

Abstract
Yeast rad50 and mre11 nuclease mutants are hypersensitive to physical and chemical agents that induce DNA double-strand breaks (DSBs). This sensitivity was suppressed by elevating intracellular levels of TLC1, the RNA subunit of telomerase. Suppression required proteins linked to homologous recombination, including Rad51, Rad52, Rad59 and Exo1, but not genes of the nonhomologous end-joining (NHEJ) repair pathway. Deletion mutagenesis experiments demonstrated that the 5′ end of TLC1 RNA was essential and a segment containing a binding site for the Yku70/Yku80 complex was sufficient for suppression. A mutant TLC1 RNA unable to associate with Yku80 protein did not increase resistance. These and other genetic studies indicated that association of the Ku heterodimer with broken DNA ends inhibits recombination in mrx mutants, but not in repair-proficient cells or in other DNA repair single mutants. In support of this model, DNA damage resistance of mrx cells was enhanced when YKU70 was co-inactivated. Defective recombinational repair of DSBs in mrx cells thus arises from at least two separate processes: loss of Mrx nuclease-associated DNA end-processing and inhibition of the Exo1-mediated secondary recombination pathway by Ku.
Keywords: telomerase, exonuclease, DNA repair, end-joining, homologous recombination
1. Introduction
Repair of spontaneous or induced DNA double-strand breaks (DSBs) is accomplished by at least two major pathways in eukaryotic organisms, called nonhomologous end-joining (NHEJ) and homologous recombination. Repair by NHEJ is mediated in S. cerevisiae (budding yeast) by the actions of the Rad50/Mre11/Xrs2 (Mrx) nuclease, the DNA end-binding Yku70/Yku80 heterodimer and DNA Ligase IV, composed of Lif1, Dnl4 and Nej1 [1,2,3]. Other proteins, including Rad27 (Fen1), Pol4, Smc cohesins and the Rsc nucleosome remodeling complex, have also been implicated in NHEJ [4,5]. In yeast cells the SIR2, SIR3 and SIR4 genes are also required for NHEJ, though their role appears to be indirect, involving regulation of NEJ1 expression [3].
Repair of DSBs by homologous recombination requires members of the Rad52 group of DNA repair proteins, including Rad51-Rad59, the Mrx complex, the Rpa single-stranded DNA binding protein, as well as several other proteins associated with strand exchange, DNA synthesis, heteroduplex strand separation and DNA ligation [6,7]. Recombination models have been proposed that involve initial resection of DSB ends by the Mrx nuclease to generate single-stranded 3′ overhangs, followed by homology search and strand exchange reactions mediated by complexes containing Rad51, Rad52, etc. [6,7]. The precise role of Mrx in resection remains obscure and the complex may perform additional functions such as recruitment of the Rad51 recombinase and/or the Rsc chromatin remodeling complex [5,6,7]. A second nuclease, Exo1, possesses a 5′-to-3′ exonuclease activity that can also generate 3′ tails at DSB ends, though this reaction is inefficient at physiological levels of the enzyme [7,8,9].
Yeast rad50, mre11 and xrs2 single mutants exhibit several similar phenotypes, including (i) defects in both NHEJ and homologous recombination assays, (ii) sensitivity to ionizing radiation and strand-breaking chemicals such as methyl methanesulfonate (MMS), bleomycin, or hydroxyurea (HU), (iii) stable but shortened telomeres, (iv) defective meiosis, (v) increased chromosome and arm loss, (vi) elevated loss-of-heterozygosity (LOH) and mutation frequencies, and (vii) defects in DNA damage-responsive cell cycle checkpoints [8,10,11,12,13,14].
The ends of chromosomes in most eukaryotic organisms contain arrays of short, repeated DNA sequences referred to as telomeres. Stable maintenance of DNA ends in dividing cells requires telomerase, an RNA-dependent DNA polymerase complex. Yeast telomerase has at least four protein subunits, called Est1, Est2, Est3 and Cdc13, as well as a 1301 nt RNA subunit encoded by the TLC1 gene [15]. Est2 is the catalytic (polymerase) subunit and Est1 and Cdc13 are DNA binding proteins thought to mediate association of the complex with chromosome ends.
The RNA subunit of telomerase acts as a scaffold for the binding of several proteins, including Est1, Est2 and Yku70/Yku80, and for association with another TLC1 RNA molecule to form dimers in vivo [16,17,18,19]. Altering cellular levels of telomerase affects cell physiology in different ways. For example, expression of TLC1 RNA at supraphysiological levels disrupts silencing of transcription at telomeres and can also suppress killing of yku70 or yku80 mutants at elevated temperatures [20,21,22,23]. Modulation of TLC1 expression also affects the proportion of Est2 molecules bound to the yeast PinX1 protein (Pxr1), a possible regulator of Est2 localization in the nucleus [24]. In the current study we have investigated the mechanism by which telomerase RNA overexpression alleviates the DNA repair defects of mrx cells, but not those of other DSB repair mutants. These experiments have revealed new connections between telomerase and DNA repair and have identified a new role for the Ku complex in regulation of DSB repair by the homologous recombination pathway.
