

Abstract
Quinolinate synthase (NadA) catalyzes a unique condensation reaction between iminoaspartate and dihydroxyacetone phosphate, affording quinolinic acid, a central intermediate in the biosynthesis of nicotinamide adenine dinucleotide (NAD). Iminoaspartate is generated via the action of l-aspartate oxidase (NadB), which catalyzes the first step in the biosynthesis of NAD in most prokaryotes. NadA from Escherichia coli was hypothesized to contain an iron–sulfur cluster as early as 1991, because of its observed labile activity, especially in the presence of hyperbaric oxygen, and because its primary structure contained a CXXCXXC motif, which is commonly found in the [4Fe-4S] ferredoxin class of iron–sulfur (Fe/S) proteins. Indeed, using analytical methods in concert with Mössbauer and electron paramagnetic resonance spectroscopies, the protein was later shown to harbor a [4Fe–4S] cluster. Recently, the X-ray structure of NadA from Pyrococcus horikoshii was solved to 2.0 Å resolution [H. Sakuraba, H. Tsuge, K. Yoneda, N. Katunuma, and T. Ohshima, (2005) J. Biol. Chem. 280, pp. 26645-26648]. This protein does not contain a CXXCXXC motif, and no Fe/S cluster was observed in the structure or even mentioned in the report. Moreover, rates of quinolinic acid production were reported to be 2.2 μmol min-1 mg-1, significantly greater than that of E. coli NadA containing an Fe/S cluster (0.10 μmol min-1 mg-1), suggesting that the [4Fe-4S] cluster of E. coli NadA may not be necessary for catalysis. In the study described herein, nadA genes from both Mycobacterium tuberculosis and Pyrococcus horikoshii were cloned, and their protein products shown to contain [4Fe–4S] clusters that are absolutely required for activity despite the absence of a CXXCXXC motif in their primary structures. Moreover, E. coli NadA, which contains nine cysteine residues, is shown to require only three for turnover (C113, C200, and C297), of which only C297 resides in the CXXCXXC motif. These results are consistent with a bioinformatics analysis of NadA sequences, which indicates that three cysteines are strictly conserved across all species. This study concludes that all currently annotated quinolinate synthases harbor a [4Fe–4S] cluster, that the crystal structure reported by Sakuraba et al does not accurately represent the active site of the protein, and that the “activity” reported does not correspond to quinolinate formation.
Nicotinamide adenine dinucleotide (NAD) is an essential and ubiquitous cofactor known primarily for its role as a co-substrate in a multitude of biological oxidation-reduction reactions that involve hydride transfers (1). It is also involved in myriad non-redox reactions such as adenylation, ADP-ribosylation, and histone deacetylation (2, 3), and in enzymes such as urocanase, in which its nicotinamide ring is proposed to form a transient covalent adduct with the substrate urocanate, facilitating catalysis via an electrophilic mechanism (4). Moreover, it serves as the initial substrate for the biosynthesis of thiamin thiazole in Saccharomyces cerevisiae (5). Quinolinic acid (QA) serves as the first common precursor to NAD and its derivatives in all organisms that synthesize NAD de novo; the subsequent steps that lead to NAD from QA are virtually identical across organisms (6). By contrast, there are two distinctly different pathways by which QA is formed. In most eukaryotes, QA is generated from the oxidative degradation of tryptophan in a series of five enzymatic steps, three of which involve molecular oxygen as a cosubstrate (6). The requirement for molecular oxygen in this pathway has led to its branding as the aerobic pathway for QA production, which distinguishes it from an alternative pathway (anaerobic pathway) found predominantly in prokaryotes, which does not display an obligate requirement for molecular oxygen. In the anaerobic pathway, l-aspartate is first converted to iminoaspartate (ImAsp) by l-aspartate oxidase (NadB); ImAsp is then condensed with dihydroxyacetone phosphate (DHAP) to form quinolinic acid in a reaction catalyzed by quinolinate synthase (NadA). The byproducts of the reaction are 2 mol of water and 1 mol of inorganic phosphate (Scheme 1) (7).
Scheme 1. Reactions catalyzed by l-aspartate oxidase and quinolinate synthase.
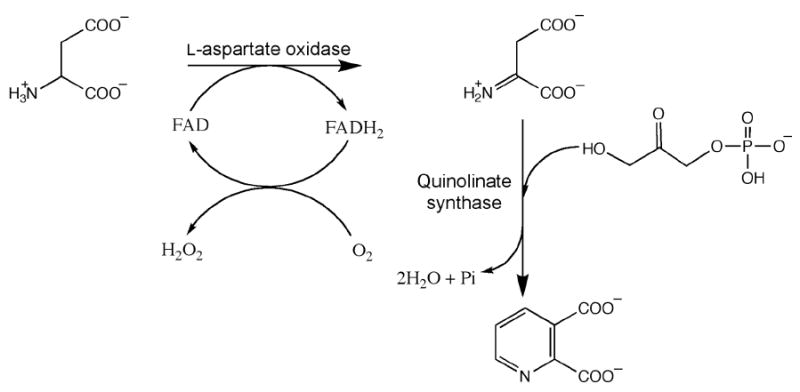
NadB is a 60 kDa flavoprotein that contains one noncovalently bound molecule of flavin adenine dinucleotide (FAD) per polypeptide (8). During catalysis, l-aspartate is transiently oxidized to ImAsp with concomitant reduction of FAD to FADH2. Under oxic conditions, FADH2 is re-oxidized to FAD by molecular oxygen, with generation of hydrogen peroxide as a byproduct (Scheme 1). Under anoxic conditions, fumarate serves as the electron acceptor, being reduced to succinate (9). ImAsp is an unstable intermediate; it hydrolyzes to ammonia and oxaloacetate (OAA) at 37 °C and pH 8.0 with a half-life of 144 s (10). In vitro the NadB protein can be replaced by conducting the reaction in the presence of OAA and an ammonia source, such as ammonium sulfate (AS), which undergo Schiff-base formation to afford an equilibrium concentration of ImAsp (11).
NadA from Escherichia coli has served as the prototype for the study of quinolinate synthases; however, detailed characterization and mechanistic analysis of the enzyme was originally hampered by its observed instability (10). The protein contains a CXXCXXC motif, which is found often in the primary structures of [4Fe-4S]-containing proteins of the ferredoxin class (12). This motif, as well as the enzyme’s reported tendency to lose catalytic competence in the presence of hyperbaric oxygen, led to the suggestion that E. coli NadA is an iron-sulfur (Fe/S) protein (13). This hypothesis was later confirmed. Analytical and spectroscopic studies by two independent laboratories showed that the protein binds and requires one [4Fe-4S]2+ cluster per polypeptide (14, 15). NadA has been purported to be a member of the Fe/S cluster-dependent class of the hydro-lyase family of enzymes (16), which includes aconitase, the prototype member (17). Aconitase contains a [4Fe-4S] cluster in which three of the iron atoms are ligated by three cysteine residues contributed by the protein scaffold. The fourth iron (Fea) provides a site for bidentate coordination by the substrate or product, with a corresponding change in its coordination geometry from tetrahedral to octahedral (18-21). Similarly, it is expected that the [4Fe-4S] cluster of NadA will also be ligated by three cysteinate protein ligands, allowing one iron site to be available for direct involvement in the reaction (16).
The X-ray crystal structure of quinolinate synthase from the hyperthermophilic archaeon Pyrococcus horikoshii (Phk) was solved to 2.0 Å in 2005. Surprisingly, the structure showed no evidence of an Fe/S cluster or any other type of cofactor, nor was mention of an Fe/S cluster made in the published report (22). Yet, the protein was stated to catalyze formation of QA with a specific activity of 2.2 μmol min-1 mg-1, an activity that is significantly higher than that reported for the Fe/S-containing E. coli enzyme (14). However, the structure contained three regions of poorly defined electron density, in which three cysteine residues that are strictly conserved among NadA proteins are found (Scheme 2). Herein, we show that the three corresponding cysteines in E. coli NadA are absolutely required for turnover in vitro, and most likely act as ligands to the [4Fe-4S] cluster. When C→S substitutions were made at each of the nine cysteine residues present in the primary structure of E. coli NadA, only substitutions at C113, C200, and C297 afforded inactive proteins when analyzed in their as-isolated (AI) or reconstituted (RCN) forms. Importantly, only C297 is present in the CXXCXXC motif (Scheme 2). In addition, we report the first published isolation of NadA from Mycobacterium tuberculosis (Mtb), and show that the protein contains a [4Fe-4S] cluster with similar spectroscopic parameters as that from E. coli. Unlike NadA from E. coli, the primary structure of the enzyme from Mtb does not contain a CXXCXXC motif, but does contain three conserved cysteine residues (Scheme 2). Last, we show that when Phk NadA is isolated under strict anaerobic conditions, it too contains a [4Fe-4S] cluster and displays a specific activity that is similar to that of the E. coli and Mtb enzymes. When it is overproduced recombinantly in the presence of o-phenanthroline, an Fe(II) chelator, or isolated aerobically, the protein is devoid of an Fe/S cluster and exhibits no catalytic activity. When this apo form of Phk NadA is reconstituted anaerobically with iron and sulfide, the [4Fe-4S] cluster reforms, and catalytic competence is regained. Since all annotated quinolinate synthases contain these three conserved cysteine residues, we argue that [4Fe-4S] clusters are common and requisite cofactors throughout this class of enzymes.
Scheme 2. Relative positioning of cysteines in NadA proteins from P horikoshii, E. coli, and M. tuberculosisa.

aThe residues that coordinate the [4Fe-4S] cluster are shown in bold face. The position of the three disordered loops of P. horikoshii NadA observed in the X-ray structure are indicated by grey bars.
MATERIALS AND METHODS
Materials
All DNA modifying enzymes and reagents, and Deep Vent DNA Polymerase and its associated 10× ThermoPol reaction buffer were purchased from New England Biolabs (Beverly, MA). PfuUltra High Fidelity DNA Polymerase and its associated 10× reaction buffer were obtained from Stratagene (La Jolla, CA). Oligonucleotide primers for cloning were obtained from Integrated DNA Technologies (Carlsbad, CA). Escherichia coli genomic DNA (strain W3110) was obtained from Sigma Corp (St. Louis, MO), and Pyrococcus horikoshii (Phk) genomic DNA was obtained from the American Type Culture Collection (Manassas, VA). Genomic DNA from Mycobacterium tuberculosis (Mtb) was a kind gift of Dr. Peter Tonge (SUNY Stony Brook). E. coli strains BL21(DE3) and Rosetta 2(DE3) were obtained from Novagen (Madison, WI), as was vector pET-28a. Isopropyl-β-d-thiogalactopyranoside (IPTG) was purchased from Biosynth International (Naperville, IL). Coomassie blue dye-binding reagent for protein concentration determination and the bovine serum albumin (BSA) standard (2 mg mL-1) were obtained from Pierce (Rockford, IL). Nickel nitrilotriacetic acid (Ni-NTA) resin was purchased from Qiagen (Valencia, CA). Sephadex G-25 resin and PD-10 pre-poured gel filtration columns were purchased from GE Biosciences (Piscataway, NJ). 2,3-Pyridinedicarboxylic acid (quinolinic acid) was obtained from Aldrich (St. Louis, MO). Dihydroxyacetone phosphate (dilithium salt), l-(+)-arabinose, ferric chloride, and α-glycerophosphate dehydrogenase were obtained from Sigma (St. Louis, MO), while o-phenanthroline was obtained from Fisher (Fair Lawn, NJ). All other buffers and chemicals were of the highest grade available.
57Fe (97-98%) metal was purchased from Isoflex USA (San Francisco, CA). It was washed with CHCl3 and dissolved with heating in an anaerobic solution of 2 N H2SO4 (1.5 mol of H2SO4 per mole of 57Fe).
