Helinski DR, DeLuca M. Cloning of firefly luciferase cDNA and the expression of active luciferase in Escherichia coli. Proc Natl Acad Sci U S A. 1985;82:7870–7873. doi: 10.1073/pnas.82.23.7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drepper T, Eggert T, Circolone F, Heck A, Krauss U, Guterl JK, Wendorff M, Losi A, Gartner W, Jaeger KE. Reporter proteins for in vivo fluorescence without oxygen. Nat Biotechnol. 2007;25:443–445. doi: 10.1038/nbt1293. [DOI] [PubMed] [Google Scholar]
- 10.Freshney RI. Culture of Animal Cells: A Manual of Basic Technique. Alan R. Liss; New York: 1987. Cloning and selection of specific cell types; pp. 137–153. [Google Scholar]
- 11.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 12.Heim R, Prasher DC, Tsien RY. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc Natl Acad Sci U S A. 1994;91:12501–12504. doi: 10.1073/pnas.91.26.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hellweg CE, Baumstark-Khan C, Horneck G. Generation of stably transfected Mammalian cell lines as fluorescent screening assay for NF-kappaB activation-dependent gene expression. J Biomol Screen. 2003;8:511–521. doi: 10.1177/1087057103257204. [DOI] [PubMed] [Google Scholar]
- 14.Hernandez-Gutierrez S, Garcia-Pelaez I, Zentella-Dehesa A, Ramos-Kuri M, Hernandez-Franco P, Hernandez-Sanchez F, Rojas E. NF-kappaB signaling blockade by Bay 11–7085 during early cardiac morphogenesis induces alterations of the outflow tract in chicken heart. Apoptosis. 2006;11:1101–1109. doi: 10.1007/s10495-006-6984-z. [DOI] [PubMed] [Google Scholar]
- 15.Karin M. How NF-kappaB is activated: the role of the IkappaB kinase (IKK) complex. Oncogene. 1999;18:6867–6874. doi: 10.1038/sj.onc.1203219. [DOI] [PubMed] [Google Scholar]
- 16.Kim JM, Oh YK, Kim YJ, Cho SJ, Ahn MH, Cho YJ. Nuclear factor-kappa B plays a major role in the regulation of chemokine expression of HeLa cells in response to Toxoplasma gondii infection. Parasitol Res. 2001;87:758–763. doi: 10.1007/s004360100447. [DOI] [PubMed] [Google Scholar]
- 17.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 18.Matz MV, Lukyanov KA, Lukyanov SA. Family of the green fluorescent protein: journey to the end of the rainbow. Bioessays. 2002;24:953–959. doi: 10.1002/bies.10154. [DOI] [PubMed] [Google Scholar]
- 19.Molestina RE, Klein JB, Miller RD, Pierce WH, Ramirez JA, Summersgill JT. Proteomic analysis of differentially expressed Chlamydia pneumoniae genes during persistent infection of HEp-2 cells. Infect Immun. 2002;70:2976–2981. doi: 10.1128/IAI.70.6.2976-2981.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Molestina RE, Payne TM, Coppens I, Sinai AP. Activation of NF-kappaB by Toxoplasma gondii correlates with increased expression of antiapoptotic genes and localization of phosphorylated IkappaB to the parasitophorous vacuole membrane. J Cell Sci. 2003;116:4359–4371. doi: 10.1242/jcs.00683. [DOI] [PubMed] [Google Scholar]
- 21.Molestina RE, Sinai AP. Detection of a novel parasite kinase activity at the Toxoplasma gondii parasitophorous vacuole membrane capable of phosphorylating host IkappaBalpha. Cell Microbiol. 2005;7:351–362. doi: 10.1111/j.1462-5822.2004.00463.x. [DOI] [PubMed] [Google Scholar]
- 22.Molestina RE, Sinai AP. Host and parasite-derived IKK activities direct distinct temporal phases of NF-kappaB activation and target gene expression following Toxoplasma gondii infection. J Cell Sci. 2005;118:5785–5796. doi: 10.1242/jcs.02709. [DOI] [PubMed] [Google Scholar]
- 23.Muller JM, Ziegler-Heitbrock HW, Baeuerle PA. Nuclear factor kappa B, a mediator of lipopolysaccharide effects. Immunobiology. 1993;187:233–256. doi: 10.1016/S0171-2985(11)80342-6. [DOI] [PubMed] [Google Scholar]
- 24.Payne TM, Molestina RE, Sinai AP. Inhibition of caspase activation and a requirement for NF-kappaB function in the Toxoplasma gondii-mediated blockade of host apoptosis. J Cell Sci. 2003;116:4345–4358. doi: 10.1242/jcs.00756. [DOI] [PubMed] [Google Scholar]
- 25.Perkins ND. The Rel/NF-kappa B family: friend and foe. Trends Biochem Sci. 2000;25:434–440. doi: 10.1016/s0968-0004(00)01617-0. [DOI] [PubMed] [Google Scholar]
- 26.Rothwarf DM, Karin M. The NF-kappa B activation pathway: a paradigm in information transfer from membrane to nucleus. Sci STKE 1999. 1999:RE1. doi: 10.1126/stke.1999.5.re1. [DOI] [PubMed] [Google Scholar]
- 27.Tenter AM, Heckeroth AR, Weiss LM. Toxoplasma gondii: from animals to humans. Int J Parasitol. 2000;30:1217–1258. doi: 10.1016/s0020-7519(00)00124-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Waes C. Nuclear factor-kappaB in development, prevention, and therapy of cancer. Clin Cancer Res. 2007;13:1076–1082. doi: 10.1158/1078-0432.CCR-06-2221. [DOI] [PubMed] [Google Scholar]
- 29.Werner SL, Barken D, Hoffmann A. Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science. 2005;309:1857–1861. doi: 10.1126/science.1113319. [DOI] [PubMed] [Google Scholar]
- 30.Wieder KJ, King KR, Thompson DM, Zia C, Yarmush ML, Jayaraman A. Optimization of reporter cells for expression profiling in a microfluidic device. Biomed Microdevices. 2005;7:213–222. doi: 10.1007/s10544-005-3028-3. [DOI] [PubMed] [Google Scholar]