2. Materials and methods
2.1. Strains and plasmids
Yeast strains used in the study included VL6α (MATα ura3-52 his3-Δ200 trp1-Δ63 lys2-801 ade2-101 met14) [25] and BY4742 (MATα ura3Δ0 leu2Δ0 his3Δ1 lys2Δ0) [26]. Gene disruptions of yeast strains were performed as described in [8]. Derivatives of VL6α included YLKL499 (rad50Δ∷hisG), YLKL500 (yku70Δ∷TRP1 rad50Δ∷hisG), YLKL529 (mre11Δ∷G418r), YLKL544 (dnl4Δ∷G418r), YLKL593 (yku70Δ∷HIS3), YLKL612 (mre11Δ∷HygBrsir4Δ∷LEU2), YLKL613 (mre11Δ∷HygBrdnl4Δ∷G418r), YLKL614 (rad50Δ∷G418rrad52Δ∷hisG), YLKL615 (mre11Δ∷HygBrrad52Δ∷hisG), YLKL618 (rad50Δ∷hisG sir4Δ∷LEU2), YLKL619 (rad50Δ∷hisG exo1Δ∷G418r), YLKL620 (rad50Δ∷hisG rad59Δ∷G418r), and YLKL725 (mre11Δ∷G418rexo1∷URA3). BY4742 derivatives included YLKL649 (rad50Δ∷G418r) and YLK963 (rad50Δ∷G418rrad51Δ∷LEU2). Vectors pRS314, pRS424, pRS315, and pRS316 have been described [27] and plasmid pTCG-3xStem was a gift from Dan Gottschling [17].
2.2. Molecular biology reagents
Methyl methanesulfonate (MMS) was obtained from Fluka and both hydroxyurea (HU) and bleomycin were purchased from CalBiochem. Geneticin/G418 (Invitrogen) and Hygromycin B (Sigma) were added to plates for selection of resistant strains at concentrations of 200 and 300 ug/ml, respectively. PCR fragment-mediated gene disruptions employed ExTaq DNA polymerase (Takara Mirus Bio) or Vent DNA polymerase (New England Biolabs) and the lithium acetate method for transformation of yeast cells [28].
2.3. Construction and assay of truncated TLC1 genes
The wildtype TLC1 gene expression plasmid pLKL74Y (2μ TRP1 GAL1p∷TLC1) was created by ligation of the 2.1 kb EcoRI/NotI GAL1p∷TLC1 gene fragment of pCDNA50.6 [23] to pRS424 that was similarly cut with EcoRI and NotI. The resulting construct has a unique BamHI site just upstream of the start site of TLC1 and an Ecl136II site immediately downstream (see Figure 2). Derivatives were created by digestion with pairs of enzymes to release a small part of TLC1, followed by gel purification of the large vector+TLC1 gene fragment and self-ligation. Enzyme sites that contained 5′ overhangs were filled in with Klenow DNA polymerase to produce flush ends prior to self-ligation. The enzymes used and the resulting plasmids were: AflII + Ecl136II (pLKL75Y), HpaI + Ecl136II (pLKL76Y), NcoI + Ecl136II (pLKL77Y), BamHI + StuI (pLKL78Y) and BamHI + NcoI (pLKL79Y). The control vector pRS424 plus each of the deletion plasmids was transformed into mre11 cells (YLKL529) to assess their ability to confer resistance to MMS or HU. Two or three independent transformants were subsequently used for survival tests.
Fig. 2.

Analysis of genetic pathways required for suppression of DNA repair by TLC1. MMS resistance of rad50 and mre11 cells containing additional mutations in DNL4 (A), and RAD51, RAD52, RAD59 or EXO1 (B-E) was analyzed as for Figure 1. Vector and TLC1 refer to pRS424 and pLKL74Y, respectively.
2.4. Dilution pronging survival assays
For the pronging experiments depicted in Figure 1, cells containing the vector pRS424 or pLKL74Y (2μ GAL1p∷TLC1 TRP1) were patched to 2% galactose plates lacking tryptophan (trp) and grown overnight at 30 °C to induce expression of TLC1. After harvesting and counting by hemacytometer, 2 × 107 total cells were placed into a microtiter dish well and serially diluted five-fold, followed by pronging to synthetic 2% galactose minus trp plates with and without each drug. Survival tests for strains YLKL649 (rad50Δ∷G418r) and YLKL963 (rad50Δ∷G418rrad51Δ∷LEU2) were performed using pRS316 and pLKL83Y (2μ ADH1p∷TLC1 URA3) with cells grown on 2% glucose plates with or without HU or MMS. Survival after induction of in vivo EcoRI expression was assayed using an mre11 strain (YLKL407) that contains a GAL1p∷EcoRI cassette integrated into the HIS3 locus on chromosome XV [29]. Cells were grown on synthetic 2% glucose plates, serially diluted 5-fold as above and pronged to the same media with or without 0.2% galactose for the induction of the endonuclease.
Fig. 1.