General Procedures
High performance liquid chromatography (HPLC) was conducted on a Beckman System Gold unit (Fullerton, CA), which was fitted with a 128 diode array detector and operated with the System Gold Nouveau software package. Iron and sulfide analyses were performed as previously described (23-25). All numbers reported are the average of four determinations, with errors reflecting one standard deviation. Sonic disruption of E. coli cells was carried out with a 550 sonic dismembrator from Fisher Scientific (Pittsburgh, PA) in combination with a horn containing a ½ inch tip. The horn was threaded through a port in an anaerobic chamber to allow the process to be conducted under anoxic conditions. The polymerase chain reaction (PCR) was performed with a Robocycler temperature cycler from Stratagene (La Jolla, CA). DNA sequencing was carried out at the Pennsylvania State University Nucleic Acid Facility.
Spectroscopic Methods
UV-vis spectra were recorded on a Cary 50 spectrometer (Varian; Walnut Creek, CA) using the associated WinUV software package. Low-temperature X-band EPR spectroscopy was carried out in perpendicular mode on a Bruker (Billerica, MA) ESP 300 spectrometer equipped with an ER 041 MR microwave bridge and an ST4102 X-band resonator (Bruker). The sample temperature was maintained with an ITC503S temperature controller and an ESR900 liquid helium cryostat (Oxford Instruments; Concord, MA).
Mössbauer spectra were recorded on a spectrometer from WEB research (Edina, MN) operating in the constant acceleration mode in transmission geometry. Spectra were recorded with the temperature maintained at 4.2 K. The sample was kept inside an SVT-400 dewar from Janis (Wilmington, MA), and a magnetic field of 53 mT was applied parallel to the γ-beam. The isomer shift is relative to the centroid of the spectrum of a metallic foil of α-Fe at room temperature. Data analysis was performed using the program WMOSS from WEB research.
Cloning of E. coli nadA and nadB Genes
The nadA gene was amplified from E. coli (strain W3110) genomic DNA by PCR technology using primers nadAfor and nadArev (Supplementary Data, Table S1). Each amplification reaction contained the following in a volume of 50 μL: 0.4 μM of each primer, 0.2 mM of each deoxynucleoside triphosphate, 100 ng of E. coli genomic DNA, 2.5 U of PfuUltra High Fidelity DNA Polymerase, and 5 μL of 10× PfuUltra™ reaction buffer. The reaction mixture was overlaid with 30 μL of mineral oil. After a 2 min denaturation step at 95 °C, 30 cycles of the following program were initiated: 30 s at 95 °C, 30 s at 70 °C, 1 min at 72 °C. Following the cycling program, the reaction was incubated further for 10 min at 72 °C. The PCR product was digested with NdeI and EcoRI and then ligated into similarly digested pET-28a by standard methods (26).
The nadB gene was amplified from E. coli (strain W3110) genomic DNA by PCR technology using primers nadBfor and nadBrev (Supplementary Data, Table S1). Each amplification reaction contained the same concentrations of reagents as described above for cloning the nadA gene, except that DMSO was added to a final concentration of 4%. After a 2 min denaturation step at 95 °C, 30 cycles of the following program were initiated: 30 s at 95 °C, 30 s at 63 °C, 3 min at 72 °C. Following the cycling program, the reaction was incubated further for 10 min at 72 °C. The PCR product was digested with NdeI and HindIII and then ligated into similarly digested pET-28a by standard methods. All other procedures were carried out by standard methods. The correct constructs were verified by DNA-sequencing and designated pET28-NadA and pET28-NadB, respectively.
Cloning of P. horikoshii nadA Gene
The nadA gene was amplified from Phk (JCM 9974) genomic DNA by PCR technology using primers PhNadA.for and PhNadA.rev (Supplementary Data, Table S1). Each amplification reaction contained the following in a volume of 50 μL: 0.48 μM of each primer, 0.25 mM of each deoxynucleoside triphosphate, 200 ng of Phk genomic DNA, 2 U of Deep Vent DNA Polymerase, and 5 μL of 10× ThermoPol reaction buffer. The reaction mixture was overlaid with 40 μL of mineral oil. After a 9 min denaturation step at 95 °C, 35 cycles of the following program were initiated: 1 min at 95 °C, 1 min at 55 °C, 2.5 min at 72 °C. Following the cycling program, the reaction was incubated further for 10 min at 72 °C. The PCR product was digested with NdeI and EcoRI and then ligated into similarly digested pET-28a by standard methods. The correct construct was verified by DNA-sequencing and designated pET28-PhNadA.
Cloning of the M. tuberculosis nadA Gene
The nadA gene from Mtb was cloned as described for the nadA gene from Phk, with the exception that the primers used for PCR amplification were MtNadA.for and MtNadA.rev (Supplementary Data, Table S1), and genomic DNA from Mtb (H37Rv) served as the template. The correct construct was verified by DNA-sequencing and designated pET28-MtNadA.
Construction of E. coli NadA Variants
E. coli NadA variants were constructed using the QuikChange II Site-directed Mutagenesis Kit (Stratagene) according to the manufacturer’s specifications, and as described previously (27), in which the cycling protocol is adapted for the Stratagene Robocycler thermocycler (28). Plasmid pET28-NadA was used as the template in conjunction with the appropriate primers for each respective amino acid substitution (Supplementary Data, Table S2). All mutations were verified by DNA-sequencing of the entire gene.
Expression of the Genes Encoding E. coli Wild-type NadA and Variants
Plasmid pET28-NadA, encoding WT or variant NadA proteins, was cotransformed into E. coli BL21(DE3) with plasmid pDB1282 as described previously (29). A single colony was selected and used to inoculate 100 mL of Luria–Bertani (LB) media containing 50 μg/mL kanamycin and 100 μg/mL ampicillin, and was cultured for ~7 h at 37 °C with shaking (180 rpm). A 60 mL portion of the culture was then evenly distributed among four 6 L Erlenmeyer flasks to inoculate 16 L of LB media containing 50 μg/mL kanamycin and 100 μg/mL ampicillin, and the bacteria were cultured further at 37 °C with shaking. At an optical density at 600 nm (OD600) of 0.3, solid l-(+)-arabinose was added to each flask at a final concentration of 0.05 % (w/v). At an OD600 of 0.6, the cultures were cooled in an ice–water bath, and solid IPTG and ferric chloride were added to each flask at final concentrations of 200 and 50 μM, respectively. The cultures were then allowed to incubate further at 18 °C with shaking for 16 h. Cells were harvested by centrifugation at 10,000 × g for 10 min at 4 °C, and the resulting cell paste was frozen in liquid N2 and stored at -80 °C until ready for use. Typical yields were 50-60 g of frozen cell paste per 16 L of culture.
Expression of the nadA Gene from M. tuberculosis
Expression of the nadA gene from Mtb was carried out similarly to that described above, except that pET28-MtNadA replaced pET28-NadA. In addition, induction was carried out a 37 °C for 4 h rather than 18 °C for 16 h.
Expression of the P. horikoshii nadA Gene
The Phk nadA gene was expressed in a similar manner, with the following exceptions. E. coli Rosetta 2(DE3) cells were cotransformed with plasmids pET28-PhNadA and pDB1282 and cultured in LB media containing 34 μg/mL chloramphenicol, 50 μg/mL kanamycin, and 100 μg/mL ampicillin. Induction of the genes on plasmid pDB1282 was initiated at an OD600 of 0.3 by addition of l-(+)-arabinose to a final concentration of 0.2%. Induction of the Phk nadA gene was initiated at an OD600 of 0.6 by addition of IPTG to a final concentration of 400 μM. Expression was allowed to proceed for 4 h at 37 °C before the cells were harvested by centrifugation at 4 °C.
Purification of E. coli and M. tuberculosis NadA Proteins
All steps of the purification were conducted inside of an anaerobic chamber from Coy Laboratory Products, Inc. (Grass Lake, MI) under an atmosphere of N2 and H2 (95%/5%), with an O2 concentration maintained below 1 ppm by the use of palladium catalysts. Steps using centrifugation were performed outside of the anaerobic chamber in centrifuge tubes that were tightly sealed before they were removed from the chamber. All buffers were prepared using distilled and deionized water that was boiled for at least 1 h and then allowed to cool with stirring uncapped in the anaerobic chamber for 48 h. All plastic ware was autoclaved and brought into the chamber hot, and allowed to equilibrate overnight before use.
Protein purification was carried out by immobilized metal affinity chromatography (IMAC) using a nickel nitrilotriacetic acid (Ni-NTA) matrix. In a typical purification, 25 g of frozen cells were resuspended in 80 mL of buffer A (50 mM HEPES, pH 7.5, 0.3 M KCl, 20 mM imidazole, and 10 mM 2-mercaptoethanol). Solid egg white lysozyme was added to a final concentration of 1 mg mL-1 and the mixture was stirred at room temperature for 30 min. After the mixture was allowed to cool in an ice–water bath to <8 °C, it was subjected to four 1 min bursts of sonic disruption (setting 7). Cellular debris was removed by centrifugation at 50,000 × g for 1 h, and the resulting supernatant was loaded onto a Ni-NTA column (2.5 × 7 cm) equilibrated in buffer A. The column was washed with 100 mL of buffer B (50 mM HEPES, pH 7.5, 0.3 M KCl, 40 mM imidazole, 10 mM 2-mercaptoethanol, and 20% glycerol), and subsequently eluted with buffer B containing 250 mM imidazole. Fractions that were brown in color were pooled and concentrated in an Amicon stirred cell (Millipore, Billerica, MA) fitted with a YM-10 membrane (10,000 Da MW cutoff). The protein was exchanged into buffer C (50 mM HEPES, pH 7.5, 0.1 M KCl, 10 mM DTT, and 20% glycerol) by anaerobic gel filtration (Sephadex G-25), concentrated, and stored in aliquots in a liquid N2 dewar until ready for use, or immediately reconstituted (vide infra). All buffers were chilled on ice prior to use, and ice packs were used to jacket the Amicon stirred cell during protein concentration steps.
Purification of P. horikoshii NadA
Phk NadA was purified as described above for the E. coli enzyme, except that two heat denaturation steps were included to eliminate additional contaminating proteins, as described by Sakuraba et al (22). After allowing the suspended cells to thaw, the suspension was incubated with lysozyme for 30 min at room temperature before heating it at 37 °C for 15 min. Following sonication, a sodium sulfate solution was added to a final concentration of 0.2 M, and the suspension of cells was heated at 85 °C for 15 min. The remainder of the purification was carried out as described for the purification of E. coli NadA, except it was performed with buffers maintained at ambient temperature.
Overproduction of E. coli and P. horikoshii Apo-NadA Proteins
E. coli and Phk NadA proteins lacking a [4Fe-4S] cluster was produced by addition of o-phenanthroline to the growth media (30). Cultures were initially grown as described above, without induction of the genes on plasmid pDB1282. At an OD600 of 0.6, 100 mM o-phenanthroline in 100 mM HCl was added to a final concentration of 100 μM. The cultures were then incubated for 15 min before addition of IPTG to a final concentration of 200 μM. Following induction, the cultures were incubated further for 4 h at 37 °C with shaking. Cells were harvested at 10,000 × g for 10 min at 4 °C, and the resulting cell paste was frozen in liquid N2 and stored at -80 °C until ready for use.
Reconstitution of NadA
Reconstitution of E. coli and Mtb NadA proteins with iron and sulfide was carried out on ice in a Coy anaerobic chamber using anaerobic buffers and solutions. A typical reconstitution contained, in a final volume of 20 mL, 100 μM NadA that was initially treated with 5 mM DTT for 20 min. Next, an 8-fold molar excess of FeCl3 was added, and the solution was allowed to sit on ice for 20 min. Finally, an 8-fold molar excess of Na2S was added over a period of 3 to 4 h, and then the mixture was allowed to incubate on ice for 5-13 h. The mixture was then concentrated in an Amicon stirred cell and subjected to centrifugation at 14,000 × g for 2 min to remove precipitate. The supernatant was removed and exchanged into buffer C by gel filtration, reconcentrated, and then stored in aliquots in a liquid N2 dewar. Reconstitution of Phk NadA was performed in a similar fashion, except that the process was conducted at room temperature.