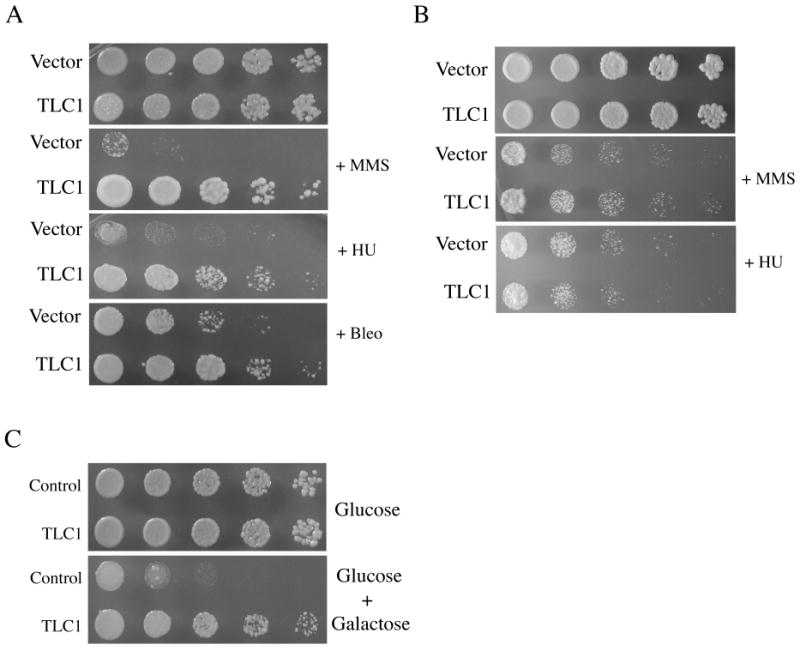
Overexpression of TLC1 RNA increases survival of mrx mutants, but not wildtype cells, after exposure to chemical clastogens. (A) YLKL503 cells (mre11) containing the vector pRS424 or plasmid pLKL74Y (2μ GAL1p∷TLC1 TRP1) were serially diluted five-fold and pronged onto 2% galactose plates with or without 0.35 mM MMS, 35 mM HU or 2 μg/ml bleomycin. (B) BY4742 (MRX) cells were grown as in (A) with or without 3 mM MMS or 200 mM HU. (C) mre11 cells containing a GAL1p∷EcoRI cassette integrated into HIS3 [29] and either pRS315 or the TLC1 overexpression plasmid pLKL64Y (ADH1p∷TLC1 LEU2) were pronged to selective plates with or without 0.2% galactose.
2.5. Genome sequence comparisons
Multiple sequence alignments were performed using ClustalW at EMBnet (www.ch.embnet.org/index) after initial BLAST searches at the Saccharomyces Genome Database (www.yeastgenome.org) and the Entrez Nucleotide database (www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=Nucleotide) to identify TLC1 gene homologs. GenBank accession numbers for the sequences used in the analyses were: S. mikatae - AY639011, S. bayanus - AY547299, S. cariocanus – AY639010 + AY737251, S. kudriavzevii – AY639012, and S. paradoxus – AY639014. Final clustal alignments were refined using the Boxshade program also present at EMBnet.
3. Results
3.1. Enhancement of resistance by TLC1 occurs uniquely in mrx mutants and requires components of the homologous recombination pathway
A previous search for overexpressed yeast genes that could increase resistance of rad50 mutants to MMS, a DNA methylating agent [30] led to the isolation of library plasmids containing EXO1 and TLC1 [23]. The earlier work, in conjunction with other studies [9], revealed that the 5′-to-3′ exonuclease encoded by EXO1 can partially substitute for Mrx in processing of DSBs and increases MMS resistance by specifically elevating repair by recombination, but not NHEJ. The second suppressor gene, TLC1, encodes the RNA subunit of telomerase.
Telomerase RNA overexpression increases resistance of mrx cells to multiple DNA damaging agents, including the S phase-dependent clastogens MMS and HU as well as the more direct DNA damaging agent bleomycin (Figure 1A). Suppression of killing by MMS and HU was similar and strong (≥125-fold in this semi-quantitative assay), though bleomycin resistance was only modestly increased at the doses tested (Figure 1A and data not shown). Bleomycin generates oxidative damage to DNA [31], similar to gamma radiation, which was also rescued less efficiently than MMS by TLC1 RNA overexpression [23]. The reduced impact with bleomycin and radiation may be due to differences in DSB end structures as radiation is known to produce “dirty ends”, i.e., ends that have base or sugar damage as well as 3′-phosphates and 3′-phosphoglycolates [32]. In contrast to mrx cells, increasing telomerase RNA levels did not affect the sensitivity of wildtype cells to high, growth-inhibitory doses of MMS (2 – 4 mM) or HU (100 – 300 mM) (Figure 1B).