Expression of the nadB Gene, and Purification of its Product
Plasmid pET28-NadB was transformed into E. coli BL21(DE3). A single colony was used to inoculate 100 mL of LB media containing 50 μg/mL kanamycin, and was cultured for 7 h at 37 °C with shaking. A 60 mL portion of the culture was evenly distributed among four 6 L Erlenmeyer flasks to inoculate 16 L of LB media containing 50 μg/mL kanamycin, and was cultured further at 37 °C. At an OD600 of 0.6, the cultures were cooled in an ice–water bath, and solid IPTG and FAD were added to each flask to final concentrations of 200 and 10 μM, respectively. The cultures were then incubated at 18 °C with shaking for 16 h. Cells were harvested at 10,000 × g for 10 min at 4 °C, and the resulting cell paste was frozen in liquid N2 and stored at -80 °C until ready for use. Typical yields of frozen cell paste were between 50 and 60 g per 16 L growth.
In a typical purification, 25 g of frozen cells were resuspended in 80 mL of buffer D (50 mM HEPES, pH 7.5, 0.2 M NaCl, 10 mM imidazole, 10 mM succinate, and 20 μM FAD). Solid egg white lysozyme and and phenylmethanesulfonyl fluoride (PMSF) were added to final concentrations of 1 mg mL-1 and 1 mM, respectively, and the mixture was stirred at room temperature for 30 min. After the mixture was allowed to cool in an ice–water bath to <8 °C, it was subjected to four 1 min bursts of sonic disruption (setting 7). Cellular debris was removed by centrifugation at 50,000 × g for 1 h, and the resulting supernatant was loaded onto a Ni-NTA column (2.5 × 7 cm) equilibrated in buffer D. The column was washed with 175 ml of buffer E (50 mM HEPES, pH 7.5, 0.2 M NaCl, 20 mM imidazole, 10 mM succinate, 20 μM FAD, and 10% glycerol) and subsequently eluted with buffer E containing 250 mM imidazole. Protein-containing fractions were pooled and concentrated in an Amicon stirred cell fitted with a YM-10 membrane (10,000 Da MW cutoff). The protein was exchanged into anaerobic buffer F (50 mM HEPES, pH 7.5, and 10% glycerol) by anaerobic gel filtration (Sephadex G-25), reconcentrated, and stored in aliquots in a liquid N2 dewar until ready for use.
Determination of Protein Concentrations
Protein concentrations were determined by the Bradford dye staining procedure with BSA as the standard (31). Quantitative amino acid analysis on parallel samples of E. coli and Mtb NadA proteins, conducted at the University of Iowa Molecular Analysis Facility, confirmed that the Bradford assay was accurate without a correction factor. Phk NadA was similarly analyzed at the University of California–Davis Molecular Structure Facility; the Bradford method overestimates its concentration by a factor of 1.54.
Activity Determinations
The activity of WT and variant NadA proteins was determined by monitoring the formation of QA over a 20 min time period at 37 °C under anaerobic conditions. The substrate ImAsp was generated enzymatically via the NadB reaction using fumarate as the electron acceptor, or chemically by replacement of NadB, l-aspartate, and fumarate in assay mixtures with OAA and AS (11). Activity determinations conducted in the presence of NadB contained, in a final volume of 1300 μL, 200 mM HEPES, pH 7.5, 25 mM l-aspartate, 25 mM fumarate, 25 μM FAD, 1 mM DHAP, 0.1 M KCl, 0.3 mM l-tryptophan (internal standard), and either 5 μM RCN NadA and 5 μM NadB or 15 μM AI NadA and 15 μM NadB. When ImAsp was generated chemically, l-aspartate, fumarate, FAD, and NadB were replaced with 30 mM OAA and 30 mM AS. The reactions were initiated by addition of either NadB or OAA after incubation of the other components of the assay mixture at 37 °C for 5 min. At designated times, 200 μL aliquots of the assay mixture were removed and added to 40 μL of 2 M trichloroacetic acid (TCA) to quench the reaction. The precipitated protein was pelleted by centrifugation, and the supernatant was analyzed by HPLC with UV detection (268 nm) using a Zorbax SB-C18 column (4.6 × 250 mm) from Agilent (Foster City, CA). The column was equilibrated in 100% Solvent A (1% trifluoroacetic acid (TFA)) at a flow rate of 1 mL min-1. These initial conditions were maintained for 10 min after injection, upon which a gradient of 0-45% Solvent B (acetonitrile) was applied over 10 min. At 20 min, a gradient of 45-90% Solvent B was applied over 3 min. Finally, at 25 min, a gradient of 90-0% Solvent B was applied over 3 min to re-establish the initial conditions until the end of the run at 32 min. Using this method, QA eluted at 8.3 min, and the tryptophan internal standard eluted at 20.3 min. The concentration of QA was determined from a calibration curve of known concentrations of the compound using the internal standard to correct for volume changes between sample injections. Activity determinations of Phk NadA were conducted at 50 °C in HEPES buffer, pH 7.0. All other components of the assay mixture were as described above.
Activity determinations were also performed by quantifying the amount of DHAP remaining as a function of time as described previously (15). Assay mixtures were as described above; however, at designated times, 50 μL aliquots of the quenched assay mixture were added to a cuvette containing 100 mM HEPES, pH 7.5, and 250 μM NADH, and the absorbance at 340 nm was measured. α-Glycerophosphate dehydrogenase (5.6 U) was added to the cuvette, and the resulting decrease in absorbance at 340 nm (ε = 6440 M-1 cm-1) was recorded and then used to calculate the concentration of DHAP present. The concentration of QA was then assumed to be the difference between the starting DHAP concentration and the concentration measured at any given time point.
Preparation of Mössbauer and EPR Samples
Samples to be analyzed by Mössbauer and EPR spectroscopies contained 425-950 μM NadA and were all prepared inside of the anaerobic chamber. For characterization by Mössbauer, bacterial growth and gene expression were carried out in M9 minimal media, and 57FeSO4 was added at induction. For the E. coli C200S variant, 57Fe was incorporated by reconstitution. Samples (400 μL) were placed in small plastic cups and frozen in liquid N2. For characterization by EPR, samples (250 μL) were treated with 2 mM sodium dithionite at room temperature for 5 min, placed in EPR tubes (2 mm i.d.), and frozen in liquid N2.
RESULTS
Cloning and Expression of the P. horikoshii nadA Gene, and Purification and Characterization of its Product
The determination of the X-ray crystal structure of NadA from Phk in 2005 produced surprising findings (22). The structure showed no presence of an Fe/S cluster; however, the protein was reported to catalyze formation of QA with a specific activity of 2.2 μmol min-1 mg-1, which is considerably greater than that reported for the recombinant E. coli enzyme (0.015 μmol min-1 mg-1) (14). This observation suggests that the presence of an Fe/S cluster is not a mechanistic imperative, but that the E. coli enzyme might simply use it to satisfy structural and/or regulatory requirements, for example. To address this issue directly, the nadA gene from Phk was amplified by PCR technology and cloned into a pET-28a expression vector. The resulting construct allowed production of an N-terminally hexahistidine-tagged form of the protein for facile and rapid purification within the confines of an anaerobic chamber.
The UV-vis spectrum of as-isolated (AI) Phk NadA is shown in Figure 1A (solid line), and is similar to the published spectrum of the E. coli enzyme isolated under similar conditions (14). Iron and sulfide analysis of the AI enzyme indicates the presence of 2.3 ± 0.4 equiv of the former and 2.6 ± 1.0 equiv of the latter per polypeptide, also similar to that obtained for the AI E. coli enzyme. The 4.2-K/53-mT Mössbauer spectrum of this sample is shown in Figure 2. It is dominated by two peaks that belong to a quadrupole doublet with parameters (isomer shift, δ, of 0.45 mm/s and quadrupole splitting parameter, ΔEQ, of 1.15 mm/s) typical of the [4Fe-4S]2+ cluster form. This quadrupole double accounts for 87 % of the total Fe, resulting in a stoichiometry of 0.5 [4Fe-4S] clusters per NadA. In addition, there is a line at +1.6 mm/s. This line is associated with a minor quadrupole doublet, the low-energy line of which overlaps with the low-energy line of the subspectrum of the [4Fe-4S]2+ clusters. This component can be simulated with the following parameters: δ = 0.65 mm/s and ΔEQ = 1.87 mm/s (13 % of total intensity). The nature of this component is currently not well understood (18, 20, 32).2 Chemical reconstitution of the AI enzyme results in an increase in the absorbance at 400 nm (Figure 1A, dashed line) and a concomitant increase in the amount of iron and sulfide bound to the protein subsequent to gel-filtration, affording 5.8 ± 0.1 equiv of iron and 4.4 ± 0.4 equiv of sulfide per polypeptide.
Figure 1.
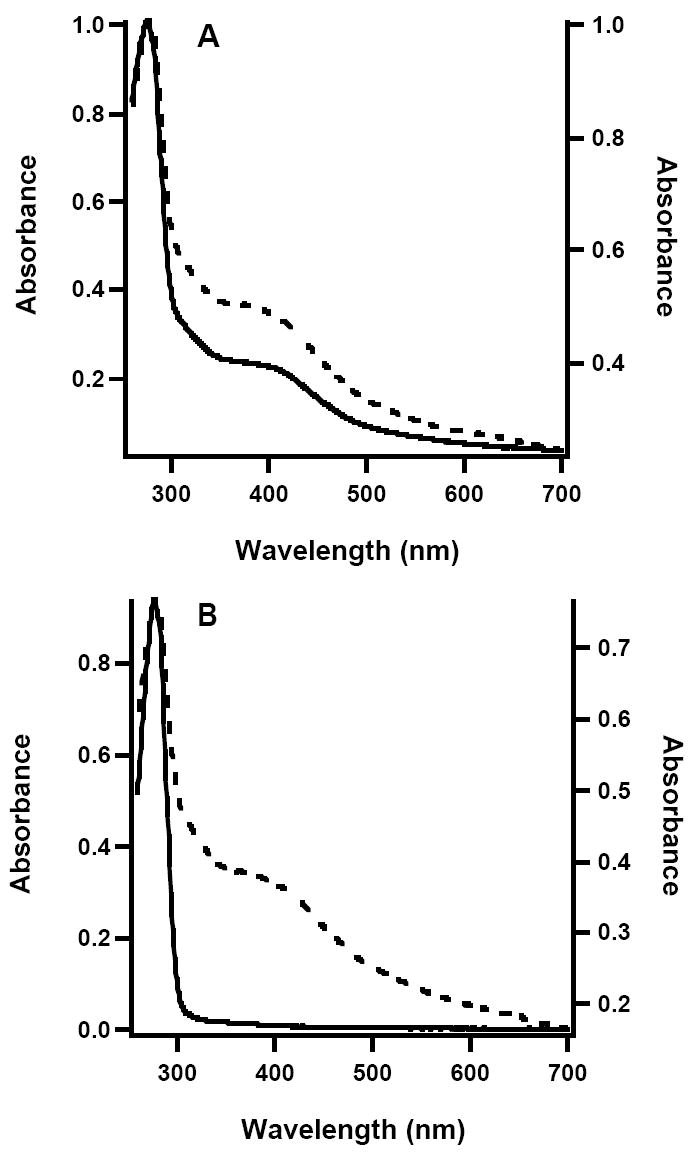
UV-vis spectra of (A) Phk WT NadA; AI (solid line, left axis); RCN (dashed line, right axis), and (B) Phk apo-NadA (solid line, left axis); reconstituted (dashed line, right axis).
Figure 2.
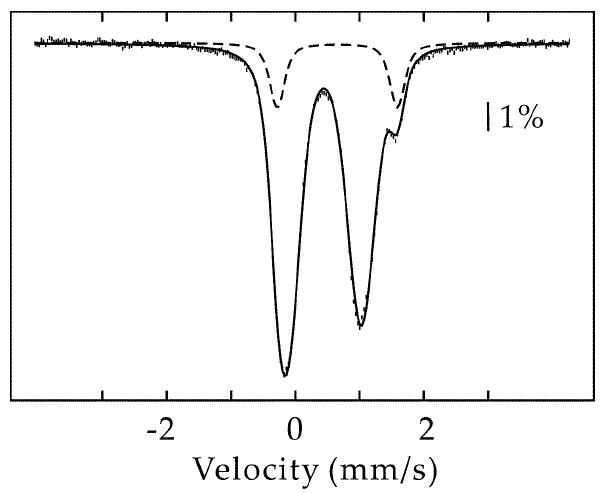
4.2-K/53-mT Mössbauer spectrum of AI Phk NadA. The spectrum can be simulated with two quadrupole doublets with the following parameters: δ(1) = 0.45 mm/s, ΔEQ(1) = 1.15 mm/s (87 % of total intensity) and δ(2) = 0.65 mm/s, ΔEQ(2) = 1.87 mm/s (13 % of total intensity). The contribution of the second quadrupole doublet is also shown individually as a dashed line.
NadA catalyzes the condensation of DHAP and ImAsp. ImAsp is an unstable species, which hydrolyzes to ammonia and OAA at 37 °C and pH 8.0 with a half-life of 144 s (10). In vivo, ImAsp is provided by the action of l-aspartate oxidase (NadB); however, an equilibrium concentration can also be generated chemically by Schiff-base formation between OAA and an ammonia source, such as AS. Using chemically generated ImAsp, AI Phk NadA catalyzed formation of QA at 50 °C with a Vmax/[ET] of 1.9 min-1, while the reconstituted enzyme displayed a Vmax/[ET] of 3.2 min-1 (specific activity = ~0.1 μmol min-1 mg-1).
Given that the activity of Phk NadA described in this work is considerably less than that reported by Sakuraba et al (22), it is conceivable that the Fe/S cluster in this enzyme acts as a negative regulator of NadA activity. To address this possibility, the apo form of Phk NadA was generated by inducing expression of the nadA gene in the absence of induction of genes on plasmid pDB1282, and adding the Fe(II) chelator o-phenanthroline to the growth medium at induction. The UV-vis spectrum of the anaerobically isolated protein is shown in Figure 1B (solid line), and is devoid of features that would indicate the presence of an Fe/S cluster. Consistent with the absence of the cofactor, the protein contained < 1.2 ± 0.2 irons per polypeptide, and sulfide was below the limit of detection (< 0.2 equiv). Activity determinations on apo Phk NadA indicated that it was unable to catalyze formation of QA; the limit of detection was 5 μM QA in a 20 min incubation. When apo Phk NadA was reconstituted with iron and sulfide, its UV-vis spectrum changed dramatically (Figure 1B, dashed line), and it displayed a Vmax/[ET] of 3.3 min-1, similar to that of the AI enzyme produced in the presence of the gene products encoded on plasmid pDB1282 and then further reconstituted. When Phk NadA was isolated aerobically by the procedure of Sakuraba et al, it was devoid of an Fe/S cluster, as evidenced by its UV-vis spectrum. It was also unable to catalyze formation of QA. Again, reconstitution of aerobically isolated Ph NadA resulted in significant gain in the protein’s catalytic competence (data not shown).
Analytical and Spectroscopic Characterization of WT and Variant E. coli NadA Proteins
The primary structure of Phk NadA contains only five cysteines, three of which (C83, C170, and C256) are conserved among all annotated NadA proteins (Scheme 2). Interestingly, these three cysteines in Phk NadA are found in or near surface loops that are not observed in the X-ray structure (residues 79-90, 165-175, and 257-260; see grey bars in Scheme 2) because of their disordered nature (22). This observation, in concert with the bioinformatics analysis, suggests that these cysteines are those that coordinate the required Fe/S cluster, and allows prediction that C→S or C→A substitutions made at equivalent positions in E. coli NadA should render the protein inactive. E. coli NadA contains a CXXCXXC motif, which is often found in Fe/S proteins of the [4Fe-4S]-containing ferredoxin class (12). The cysteines in this motif typically contribute three ligands to the Fe/S cluster, while the fourth is provided by a cysteine that can be considerably further away in sequence space. This motif is found in a large fraction, if not a majority, of NadA proteins, and was one of the initial indications that these proteins might contain Fe/S clusters (13). However, Phk and Mtb NadA proteins do not contain the motif, and only the last cysteine within it is strictly conserved. Therefore, serine substitutions at the cysteines in this motif were also made to assess their effect on cluster formation and turnover.
WT and variant E. coli hexahistidine-tagged NadA proteins were produced in the presence of the gene products from plasmid pDB1282 as described previously (29). Typical purifications from 20 g of cell paste yielded 200-250 mg of protein that was ≥95% pure. The UV-vis spectrum of AI WT E. coli NadA has been reported (14). It is shown again here in Figure 3A (solid line) to allow visual comparison with UV-vis spectra of variant proteins containing targeted C→S substitutions. The AI WT protein contains 3.0 ± 0.1 irons and 2.3 ± 0.1 sulfides, and the ratio of the absorbance at 280 nm to that at 400 nm, a qualitative characterization of cluster content (lower numbers indicate better cluster incorporation), is 4.5 (Table 1). After reconstitution with an 8-fold excess of iron and an 8-fold excess of sulfide, the protein is found to contain 10.0 ± 0.7 equiv of Fe and 8.2 ± 1.2 equiv of S2- per polypeptide, indicating the presence of adventitiously bound iron and sulfide, as has been reported in a previous Mössbauer analysis of E. coli NadA (14). Reconstitution with a 3-fold excess of iron and an 8-fold excess of sulfide allows for more reasonable Fe/S numbers (3.5 ± 0.5 equiv of Fe and 3.6 ± 0.3 equiv of S2- per polypeptide), which are in the range of the expected values for a [4Fe-4S] cluster. Reconstitution also increases the amplitude of the peak at 400 nm in the UV-vis spectrum (Figure 3A, dashed line), as well as the activity of the protein (Table 1, and vide infra).
Figure 3.
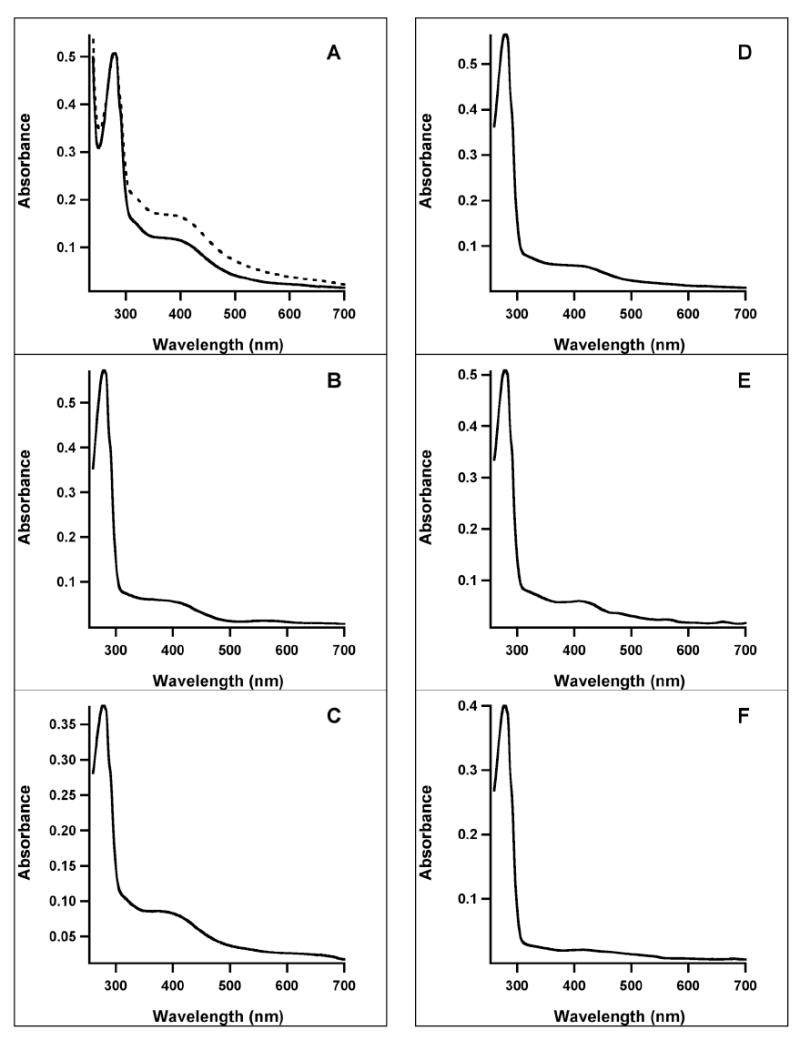
UV-vis spectra of AI (solid line) and RCN (dashed line) WT and variant E. coli NadA proteins (A) WT; (B) C291S; (C) C294S; (D) C113S; (E) C200S; (F) C297S.
Table 1.
Properties of E. coli Wild-Type and Variant NadA Proteinsa
| Sample | Iron per polypeptide | Sulfide per polypeptide | Ratioc | Turnover number (U)b | |||
|---|---|---|---|---|---|---|---|
| As Isolated | Reconstituted | ||||||
| NadB | OAA/AS | NadB | OAA/AS | ||||
| WT | 3.0 ± 0.1 | 2.3 ± 0.1 | 4.5 | 0.30 | 2.2 | 1.8 | 4.1 |
| C64S | 1.4 ± 0.1 | 0.73 ± 0.2 | 5.5 | 0.15 | 0.52 | 1.4 | 1.9 |
| C113A | 0.84 ± 0.1 | 0.9 ± 0.1 | 8.2 | ND | ND | ND | ND |
| C113S | 1.3 ± 0.4 | 0.4 ± 0.1 | 9.8 | ND | ND | ND | ND |
| C119S | 1.0 ± 0.4 | 0.6 ± 0.1 | 8.7 | 0.01 | 0.01 | 0.20 | 0.28 |
| C128S | 2.7 ± 0.1 | 1.1 ± 0.1 | 3.6 | 0.30 | 0.26 | 0.97 | 0.66 |
| C195S | 1.5 ± 0.1 | 1.5 ± 0.1 | 5.5 | 0.22 | 0.69 | 2.8 | 3.2 |
| C200A | 1.5 ± 0.3 | 1.2 ± 0.1 | 7.1 | ND | ND | ND | ND |
| C200S | 1.0 ± 0.3 | 0.5 ± 0.1 | 8.6 | ND | ND | ND | ND |
| C291S | 0.8 ± 0.1 | 0.5 ± 0.1 | 10.2 | 0.09 | 0.33 | 1.6 | 3.3 |
| C294S | 2.1 ± 0.1 | 1.8 ± 0.2 | 4.6 | 1.1 | 2.0 | 4.2 | 3.0 |
| C297A | 0.3 ± 0.1 | 0.5 ± 0.1 | 15.3 | ND | ND | ND | ND |
| C297S | 0.3 ± 0.2 | 0.1 ± 0.1 | 19.4 | ND | ND | ND | ND |
ND, activity not detected. Reported errors reflect one standard deviation
Unit (U) defined as μM quinolinic acid per μM NadA, per min
Defined as A280 nm/A400 nm
In the presence of chemically generated ImAsp, RCN WT NadA catalyzed formation of QA with a Vmax/[ET] of 4.1 min-1, a 1.9-fold increase from that of the AI enzyme (Figures 4A and 4B, closed circles, and Table 1). The activity determined using chemically generated ImAsp is significantly higher than that obtained when using NadB and l-aspartate to generate the unstable substrate (Table 1, and Figures 4A and 4B, open circles). The lower activity may result from the finding that fumarate, the electron acceptor used to re-oxidize the flavin cofactor of NadB under anaerobic conditions, displays competitive inhibition with respect to ImAsp, and that ImAsp binds tightly to NadB (8). Therefore, ImAsp must be released from the enzyme prior to fumarate binding to allow re-oxidation of the flavin for subsequent rounds of turnover. To substantiate the accuracy of QA determination by HPLC, a second, independent method for activity determination was also employed, in which the amount of DHAP consumed was quantified spectrophotometrically (15). As shown in Figure 5, these two different methods correlate well.
Figure 4.