Expression of the bacterial endonuclease EcoRI in yeast cells has been employed for several studies of DSB repair [14,33,34,35]. This enzyme generates DSBs at its recognition sequence GˆAATTC that retain 5′ overhangs which are 4 nucleotides long. For the experiments shown in Figure 1C, mre11 cells containing a chromosomal GAL1p∷EcoRI cassette and either the vector pRS315 or the TLC1 overexpression plasmid pLKL64Y (ADH1p∷TLC1 LEU2) were pronged to selective plates with or without added galactose to induce EcoRI. Increased intracellular levels of telomerase RNA strongly suppressed killing by EcoRI. Survival was increased approximately 125-fold in the assay, comparable to the effects with MMS and HU, suggesting that DSB repair is the specific process being enhanced.
The possibility that TLC1 might reactivate either the recombination or NHEJ repair pathway, which are each compromised in mrx mutants, was assessed in Figure 2. NHEJ repair requires DNA Ligase IV, whose catalytic subunit is encoded by DNL4. The MMS sensitivity of mre11 dnl4 mutants (YLKL613) was still suppressed by TLC1 (Figure 2A). In contrast, resistance of double mutants combining mrx deletions with either rad51, rad52 or rad59, impairing strand exchange, was not increased (Figure 2 B-D). mre11 cells lacking the 5′-to-3′ exonuclease encoded by EXO1, which can substitute, albeit inefficiently, for Mrx were also not rescued by TLC1, even at very low doses of MMS (Figure 2E).
Results of tests with these mutants and several others are summarized in Table 1. Telomerase RNA overexpression in each strain combining rad50 or mre11 with deletions of genes affecting NHEJ (dnl4 or sir4) still exhibited increased MMS resistance. sir4 strains do not express Nej1, another component of the DNA ligase IV complex [2,3]. Rescue was abolished when genes associated with recombination such as RAD51, RAD52, RAD59 and EXO1 were co-inactivated. Importantly, DSB repair single mutants such as rad51, rad52, rad54 and rad59 strains, as well as yku70 and dnl4 mutants, did not show enhanced resistance. Finally, resistance of two different wildtype (repair-proficient) strain backgrounds (VL6α and BY4742) was not supranormal when TLC1 was overexpressed. These results demonstrate the specificity of the phenomenon for mrx cells and suggest that TLC1 enhances repair of DSBs via the recombination pathway.
Table 1.
Genetic requirements for elevation of MMS resistance by TLC1 RNA
| TLC1-induced | TLC1-induced | ||
|---|---|---|---|
| Strain | MMS resistance | Strain | MMS resistance |
| wt (VL6α or BY4742)a | - | ||
| rad50 (YLKL499) | + | mre11 (YLKL529) | + |
| rad50 sir4 (YLKL618) | + | mre11 sir4 (YLKL612) | + |
| mre11 dnl4 (YLKL613) | + | ||
| rad50 rad51 (YLKL963) | - | mre11 rad52 (YLKL615) | - |
| rad50 rad52 (YLKL614) | - | mre11 exo1 (YLKL725) | - |
| rad50 rad59 (YLKL620) | - | ||
| rad50 exo1 (YLKL619) | - | ||
| rad51 (YLKL532)b | - | rad54 (YLKL531) b | - |
| rad52 (YLKL276) b | - | rad59 (YLKL514) b | - |
| dnl4 (YLKL544) | - | yku70 (YLKL593) | - |
3.2. Suppression requires sequences at the 5′ end of TLC1
TLC1 RNA forms a complex tertiary structure that serves as a scaffold for the binding of several proteins [18]. The locations of known binding sites for Est1, Est2 and the Yku70/Yku80 complex are depicted schematically in Figure 3A [16,18,19]. To identify the region(s) of TLC1 needed for enhancement of repair, unique restriction sites present in the GAL1p∷TLC1 plasmid pLKL74Y (Figure 3A) were used as breakpoints to create the deletion derivatives pLKL75Y-79Y shown in Figure 3B and 3C (see Materials and methods). mre11 cells (YLKL529) containing vector or TLC1 deletion plasmids were pronged to galactose plates with or without 0.35 mM MMS or 35 mM HU. Each of the truncated forms of TLC1 was able to increase resistance to MMS and HU except pLKL79Y, which contained a deletion of nt 1-450 (up to NcoI) (Figure 3C). The derivative missing nt 1 – 128 (pLKL78Y) was still able to suppress killing, suggesting that the critical region lies between nt 128 and 450.
Fig. 3.

Identification of the region(s) within TLC1 RNA required for stimulation of DSB repair in mrx mutants. (A) Functional regions and protein binding sites on TLC1 RNA. (B) Deleted regions within five truncated derivatives of the TLC1 gene (shown as single lines between unique restriction sites). (C) MMS- and HU-resistance of mre11 cells (YLKL529) expressing wt or truncated forms of TLC1.