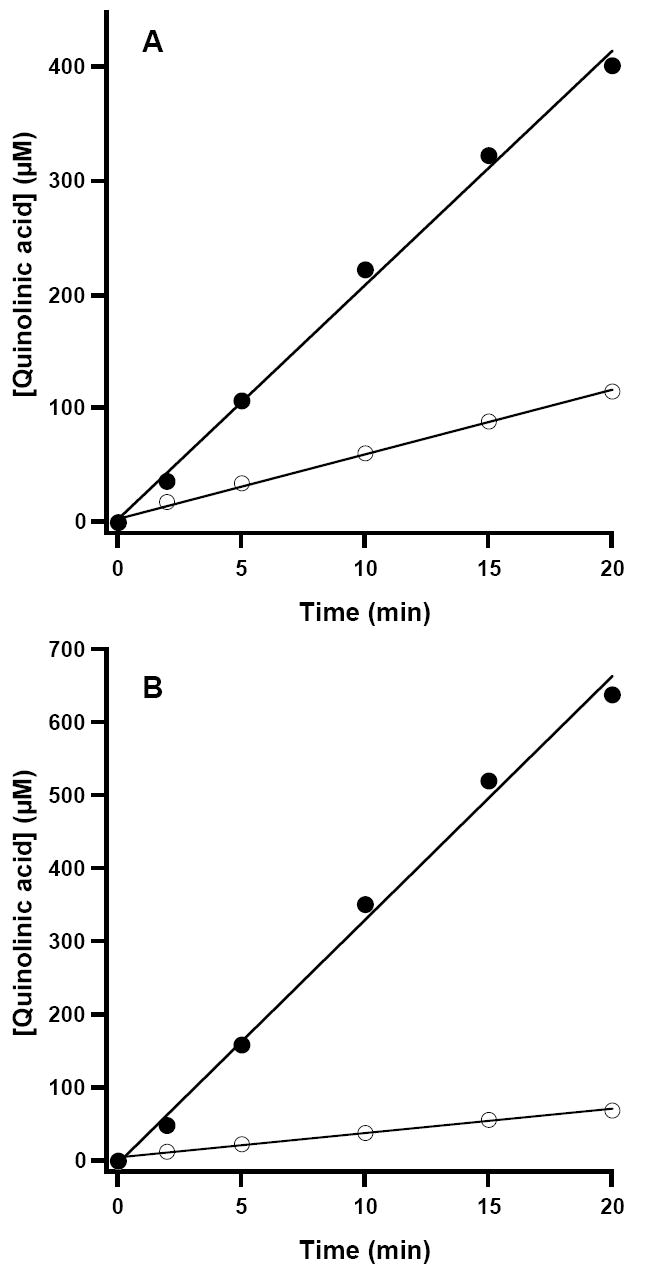
Time-dependent formation of QA catalyzed by WT E. coli NadA. The reaction was carried out as described in Materials and Methods and contained either 15 μM AI NadA (A) or 5 μM RCN NadA (B). Open circles represent assays with NadB and l-aspartate as the ImAsp source; closed circles represent assays with OAA and AS as the ImAsp source. Solid lines correspond to linear fits to the data over the entire range.
Figure 5.
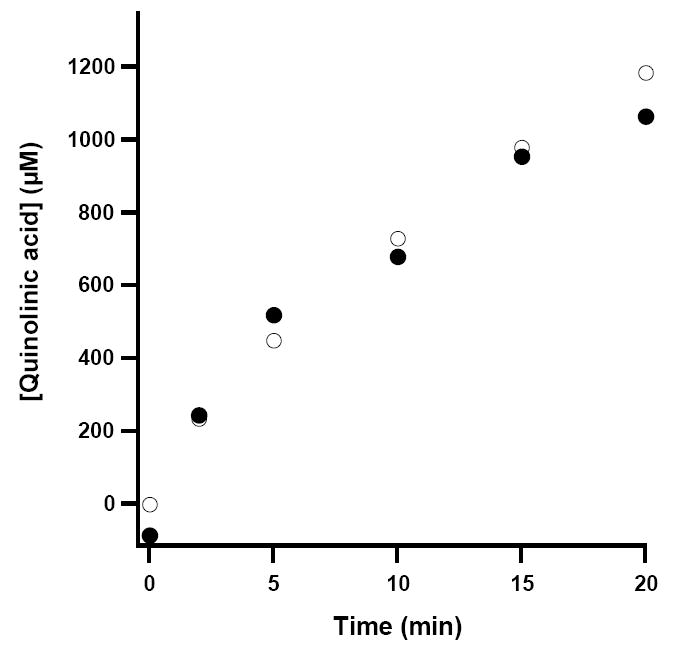
Correlation of two methods for the time-dependent determination of QA. HPLC method (open circles); enzymatic method (closed circles). The curvature associated with the plots derives from the almost complete turnover of the DHAP substrate in the assay (~1 mM).
The C113S, C200S, C291S, C294S and C297S variants were purified anaerobically—under the conditions described for WT NadA—and characterized. Cysteines 291 and 294 are the first two cysteines in the CXXCXXC motif, while cysteines 113, 200, and 297 are those that are strictly conserved among NadA proteins (Scheme 2). The C291S and C294S NadA variants are brown in color and display UV-vis spectra that are similar to that of the WT protein (Figures 3B & 3C, respectively). The A280/A400 ratios, 10.2 and 4.6, respectively, indicate varying degrees of cluster incorporation, which is consistent with the amount of iron and sulfide associated with the AI proteins (Table 1). Although the C291S variant appears to be seriously deficient in cluster content, the AI protein is capable of catalyzing formation of QA, displaying a Vmax/[ET] that is ~3-fold lower than that of the WT protein when using NadB to generate ImAsp, and ~7-fold lower when using chemically generated ImAsp. Upon reconstitution of this variant, its activity increased dramatically, and was comparable to that of WT NadA (Table 1). The AI and RCN C294S variant displayed higher activity than the WT protein when NadB was used to generate ImAsp, and comparable activity in the presence of chemically generated ImAsp (Table 1).
The UV-vis spectra of the AI C113S, C200S, and C297S variants are displayed in Figures 3D, 3E, and 3F, respectively. Their A280/A400 ratios varied from 8.6 to 19.4, and paralleled the amount of iron and sulfide found associated with the particular protein, which was consistently lower than that of WT NadA (Table 1). In addition, neither of these three variants, in its AI or RCN state, was capable of catalyzing formation of QA, regardless of the method used to generate ImAsp, suggesting that they are prime candidates for ligands to the Fe/S cluster. After reconstitution, all three variants showed an increase in the peak at 400 nm (not shown), yet there was no concomitant gain in observed activity (vide infra) (Table 1).
In addition to the five cysteine residues that are conserved and/or present in the CXXCXXC motif, the four remaining Cys residues in the E. coli protein (C64, C119, C128, and C195) were also each individually changed to Ser, and the corresponding variants characterized. All of these AI variants, except for C119S, had only slightly lower iron and sulfide content, and displayed UV-vis spectra with slightly higher A280/A400 ratios (Table I). The C119S variant had significantly lower iron and sulfide content and displayed a significantly higher A280/A400 ratio; however, the enzyme displayed catalytic competence, though significantly low, in both the AI and RCN forms, suggesting that this residue does not ligate the cluster (Table 1). The activities of the C64S, C128S, C195S, C291S, and C294S variants varied considerably depending on the particular variant and the conditions of the assay. The activity of AI or RCN WT E. coli NadA is higher when ImAsp is generated chemically than when ImAsp is generated using NadB and l-aspartate. Only one of the variants responded similarly (C291S), while most of the others displayed comparable activities under both conditions. The RCN C294S variant, however, displayed slightly lower activity in the presence of chemically generated ImAsp (Table 1).
EPR and Mössbauer Spectroscopy of the C200S variant of E. coli NadA
As shown in Table 1, iron and sulfide analyses, and UV-vis spectra of E. coli NadA variants C113S, C200S, and C297S, in which each of the three proposed cysteine ligands to the Fe/S cluster has been changed, suggest the presence of small amounts of Fe/S clusters despite having one of the purported ligands to the cluster removed. The assignment as true [4Fe-4S] clusters rather than adventitiously bound iron and sulfide was verified by further spectroscopic studies on the C200S variant. The RCN C200S variant contained 5.7 ± 0.4 irons and 2.9 ± 0.2 sulfides per polypeptide. The 4.2-K/53-mT Mössbauer spectrum of this sample is shown in Figure 6A. In addition to the broad and featureless absorption extending from −5 mm/s to +5 mm/s (~40 % of total Fe), there is a pronounced quadrupole doublet with parameters typical of a [4Fe-4S]2+ cluster: δ = 0.44 mm/s and ΔEQ, = 1.13 mm/s. This quadrupole doublet accounts for 55% of total Fe. The EPR spectrum of RCN C200S NadA, which is reduced in the presence of 2 mM sodium dithionite corroborates the presence of reduced [4Fe-4S]+ clusters (Figure 6B). The spectrum, obtained at 13 K and 5 mW power, shows an axial signal with approximate g-values (g1 = 2.04 and g2 = 1.93) that are nearly identical to the g-values observed for the WT protein (14, 15).
Figure 6.
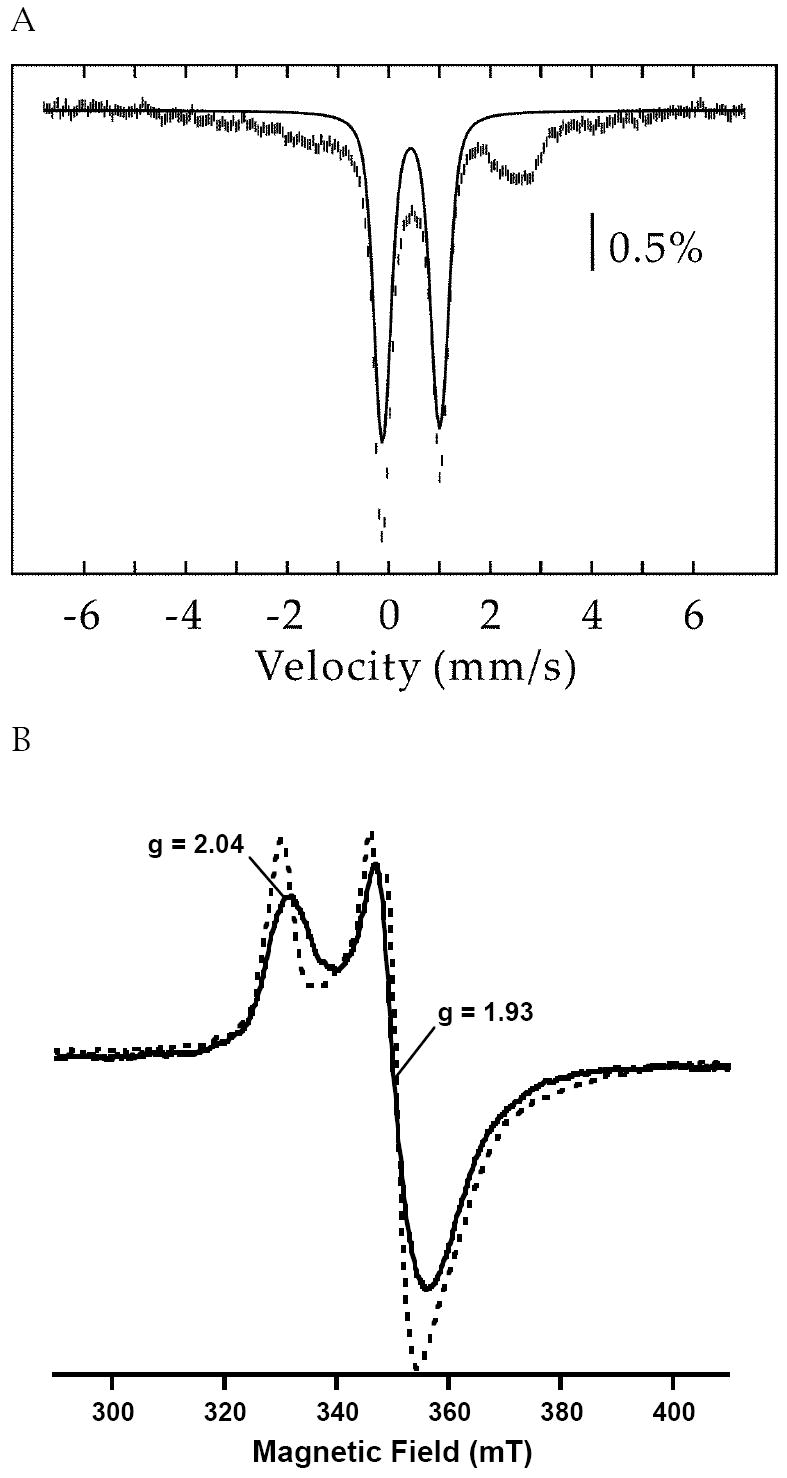
(A) 4.2-K/53-mT Mössbauer spectrum of C200S E. coli NadA. The solid line is a quadrupole doublet (δ = 0.44 mm/s and ΔEQ = 1.13 mm/s, 55% of total intensity) representing the fraction of [4Fe-4S]2+ clusters. (B) EPR spectrum of RCN C200S NadA (581 μM, solid line) overlaid with spectrum of RCN WT E. coli NadA (300 μM, dashed line), both reduced in the presence of 2 mM sodium dithionite. Obtained at 5 mW power, 13 K and 10 G modulation amplitude.