3.3. Stimulation of repair by TLC1 involves association with the Ku complex
The segment of TLC1 RNA residing between nt 128 and 450 (from StuI to NcoI) includes a suggested binding site for the Yku70/Yku80 complex (Figure 3A). Work in the Gottschling laboratory previously demonstrated that a 48 nt segment of TLC1 within this region (nt 288 – 335) forms tight associations with Yku80 protein in in vitro gel binding assays [17]. To test the role of Ku, a plasmid containing three copies of the 48 nt Ku-binding segment (pTCG-3xStem) was overexpressed in mre11 cells and found to be sufficient for suppression of killing, though resistance did not reach levels seen with the full-length allele (Figure 4B). A mutant version of the stem segment that is unable to bind Ku proteins in gel binding assays [17] was also tested. This allele has three nucleotide substitutions (T301A, T307G and T324G), shown above the gene sequences in Figure 4A, including two changes at strongly conserved positions (shown as black boxes). Overexpression of this mutant RNA (called pTCG-3xStemm) did not suppress killing by MMS or HU (Figure 4B).
Fig. 4.

Role of Ku in enhancement of DSB repair by TLC1. (A) Alignment of S. cerevisiae TLC1 sequence (nt 281-335) with TLC1 genes of five other yeasts. Black boxes indicate perfectly conserved positions. The Stem region is shown above the alignments, with nucleotide substitutions that abolish Ku binding indicated by vertical lines. (B) Assessment of resistance to MMS (0.35 mM) and HU (35 mM) in mre11 cells expressing TLC1 (pLKL74Y), TLC1-Stem or TLC1-Stemm plasmids. #1 and #2 indicate independent isolates. (C) Resistance of rad50 versus rad50 yku70 cells (YLKL499 and YLKL500) to 0.35 mM MMS.
3.4. Inactivation of the YKU70 gene mimics the effects of TLC1 overexpression
Results obtained with the truncated TLC1 alleles suggested that enhanced repair in mrx cells involves recruitment of Ku proteins onto telomerase RNA molecules. It is possible that binding to telomerase RNA might improve resistance by preventing Ku from acting as an inhibitor of repair. If this idea is correct, then one would predict that an mrx ku double mutant strain lacking the inhibitory Ku complex would exhibit increased repair proficiency. A rad50 yku70 strain (YLKL500) was constructed and DNA damage sensitivity compared to that of a rad50 single mutant as before. MMS resistance was increased in rad50 yku70 strains compared to rad50 cells (Figure 4C). This finding, in conjunction with all of the survival and DSB repair assays using DNA repair single and double mutants performed here and in our previous paper [23] as well as the TLC1 RNA deletion and site-directed mutagenesis experiments, are consistent. The aggregate data indicate that the presence of Ku in vivo is strongly inhibitory to recombinational repair of induced DSBs in mrx mutants, but not in wt cells or in other DSB repair mutants.
4. Discussion
In the current study we have identified an additional major mechanism responsible for reduction of recombinational repair of DSBs in mrx mutants. This inhibitory process was identified through an investigation of the means by which overexpression of telomerase RNA enhances resistance to DNA damage. Elevated intracellular levels of TLC1 RNA were found to increase the resistance of mrx mutants to multiple DNA damaging agents, including the S phase-dependent clastogens MMS and HU, as well as the more direct DNA strand-breaking agent bleomycin. In addition, the lethality arising from in vivo expression of EcoRI endonuclease was also abrogated, indicating that repair of DNA DSBs was specifically increased by TLC1. Bleomycin resistance, similar to the ionizing radiation resistance results from our initial study [23], was only weakly rescued, possibly because these two treatments produce more broken ends with unusual structures than the other DNA damaging agents [31,32].
Genetic analysis using several single and double mutant strains demonstrated that (a) resistance was not increased by TLC1 in any other DSB repair single mutants, pointing to the specificity of the process for rad50, mre11 and xrs2 cells, (b) rescue was unaffected in mre11 and rad50 strains in which either DNL4 or SIR4 were also inactivated, indicating that increased repair by NHEJ was not involved; this conclusion is in accord with our previous observation that overexpression of TLC1 RNA does not increase NHEJ in a plasmid repair assay [23], and (c) the positive effect of TLC1 was abolished in all double mutants combining mre11 or rad50 mutations with rad51, rad52, rad59 or exo1 deletions. Thus, a functional recombination pathway is essential for stimulation of repair. RAD51 and RAD52 are critical components of this pathway and inactivation of either gene causes a strong reduction in most assays of recombination [6,7]. In contrast, rad59 single mutants exhibit only modest defects in recombination assays, but the gene was found to be essential here. Past work has identified a role for Rad59, a Rad52 homologue, in a subset of homology-dependent repair events such as break-induced DNA replication (BIR) and single-strand annealing (SSA) [36,37]. The Rad51 and Rad59 requirements for efficient stimulation of DSB repair by TLC1 RNA may point to important roles for both conventional gene conversion involving Rad51 and Rad52, as well as BIR-SSA events requiring Rad59 and Rad52 [36].
Analysis of several truncated forms of TLC1 demonstrated that only the 5′ end was required and the region from nt 288 – 335, previously shown to be a binding site for Yku80 protein in vitro [38], was sufficient for resistance to MMS and HU. Overexpression of a mutant form of this short RNA unable to associate with Yku80 protein did not increase resistance, suggesting that binding of Ku is a requirement.