The results of EPR and Mössbauer spectroscopies suggest that despite the substitution of one of the Fe/S cluster ligands with Ser, a substoichiometric amount of [4Fe-4S] cluster is still formed; however, the resulting enzyme is inactive. To assess whether the ability to form a cluster on this variant protein derives from possible Ser ligation, variants containing C→A substitutions at C113, C200, and C297 were also constructed. As expected, the resulting Ala variants were also inactive, but displayed similar UV-vis spectra and iron and sulfide content, suggesting that they too were able to support formation of a [4Fe-4S] cluster (Table 1).
Analysis of WT E. coli Apo-NadA
In previous studies of E. coli NadA, the protein was produced largely in the apo form, containing 0.15-0.2 iron and sulfide atoms per polypeptide when it was isolated under aerobic conditions (15). When the apo protein was reconstituted with iron and sulfide in the presence of DTT, approximately one [4Fe-4S] cluster was found to assemble, and the protein gained catalytic activity. To obviate the possibility that the apo protein may have lost activity via oxidative processes that result in structural changes (e.g. disulfide or sulfenic acid formation) that are reversed upon incubation under reconstitution conditions, WT E. coli NadA was produced recombinantly in the presence of o-phenanthroline, and then isolated under anaerobic conditions. Similarly to Phk NadA produced under the same conditions, the E. coli enzyme was isolated largely in an apo form as determined from its UV-vis spectrum (Figure 7, solid line) and iron and sulfide content (less than 0.4 equiv each per polypeptide). Reconstitution with a 5-fold excess of iron and sulfide afforded a protein exhibiting UV-vis spectral features that are consistent with the presence of a [4Fe-4S] cluster (Figure 7, dashed line). Iron and sulfide analyses of the reconstituted apo protein indicated the presence of 5.8 ± 0.4 irons and 5.0 ± 0.2 sulfides per polypeptide, while activity assays in the presence of NadB showed it to catalyze formation of QA with a Vmax/[ET] of 0.34 min-1, similar to that obtained for the AI WT enzyme produced in the absence of o-phenanthroline supplementation (Table 1). Importantly, when apo E. coli NadA was reconstituted with DTT alone, with DTT plus sulfide, or with DTT plus iron, the activity of the protein remained below the limit of detection of the assay, suggesting that indeed the presence of the cluster is necessary for activity rather than treatment of the protein with any one of the components of the reconstitution mixture.
Figure 7.
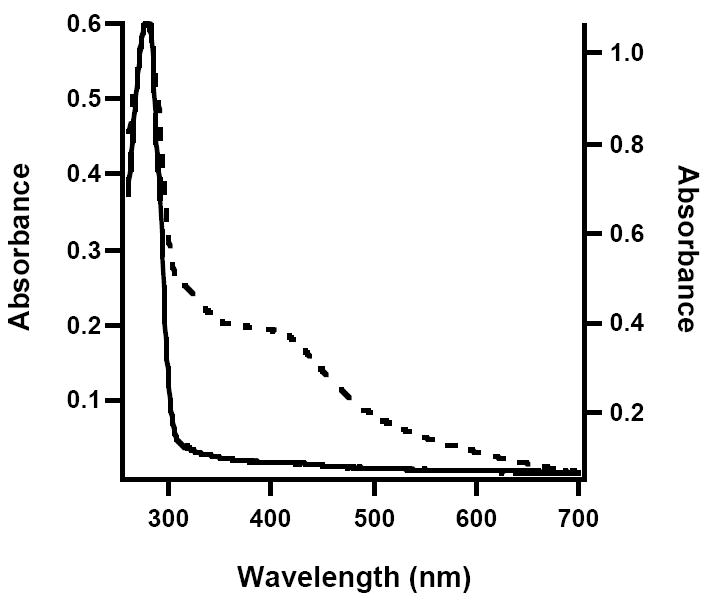
UV-vis spectra of AI (solid line, left axis) and RCN (dashed line, right axis) apo-NadA from E. coli.
Cloning and Expression of the M. tuberculosis nadA Gene, and Purification and Characterization of its Product
The enzymes involved in NAD biosynthesis and recycling have been suggested to be noteworthy targets for the design of antibacterial agents against a number of bacteria, including M. tuberculosis (Mtb) (33, 34). To this end, the nadA gene from Mtb was cloned and co-expressed in E. coli along with genes of the isc operon from A. vinelandii, encoded on plasmid pDB1282. NadA from Mtb lacks the CXXCXXC motif found in the E. coli enzyme, but contains three conserved cysteines in a total of ten. Consistent with the hypothesis that these three cysteines coordinate an Fe/S cluster required for turnover, as-isolated (AI) Mtb NadA was brown in color and displayed a UV-vis spectrum that is in line with the presence of [4Fe-4S] clusters (Figure 8, solid line). Chemical reconstitution of AI Mtb NadA results in an increase in the absorbance at 400 nm (Figure 8, dashed line), suggestive of additional cluster incorporation. Iron and sulfide analyses indicate that AI Mtb NadA contains 4.7 ± 0.2 equiv of Fe and 2.6 ± 0.1 equiv of S2- per polypeptide, while the RCN protein contains 6.4 ± 0.3 equiv of Fe and 2.7 ± 0.1 equiv of S2- per polypeptide. Under anaerobic conditions, AI Mtb NadA catalyzed formation of QA with a Vmax/[ET] of 0.24 min-1; reconstitution resulted in rates that were typically two-fold higher. The presence of one [4Fe-4S] cluster on Mtb NadA is substantiated by Mössbauer and EPR spectroscopies. The Mössbauer spectrum of AI Mtb NadA is dominated by a quadrupole doublet with parameters typical of [4Fe-4S]2+ clusters (δ = 0.46 mm/s and ΔEQ = 1.14 mm/s, 91% of total intensity) (Figure 9). In conjunction with the results from iron analysis, the Mössbauer spectrum indicates the presence of one [4Fe-4S] cluster per polypeptide; the remaining iron in excess of 4 equiv is predominantly associated with adventitiously bound ferrous species. A sample of AI Mtb NadA reduced with 2 mM dithionite exhibited a spectrum similar to that observed for E. coli NadA treated in the same fashion, displaying g-values of approximately g1 = 2.06 and g2 = 1.92 (Figure 10).
Figure 8.
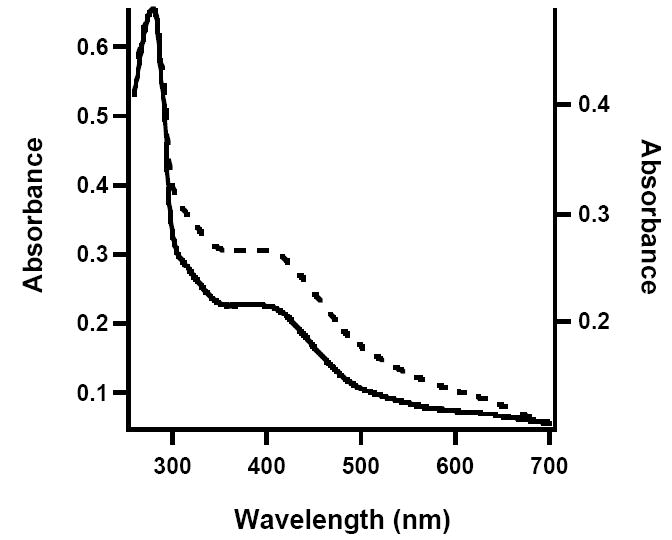
UV-vis spectra of Mtb NadA. AI (solid line, left axis), RCN (dashed line, right axis).
Figure 9.
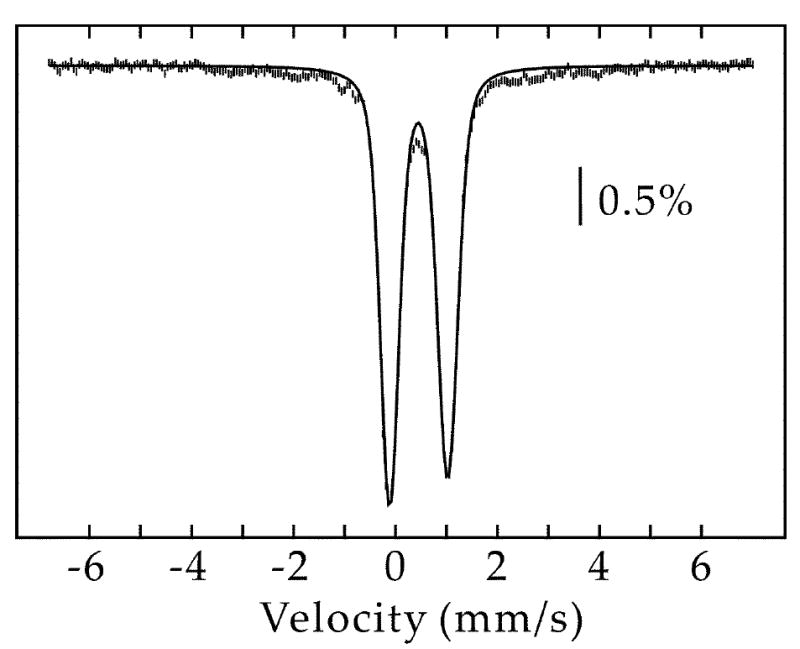
4.2-K/53-mT Mössbauer spectrum of Mtb NadA. The solid line is a quadrupole doublet (δ = 0.46 mm/s and ΔEQ = 1.14 mm/s, 91% of total intensity) representing the fraction of [4Fe-4S]2+ clusters.
Figure 10.
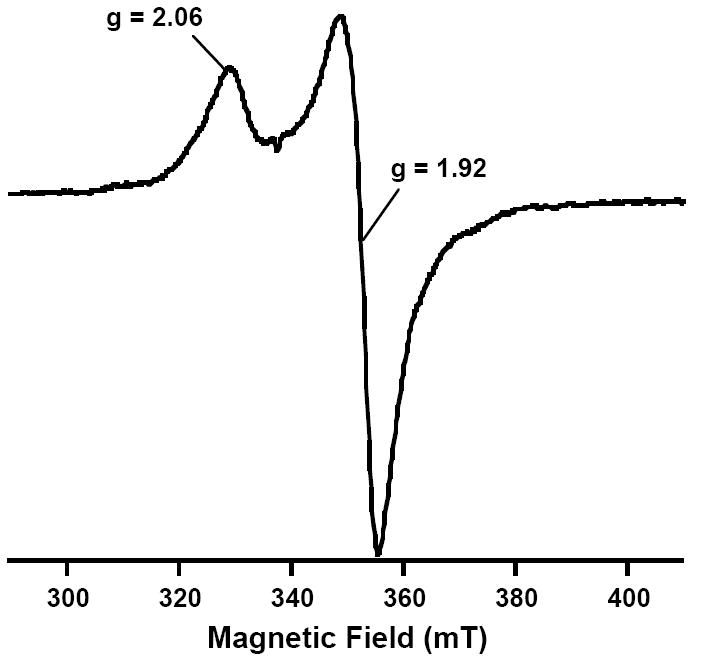
EPR spectrum of dithionite-reduced AI Mtb NadA. Parameters: 13 K, 5 mW power, and 10 G modulation amplitude.
DISCUSSION
Quinolinate synthase has suffered a sinuous history in regard to its cofactor requirement in catalysis. The first indication that it might require an Fe/S cluster arose from studies by Gardner and Fridovich, in which the protein was shown to be inactivated by exposure to hyperbaric oxygen (13). In E. coli cell extracts, this oxygen-dependent inactivation was partially reversed upon incubation under anaerobiosis; however, reversal was blocked in the presence of iron chelators such as α,α’-dipyridyl or 1,10-phenanthroline. Moreover, it was noticed that the primary structure of E. coli NadA contained a CXXCXXC motif, which is often found in proteins that contain Fe/S clusters, especially [4Fe-4S]-containing ferredoxins (12). The cysteines lying in the motif contribute three protein-thiolate ligands to the cluster, while the fourth cysteine typically resides 20 to 40 amino acids away. Additional evidence in support of an Fe/S cluster associated with E. coli NadA emanated from genetic studies in E. coli, wherein an iscS deletion mutant was found to be auxotrophic for nicotinic acid when grown on minimal medium (35). The iscS gene encodes a cysteine desulfurase, a pyridoxal 5’-phosphate-dependent enzyme that liberates elemental sulfur in the form of a cysteine persulfide to be used in the biosynthesis of Fe/S clusters and other important biological compounds (36, 37). E. coli NadA was subsequently shown by two independent groups to contain [4Fe–4S] clusters. In one study the protein was overproduced in the presence of proteins that participate in various aspects of Fe/S cluster biosynthesis, and was found to contain 5.0 ± 1.4 irons and 2.8 ± 0.3 sulfides per polypeptide (14). In another study it was overproduced in the absence of accessory proteins, and contained 3.1 irons and 3.0 sulfides per polypeptide (15). Upon exposure of the protein to oxygen, UV-vis signatures that indicate the presence of Fe/S clusters bleached concomitantly with loss of activity (15).
Results from the Fontecave and Booker laboratories were in contrast to those of Ceciliani et al, who reported the first purification of recombinant NadA to homogeneity, in which the E. coli enzyme was isolated from inclusion bodies obtained during overproduction (38). The purification included solubilization in the presence of 4 M urea and 50 mM EDTA, and dialysis in the presence of 10 mM EDTA, conditions not typically conducive to maintaining Fe/S clusters within a protein scaffold. Moreover, their reported specific activity (600 μmol min-1 mg-1) was significantly greater than that reported by Cicchillo et al for the Fe/S-containing enzyme (0.015 μmol min-1 mg-1) (14), or that reported in this study for reconstituted E. coli NadA (0.10 μmol min-1 mg-1). It is possible that there was a typographical error in their definition of a unit, which was 1 mmol min-1 instead of 1 μmol min-1, the accepted international unit definition. However, their reported activity would still be considerably higher than that observed for the enzyme containing a [4Fe-4S] cluster.
Surprisingly, the X-ray structure determination of NadA from Phk, solved in 2005, seemed to lend support to the results of Ceciliani et al (22). The structure was solved to 2.0 Å resolution, and described as monomeric, but exhibiting pseudo-3-fold symmetry involving three repeating αβα sandwich domains. The protein was crystallized in the presence of malate, which is found in the structure at the interface of the three domains, suggesting the location of the active site. Interestingly, no metal or metallocofactor is found in the structure, and the possibility that the enzyme might contain an Fe/S cluster was not even addressed. Most surprisingly, however, this enzyme was stated to catalyze formation of QA with a specific activity of 2.2 μmol min-1 mg-1 (22).
Given the inconsistencies listed above we initiated efforts to conduct a comprehensive analysis of the requirement for Fe/S clusters in several quinolinate synthases. In light of the suggestion that NadA is a member of the Fe/S-dependent class of the hydro-lyase family of enzymes (16), we posited that similarly to aconitase, each of three of the four iron atoms in the [4Fe-4S] cluster would be ligated by one Cys residue, allowing the non-cysteinate-ligated iron to participate directly in the reaction. This premise allows prediction that NadA should contain at least three conserved Cys residues in its primary structure. Indeed bioinformatics analyses conducted by us (39) and others (40) indicate that three cysteines are conserved throughout over 700 annotated NadA proteins. Using site-directed mutagenesis, we changed each of the nine Cys residues in the primary structure of E. coli NadA to Ser or both Ser and Ala, and isolated and characterized each of the variant proteins. Only six of the variants (C113S, C113A, C200S, C200A, C297S, C297A) displayed complete loss of activity, suggesting that C113, C200, and C297 are viable candidates for ligands to the cluster. Initially we had predicted that removal of one of three Cys ligands to a [4Fe-4S] cluster would yield a protein that was completely devoid of cluster. However, iron and sulfide analyses in concert with UV-vis spectroscopy suggested the presence of small amounts of bound cluster, which was verified by Mössbauer spectroscopy on the 57FeSO4-RCN C200S variant. We would predict, therefore, that the [4Fe-4S] cluster in the C200S variant is ligated by only two Cys residues as has been shown previously for the RCN C30A variant of the activase protein of the anaerobic ribonucleotide reductase (41). Interestingly, only one of the three conserved Cys residues (C297) is found in the CXXCXXC motif that was predicted to house the ligands to the cluster. Recently, we have shown that the remaining two cysteines (C291 and C294) can be oxidized to a disulfide bond by molecular oxygen or oxidized thioredoxin, and that the dithiol/disulfide transition regulates the activity of the protein (42).
Although it was shown that E. coli NadA could bind a [4Fe-4S] cluster (14, 15), given the differences in the specific activity between presumably apo NadA, isolated by Ceciliani et al (38), and NadA containing a [4Fe-4S] cluster (14), we suggested that (1) the cluster might not be obligate for turnover, but its presence might down-regulate the activity of the protein; or (2) it is not the presence of the Fe/S cluster that gives rise to activity, but rather treatment of the protein with one of the components of the reconstitution mixture. We produced apo forms of NadA from both Phk and E. coli by addition of o-phenanthroline to the media at induction, a procedure devised by Parkin et al to generate an apo form of an F208Y variant of the R2 subunit of the class I ribonucleotide reductase from E. coli (30). The proteins were then isolated under anaerobic conditions and characterized. In both cases, the resulting apo proteins did not display observable turnover; however, activity was restored upon incubation under conditions that result in formation of [4Fe-4S] clusters on the proteins. To show that indeed the Fe/S cluster is required for turnover, apo E. coli NadA was incubated with individual components of the reconstitution mixture and then used in activity determinations. Incubation with neither DTT alone, nor DTT and FeCl3, nor DTT and sodium sulfide resulted in observable turnover. All three key components of the reconstitution mixture were necessary to bring about catalytic competence.
As proof of principle, we cloned another nadA gene that did not encode a protein containing a CXXCXXC motif. The enzyme from Mtb was chosen, because it had been reported that it is essential for in vitro growth of the organism (34), suggesting that it might be a viable target for the design of antituberculosis agents. Recently, however, it was shown that, in contrast to prevailing thought, Mtb can scavenge nicotinamide from its infected host and construct NAD via the Preiss–Handler recycling pathway, suggesting that it is not essential in vivo (43). We cloned and expressed the nadA gene from Mtb and isolated and characterized its product. Like the enzymes from E. coli and Phk, AI Mtb NadA contained a [4Fe-4S] cluster, as determined by chemical and spectroscopic analyses. In addition, it catalyzed formation of QA with Vmax/[ET] values that are similar to those from Phk and E. coli NadA proteins. Efforts were made to generate C→S variants at the three conserved cysteines in Mtb NadA (C114, C201, and C300); however, each of the resulting proteins was highly unstable and could not be isolated in soluble form.
Finally, we believe that our findings and the report of Sakuraba et al can be brought into accord. Sakuraba et al note that there are three regions that are not visible in electron density maps due to their disordered nature (22). These regions constitute three surface loops that separate each of the domains, spanning amino acids 79-90, 165-175, and 257-260. Our bioinformatics analysis indicates that two of these disordered loops contain C83 and C170, two of the strictly conserved cysteine residues postulated to ligate a [4Fe-4S] cluster. The third conserved cysteine (C256) is visible in the structure; however, it is immediately adjacent to the disordered loop spanning amino acids 257-260. It is likely that the absence of the [4Fe-4S] cluster, to which the three cysteines coordinate, renders the loops mobile, giving rise to their disordered nature.
The observed high activity displayed by Phk NadA can also be rationalized. In the report by Sakuraba et al, an HPLC method was used to quantify QA, employing the NadB reaction to generate the required ImAsp. We use a similar HPLC method to quantify QA, which is described herein. QA displays a maximum absorbance at 268 nm; however, in the report by Sakuraba et al, formation of QA was monitored at 254 nm. OAA displays a λmax at 255 nm, and we routinely observe time-dependent formation of this compound when conducting assays in which the NadB reaction is used to generate ImAsp, and observe it instantaneously in assays in which ImAsp is generated chemically. Therefore, it appears that the numbers reported by Sakuraba et al reflect formation of OAA from ImAsp (t1/2 = 144 s) generated in the NadB reaction, rather than turnover by NadA. We believe, therefore, that crystallization of Phk NadA in the presence of its [4Fe-4S] cofactor will give a much clearer picture as to the organization of the active site of the enzyme, and the manner in which this enzyme catalyzes its reaction.
Supplementary Material
Tables S1 and S2. This material is available free of charge via the Internet at http:/pubs.acs.org
Footnotes
This work was supported by NIH Grant GM-63847 (S.J.B.), the Dreyfus Foundation (Teacher Scholar Award to C.K.), and the Beckman Foundation (Young Investigator Award to C.K.)
Abbreviations: AS, ammonium sulfate; AI, as-isolated; BSA, bovine serum albumin; DHAP, dihydroxyacetone phosphate; DTT, dithiothreitol; EDTA; ethylenediaminetetraacetic acid; EPR, electron paramagnetic resonance; FAD, flavin adenine dinucleotide; Fe/S, iron–sulfur; HEPES, N-(2-hydroxyethyl)piperizine-N’-(2-ethanesulfonic acid); HPLC, high-performance liquid chromatography; IMAC, immobilized metal affinity chromatography; ImAsp, iminoaspartate; IPTG, isopropyl-β-d-thiogalactopyranoside; IS, internal standard; LB, Luria–Bertani; Mtb, Mycobacterium tuberculosis; MW, molecular weight; NAD, nicotinamide adenine dinucleotide; NadA, quinolinate synthase; NadB, l-aspartate oxidase; Ni-NTA, nickel nitrilotriacetic acid; OAA, oxaloacetate; PCR, polymerase chain reaction; Phk, Pyrococcus horikoshii; PMSF, phenylmethanesulfonyl fluoride; QA, quinolinic acid; RCN, reconstituted; SDS-PAGE, sodium dodecylsulfate-polyacrylamide gel electrophoresis; TCA, trichloroacetic acid; TFA, trifluoroacetic acid; UV-vis, UV-visible; WT, wild-type
The position of the low-energy line of the novel quadrupole doublet cannot be determined accurately. The spectrum can also be simulated with the following parameters: δ(1) = 0.43 mm/s, ΔEQ(1) = 1.19 mm/s (87% intensity); and δ(2) = 0.82 mm/s, ΔEQ(2) = 1.53 mm/s (13 % intensity). The parameters of the major component are nearly identical, and indicate the presence of [4Fe-4S]2+ clusters, regardless of the fitting model used. We speculate that the larger isomer shift of the minor component may indicate multidentate binding of an organic molecule with hard N/O donor atoms to the unique Fe site of the [4Fe-4S]2+ cluster, as was observed previously for aconitase (17) and pyruvate formate–lyase activase (31), a member of the radical SAM enzyme family. Addition of substrates, product, or analogs thereof to AI P. horikoshii NadA did thus far not yield a larger fraction of this component for more detailed spectroscopic characterization.