The Ku proteins are known to associate in a sequence-independent manner with induced DSB ends in yeast cells after exposure to DNA damaging agents [34,39]. In a wildtype cell the binding of Ku to DSB ends has the potential to be the initial step in repair by NHEJ, in conjunction with Rad50/Mre11/Xrs2 and Dnl4/Lif1/Nej1. However, in an mre11 mutant (or rad50), there is no functional Mrx complex to participate with in NHEJ so binding of Ku is unproductive. The evidence presented here suggests that, not only is such binding of Ku to DSBs unproductive for NHEJ, but it is also inhibitory toward repair by the backup Exo1 pathway of recombination.
A model for the interplay between Mrx, Exo1, TLC1 RNA and the Ku complex in homologous recombination is presented in Figure 5. In wildtype S. cerevisiae cells both NHEJ and recombination are active, but most DSBs are repaired by recombination. Thus, most induced DSB ends are processed by Mrx (possibly in conjunction with other proteins) to create tailed substrates for binding of Rpa (single-stranded DNA binding protein) and the Rad51-Rad52 strand exchange complex, with some processing by the backup nuclease Exo1 (Figure 5A). Haploid mrx mutants (Figure 5B) become hypersensitive to DSB-inducing agents because of at least two distinct changes in repair capacity: (1) recombination is reduced because Mrx-associated end-processing is absent, and (2) DNA binding by enzymes of the backup recombination pathway, including Exo1 and possibly other proteins, is inhibited when Ku binds to the broken ends first (Figure 5B). Mrx binds to induced DSBs early [7] and may compete effectively with Ku in wildtype cells, allowing efficient processing. Alternatively, Ku proteins may bind to DSB ends first even in normal cells, but be subsequently displaced by Mrx or Mrx-associated proteins. It is formally possible that Ku may also bind to DNA structures that are downstream intermediates of recombination when Mrx is absent.
Fig. 5.

Proposed model for stimulation of recombination uniquely in mrx cells by TLC1. (A) In normal cells, induced DSB ends are processed efficiently. For simplicity, resection of only one end is shown. (B) In mrx nuclease mutants, recombination is compromised because of a loss of Mrx-associated processing and also because of the inhibitory effects of Yku70/Yku80 binding to the DSB ends. (C) Overexpression of TLC1 RNA molecules results in titration of Ku dimers away from the broken ends, allowing greater access for enzymes of the backup recombinational pathway.
According to this model (Figure 5C) Ku proteins become bound to TLC1 RNA, reducing the number of heterodimers available for association with induced DSB ends. This titration increases access of Exo1 and possibly other proteins to the broken ends and repair is enhanced. Consistent with the idea of a competition for ends, overexpression of Exo1 stimulates recombinational repair of DSBs in mrx mutants [8,9]. This rescue effect, presumably due primarily to an increase in the number of active enzyme molecules, may also involve an increased ability of Exo1 to compete for ends with Ku.
In support of the model, we observed that MMS resistance is strongly increased in rad50 yku70 double mutants, which lack the inhibitory Ku complex, compared to rad50 cells. Increased damage resistance in mrx ku mutants has also been observed in past studies in both S. cerevisiae and S. pombe [40,41], though another report saw no effect [42].
Additional support for the concept of Ku as a negative regulator comes from studies which observed that inactivation of Ku genes leads to a modest increase in some classes of recombination events in yeast cells [35,41,43]. Repair at a single DSB produced by HO endonuclease is also slightly faster in cells lacking a functional Ku complex. Interestingly, mrx mutants are not defective in repair of an HO-induced DSB, but resection is slightly slower than in wt cells (∼ 50% of the rate of wt cells) [44]. Consistent with this, we observed that while TLC1 overexpression increased growth rates of mre11 cells producing EcoRI endonuclease, it did not affect growth of mre11 cells expressing HO endonuclease from a GAL promoter (Figure 1 and data not shown).
The concept of titration of Ku complexes in vivo by binding to TLC1 RNAs was previously postulated by the Gottschling laboratory [38], which observed that TLC1 overexpression reduced telomere silencing. It was suggested that the RNAs titrated Ku heterodimers away from telomeric chromatin. Subsequent work [17,45] indicated a role for Ku:TLC1 interaction in recruitment of telomerase to chromosome ends. Stellwagon et al. [17] suggested that cells became effectively like ku mutants upon TLC1 overexpression. We did not detect either telomere shortening or growth defects at 37 °C in wildtype or mre11 cells when TLC1 was overexpressed (phenotypes of ku strains; data not shown), possibly because steady state RNA levels achieved in each strain were different. A recent study [46] demonstrated that overexpression of the Yku70/Yku80 complex slows the growth of normal cells but is alleviated by inactivation of RAD27. The authors speculate that this is because rad27 mutants have increased ssDNA at the telomeres that may titrate Ku proteins, preventing them from binding elsewhere, a concept with similarities to the mechanisms discussed above. mrx cell cultures exhibit strong DNA damage-induced checkpoint responses [8], but events such as phosphorylation of Rad53, Chk1 and Rad9 checkpoint proteins are reduced [13,14]. The contribution of these defects to the MMS sensitivity of mrx mutants and the possible impact of TLC1 on checkpoint controls is unclear.
Evidence presented in this study suggests that elevated levels of telomerase RNA can enhance DNA repair by titration of Ku proteins away from broken DNA ends. Human KU70/KU80 also associates with telomerase, forming interactions with both RNA and protein subunits [47,48,49]. Thus, it is possible that alteration of telomerase levels within human cells may also impact the DNA repair and telomere stabilization functions of Ku. One implication of these results is that cancer cells, which frequently produce high levels of telomerase [50], might exhibit altered resistance to X-rays and clastogenic anti-tumor drugs such as bleomycin because of such Ku:telomerase interactions. In support of this idea, links between telomerase levels and DNA repair have been identified in human cells [51,52], though the mechanisms involved have not been determined.
Acknowledgments
The authors wish to thank Dan Gottschling and Nancy Kleckner for gifts of plasmids and Shanna Calero for expert technical assistance. KL was supported in part by Research Corporation grant CC5767 and National Institutes of Health grant 1R15AG028520-01A1.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lewis LK, Resnick MA. Tying up loose ends: nonhomologous end-joining in Saccharomyces cerevisiae. Mutat Res. 2000;451:71–80. doi: 10.1016/s0027-5107(00)00041-5. [DOI] [PubMed] [Google Scholar]
- 2.Hefferin ML, Tomkinson AE. Mechanism of DNA double-strand break repair by non-homologous end joining. DNA Repair. 2005;4:639–648. doi: 10.1016/j.dnarep.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 3.Daley JM, Palmbos PL, Wu D, Wilson TE. Nonhomologous end joining in yeast. Annu Rev Genet. 2005;39:431–451. doi: 10.1146/annurev.genet.39.073003.113340. [DOI] [PubMed] [Google Scholar]
- 4.Dudásová Z, Dudás A, Chovanec M. Non-homologous end-joining factors of Saccharomyces cerevisiae. FEMS Microbiol Rev. 2004;28:581–601. doi: 10.1016/j.femsre.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 5.Shim EY, Hong SJ, Oum JH, Yanez Y, Zhang Y, Lee SE. RSC mobilizes nucleosomes to improve accessibility of repair machinery to the damaged chromatin. Mol Cell Biol. 2007;27:1602–1613. doi: 10.1128/MCB.01956-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sung P, Klein H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol. 2006;7:739–750. doi: 10.1038/nrm2008. [DOI] [PubMed] [Google Scholar]
- 7.Wyman C, Kanaar R. DNA double-strand break repair: all's well that ends well. Annu Rev Genet. 2006;40:363–383. doi: 10.1146/annurev.genet.40.110405.090451. [DOI] [PubMed] [Google Scholar]
- 8.Lewis LK, Storici F, Van Komen S, Calero S, Sung P, Resnick MA. Role of the nuclease activity of Saccharomyces cerevisiae Mre11 in repair of DNA double-strand breaks in mitotic cells. Genetics. 2004;166:1701–1713. doi: 10.1534/genetics.166.4.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tran PT, Erdeniz N, Symington LS, Liskay RM. EXO1-A multi-tasking eukaryotic nuclease. DNA Repair. 2004;3:1549–1559. doi: 10.1016/j.dnarep.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 10.D'amours D, Jackson SP. The Mre11 complex: at the crossroads of DNA repair and checkpoint signalling. Nat Rev Mol Cell Biol. 2002;3:317–327. doi: 10.1038/nrm805. [DOI] [PubMed] [Google Scholar]
- 11.Kolodner RD, Putnam CD, Myung K. Maintenance of genome stability in Saccharomyces cerevisiae. Science. 2002;297:552–557. doi: 10.1126/science.1075277. [DOI] [PubMed] [Google Scholar]
- 12.Yoshida J, Umezu K, Maki H. Positive and negative roles of homologous recombination in the maintenance of genome stability in Saccharomyces cerevisiae. Genetics. 2003;164:31–46. doi: 10.1093/genetics/164.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakada D, Hirano Y, Sugimoto K. Requirement of the Mre11 complex and exonuclease 1 for activation of the Mec1 signaling pathway. Mol Cell Biol. 2004;24:10016–10025. doi: 10.1128/MCB.24.22.10016-10025.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grenon M, Magill CP, Lowndes NF, Jackson SP. Double-strand breaks trigger MRX- and Mec1-dependent, but Tel1-independent, checkpoint activation. FEMS Yeast Res. 2006;6:836–847. doi: 10.1111/j.1567-1364.2006.00076.x. [DOI] [PubMed] [Google Scholar]
- 15.Smogorzewska A, De Lange T. Regulation of telomerase by telomeric proteins. Annu Rev Biochem. 2004;73:177–208. doi: 10.1146/annurev.biochem.73.071403.160049. [DOI] [PubMed] [Google Scholar]