References
- 1.Frey PA, Hegeman AD. Enzymatic Reaction Mechanisms. Oxford University Press; Oxford: 2007. [Google Scholar]
- 2.Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci. 2007;32:12–19. doi: 10.1016/j.tibs.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 3.Pollak N, Dölle C, Ziegler M. The power to reduce: pyridine nucleotides–small molecules with a multitude of functions. Biochem J. 2007;402:205–218. doi: 10.1042/BJ20061638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kessler D, Rétey J, Schulz GE. Structure and action of urocanase. J Mol Biol. 2004;342:183–194. doi: 10.1016/j.jmb.2004.07.028. [DOI] [PubMed] [Google Scholar]
- 5.Begley TP, Chatterjee A, Hanes JW, Hazra A, Ealick SE. Cofactor biosynthesis—still yielding fascinating new biological chemistry. Curr Opin Chem Biol. 2008;12:118–125. doi: 10.1016/j.cbpa.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foster JW, Moat AG. Nicotinamide adenine dinucleotide biosynthesis and pyridine nucleotide cycle metabolism in microbial systems. Microbiol Rev. 1980;44:83–105. doi: 10.1128/mr.44.1.83-105.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chandler JL, Gholson RK. Studies on the de novo biosynthesis of NAD in Escherichia coli. Anal Biochem. 1972;48:529–535. doi: 10.1016/0003-2697(72)90108-x. [DOI] [PubMed] [Google Scholar]
- 8.Mortarino M, Negri A, Tedeschi G, Simonic T, Duga S, Gassen HG, Ronchi S. l-Aspartate oxidase from Escherichia coli. I. Characterization of coenzyme binding and product inhibition. Eur J Biochem. 1996;239:418–426. doi: 10.1111/j.1432-1033.1996.0418u.x. [DOI] [PubMed] [Google Scholar]
- 9.Tedeschi G, Negri A, Mortarino M, Ceciliani F, Simonic T, Faotto L, Ronchi S. l-Aspartate oxidase from Escherichia coli. II. Interaction with C4 dicarboxylic acids and identification of a novel l-aspartate:fumarate oxidoreductase activity. Eur J Biochem. 1996;239:427–433. doi: 10.1111/j.1432-1033.1996.0427u.x. [DOI] [PubMed] [Google Scholar]
- 10.Nasu S, Wicks FD, Gholson RK. l-Aspartate oxidase, a newly discovered enzyme of Escherichia coli, is the B protein of quinolinate synthetase. J Biol Chem. 1982;257:626–632. [PubMed] [Google Scholar]
- 11.Nasu S, Gholson RK. Replacement of the B protein requirement of the E. coli quinolinate synthetase system by chemically-generated iminoaspartate. Biochem Biophys Res Commun. 1981;101:533–539. doi: 10.1016/0006-291x(81)91292-4. [DOI] [PubMed] [Google Scholar]
- 12.Yasunobu KT, Tanaka M. The types, distribution in nature, structure-function, and evolutionary data of the iron-sulfur proteins. In: Lovenberg W, editor. Iron-Sulfur Proteins. II. Academic Press; New York: 1973. [Google Scholar]
- 13.Gardner PR, Fridovich I. Quinolinate synthetase: The oxygen-sensitive site of de novo NAD(P)+ biosynthesis. Arch Biochem Biophys. 1991;284:106–111. doi: 10.1016/0003-9861(91)90270-s. [DOI] [PubMed] [Google Scholar]
- 14.Cicchillo RM, Tu L, Stromberg JA, Hoffart LM, Krebs C, Booker SJ. Escherichia coli quinolinate synthetase does indeed harbor a [4Fe–4S] cluster. J Am Chem Soc. 2005;127:7310–7311. doi: 10.1021/ja051369x. [DOI] [PubMed] [Google Scholar]
- 15.Ollagnier-de Choudens S, Loiseau L, Sanakis Y, Barras F, Fontecave M. Quinolinate synthetase, an iron–sulfur enzyme in NAD biosynthesis. FEBS Lett. 2005;579:3737–3743. doi: 10.1016/j.febslet.2005.05.065. [DOI] [PubMed] [Google Scholar]
- 16.Flint DH, Allen RM. Iron–sulfur proteins with nonredox functions. Chem Rev. 1996;96:2315–2334. doi: 10.1021/cr950041r. [DOI] [PubMed] [Google Scholar]
- 17.Beinert H, Kennedy MC, Stout CD. Aconitase as iron-sulfur protein, enzyme, and iron-regulatory protein. Chem Rev. 1996;96:2335–2373. doi: 10.1021/cr950040z. [DOI] [PubMed] [Google Scholar]
- 18.Emptage MH, Kent TA, Kennedy MC, Beinert H, Münck E. Mössbauer and EPR studies of activated aconitase: Development of a localized valence state at a subsite of the [4Fe-4S] cluster on binding of citrate. Proc Natl Acad Sci USA. 1983;80:4674–4678. doi: 10.1073/pnas.80.15.4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kennedy MC, Werst M, Telser J, Emptage MH, Beinert H, Hoffman BM. Mode of substrate carboxyl binding to the [4Fe-4S]+ cluster of reduced aconitase as studied by 12O and 13C electron–nuclear double resonance spectroscopy. Proc Natl Acad Sci USA. 1987;84:8854–8858. doi: 10.1073/pnas.84.24.8854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kent TA, Emptage MH, Merkle H, Kennedy MC, Beinert H, Münck E. Mössbauer studies of aconitase. Substrate and inhibitor binding, reaction intermediates, and hyperfine interactions of reduced 3Fe and 4Fe clusters. J Biol Chem. 1985;260:6871–6881. [PubMed] [Google Scholar]
- 21.Lauble H, Kennedy MC, Beinert H, Stout CD. Crystal structures of aconitase with isocitrate and nitroisocitrate bound. Biochemistry. 1992;31:2735–2748. doi: 10.1021/bi00125a014. [DOI] [PubMed] [Google Scholar]
- 22.Sakuraba H, Tsuge H, Yoneda K, Katunuma N, Ohshima T. Crystal structure of the NAD biosynthetic enzyme quinolinate synthase. J Biol Chem. 2005;280:26645–26648. doi: 10.1074/jbc.C500192200. [DOI] [PubMed] [Google Scholar]
- 23.Beinert H. Micro methods for the quantitative determination of iron and copper in biological material. Methods Enzymol. 1978;54:435–445. doi: 10.1016/s0076-6879(78)54027-5. [DOI] [PubMed] [Google Scholar]
- 24.Beinert H. Semi-micro methods for analysis of labile sulfide and of labile sulfide plus sulfane sulfur in unusually stable iron-sulfur proteins. Anal Biochem. 1983;131:373–378. doi: 10.1016/0003-2697(83)90186-0. [DOI] [PubMed] [Google Scholar]
- 25.Kennedy MC, Kent TA, Emptage M, Merkle H, Beinert H, Münck E. Evidence for the formation of a linear [3Fe-4S] cluster in partially unfolded aconitase. J Biol Chem. 1984;259:14463–14471. [PubMed] [Google Scholar]
- 26.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Vol. 3. Cold Spring Harbor Laboratory Press; Plainview, New York: 1989. [Google Scholar]
- 27.Cicchillo RM, Lee K-H, Baleanu-Gogonea C, Nesbitt NM, Krebs C, Booker SJ. Escherichia coli lipoyl synthase binds two distinct [4Fe–4S] clusters per polypeptide. Biochemistry. 2004;43:11770–11781. doi: 10.1021/bi0488505. [DOI] [PubMed] [Google Scholar]
- 28.Ramamurthy V, Swann SL, Paulson JL, Spedaliere CJ, Mueller EG. Critical aspartic acid residues in pseudouridine synthases. J Biol Chem. 1999;274:22225–22230. doi: 10.1074/jbc.274.32.22225. [DOI] [PubMed] [Google Scholar]
- 29.Cicchillo RM, Iwig DF, Jones AD, Nesbitt NM, Baleanu-Gogonea C, Souder MG, Tu L, Booker SJ. Lipoyl synthase requires two equivalents of S-adenosyl-l-methionine to synthesize one equivalent of lipoic acid. Biochemistry. 2004;43:6378–6386. doi: 10.1021/bi049528x. [DOI] [PubMed] [Google Scholar]
- 30.Parkin SE, Chen S, Ley BA, Mangravite L, Edmondson DE, Huynh BH, Bollinger JM., Jr Electron injection through a specific pathway determines the outcome of oxygen activation at the diiron cluster in the F208Y mutant of Escherichia coli ribonucleotide reductase Protein R2. Biochemistry. 1998;37:1124–1130. doi: 10.1021/bi9723717. [DOI] [PubMed] [Google Scholar]
- 31.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical biochemistry. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 32.Krebs C, Broderick WE, Henshaw TF, Broderick JB, Huynh BH. Coordination of adenosylmethionine to a unique iron site of the [4Fe–4S] of pyruvate formate–lyase activating enzyme: a Mössbauer spectroscopic study. J Am Chem Soc. 2002;124:912–913. doi: 10.1021/ja017562i. [DOI] [PubMed] [Google Scholar]
- 33.Gerdes SY, Scholle MD, D’Souza M, Bernal A, Baev MV, Farrell M, Kurnasov OV, Daugherty MD, Mseeh F, Polanuyer BM, Campbell JW, Anantha S, Shatalin KY, Chowdhury SA, Fonstein MY, Osterman AL. From genetic footprinting to antimicrobial drug targets: examples in cofactor biosynthetic pathways. J Bacteriol. 2002;184:4555–4572. doi: 10.1128/JB.184.16.4555-4572.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 35.Lauhon CT, Kambampati R. The iscS gene in Escherichia coli is required for the biosynthesis of 4-thiouridine, thiamin, and NAD. J Biol Chem. 2000;275:20096–20103. doi: 10.1074/jbc.M002680200. [DOI] [PubMed] [Google Scholar]
- 36.Zheng L, White RH, Cash VL, Dean DR. Mechanism for the desulfurization of l-cysteine catalyzed by the nifS gene product. Biochemistry. 1994;33:4714–4720. doi: 10.1021/bi00181a031. [DOI] [PubMed] [Google Scholar]
- 37.Mueller EG. Trafficking in persulfides: delivering sulfur in biosynthetic pathways. Nat Chem Biol. 2006;2:185–194. doi: 10.1038/nchembio779. [DOI] [PubMed] [Google Scholar]
- 38.Ceciliani F, Camori T, Ronchi S, Tedeschi G, Mortarino M, Galizzi A. Cloning, overexpression, and purification of Escherichia coli quinolinate synthetase. Prot Express Purif. 2000;18:64–70. doi: 10.1006/prep.1999.1153. [DOI] [PubMed] [Google Scholar]
- 39.The_UniProt_Consortium. The Universal Protein Resource (UniProt) Nucleic Acids Res. 2007;35:D193–D197. doi: 10.1093/nar/gkl929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murthy UMN, Ollagnier-de Choudens S, Sanakis Y, Abdel-Ghany SE, Rousset C, Ye H, Fontecave M, Pilon-Smits EAH, Pilon M. Characterization of Arabidopsis thaliana SufE2 and SufE3. Functions in chloroplast iron-sulfur cluster assembly and NAD synthesis. J Biol Chem. 2007;282:18254–18264. doi: 10.1074/jbc.M701428200. [DOI] [PubMed] [Google Scholar]
- 41.Tamarit J, Gerez G, Meier C, Mulliez E, Trautwein A, Fontecave M. The activating component of the anaerobic ribonucleotide reductase from Escherichia coli: An iron-sulfur center with only three cysteines. J Biol Chem. 2000;275:15669–15675. doi: 10.1074/jbc.275.21.15669. [DOI] [PubMed] [Google Scholar]
- 42.Saunders AH, Booker SJ. Regulation of the activity of Escherichia coli quinolinate synthase by reversible disulfide-bond formation. Biochemistry. 2008;47:8467–8469. doi: 10.1021/bi801135y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boshoff HIM, Xu X, Tahlan K, Dowd CS, Pethe K, Camacho LR, Park T-H, Yun C-S, Schnappinger D, Ehrt S, Williams KJ, Barry CE., III Biosynthesis and recylcing of nicotinamide cofactorsin Mycobacterium tuberculosis: an essential role for NAD in nonreplicating bacilli. J Biol Chem. 2008;283:19329–19341. doi: 10.1074/jbc.M800694200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1 and S2. This material is available free of charge via the Internet at http:/pubs.acs.org