- 16.Livengood AJ, Zaug AJ, Cech TR. Essential regions of Saccharomyces cerevisiae telomerase RNA: separate elements for Est1p and Est2p interaction. Mol Cell Biol. 2002;22:2366–2374. doi: 10.1128/MCB.22.7.2366-2374.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stellwagen AE, Haimberger ZW, Veatch JR, Gottschling DE. Ku interacts with telomerase RNA to promote telomere addition at native and broken chromosome ends. Genes Dev. 2003;17:2384–2395. doi: 10.1101/gad.1125903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zappulla DC, Cech TR. Yeast telomerase RNA: a flexible scaffold for protein subunits. Proc Natl Acad Sci U S A. 2004;101:10024–10029. doi: 10.1073/pnas.0403641101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gipson CL, Xin ZT, Danzy SC, Parslow TG, Ly H. Functional characterization of yeast telomerase RNA dimerization. J Biol Chem. 2007;282:18857–18863. doi: 10.1074/jbc.M700057200. [DOI] [PubMed] [Google Scholar]
- 20.Singer MS, Gottschling DE. TLC1: template RNA component of Saccharomyces cerevisiae telomerase. Science. 1994;266:404–409. doi: 10.1126/science.7545955. [DOI] [PubMed] [Google Scholar]
- 21.Nugent CI, Bosco G, Ross LO, Evans SK, Salinger AP, Moore JK, Haber JE, Lundblad V. Telomere maintenance is dependent on activities required for end repair of double-strand breaks. Curr Biol. 1998;8:657–660. doi: 10.1016/s0960-9822(98)70253-2. [DOI] [PubMed] [Google Scholar]
- 22.Teo SH, Jackson SP. Telomerase subunit overexpression suppresses telomere-specific checkpoint activation in the yeast yku80 mutant. EMBO Rep. 2001;2:197–202. doi: 10.1093/embo-reports/kve038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewis LK, Karthikeyan G, Westmoreland JW, Resnick MA. Differential suppression of DNA repair deficiencies of yeast rad50, mre11 and xrs2 mutants by EXO1 and TLC1 (the RNA component of telomerase) Genetics. 2002;160:49–62. doi: 10.1093/genetics/160.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin J, Blackburn EH. Nucleolar protein PinX1p regulates telomerase by sequestering its protein catalytic subunit in an inactive complex lacking telomerase RNA. Genes Dev. 2004;18:387–396. doi: 10.1101/gad.1171804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Larionov V, Kouprina N, Nikolaishvili N, Resnick MA. Recombination during transformation as a source of chimeric mammalian artificial chromosomes in yeast (YACs) Nucleic Acids Res. 1994;22:4154–4162. doi: 10.1093/nar/22.20.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 27.Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gietz RD, Woods RA. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002;350:87–96. doi: 10.1016/s0076-6879(02)50957-5. [DOI] [PubMed] [Google Scholar]
- 29.Lewis LK, Kirchner JM, Resnick MA. Requirement for end-joining and checkpoint functions, but not RAD52-mediated recombination, after EcoRI endonuclease cleavage of Saccharomyces cerevisiae DNA. Mol Cell Biol. 1998;18:1891–1902. doi: 10.1128/mcb.18.4.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wyatt MD, Pittman DL. Methylating agents and DNA repair responses: Methylated bases and sources of strand breaks. Chem Res Toxicol. 2006;19:1580–1594. doi: 10.1021/tx060164e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramotar D, Wang H. Protective mechanisms against the antitumor agent bleomycin: lessons from Saccharomyces cerevisiae. Curr Genet. 2003;43:213–224. doi: 10.1007/s00294-003-0396-1. [DOI] [PubMed] [Google Scholar]
- 32.Cadet J, Bellon S, Douki T, Frelon S, Gasparutto D, Muller E, Pouget JP, Ravanat JL, Romieu A, Sauvaigo S. Radiation-induced DNA damage: formation, measurement, and biochemical features. J Environ Pathol Toxicol Oncol. 2004;23:33–43. doi: 10.1615/jenvpathtoxoncol.v23.i1.30. [DOI] [PubMed] [Google Scholar]
- 33.Lewis LK, Westmoreland JW, Resnick MA. Repair of endonuclease-induced double-strand breaks in Saccharomyces cerevisiae: essential role for genes associated with nonhomologous end-joining. Genetics. 1999;152:1513–1529. doi: 10.1093/genetics/152.4.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mills KD, Sinclair DA, Guarente L. MEC1-dependent redistribution of the Sir3 silencing protein from telomeres to DNA double-strand breaks. Cell. 1999;97:609–620. doi: 10.1016/s0092-8674(00)80772-2. [DOI] [PubMed] [Google Scholar]
- 35.Yamana Y, Maeda T, Ohba H, Usui T, Ogawa HI, Kusano K. Regulation of homologous integration in yeast by the DNA repair proteins Ku70 and RecQ. Mol Genet Genomics. 2005;273:167–176. doi: 10.1007/s00438-005-1108-y. [DOI] [PubMed] [Google Scholar]
- 36.Ira G, Haber JE. Characterization of RAD51-independent break-induced replication that acts preferentially with short homologous sequences. Mol Cell Biol. 2002;22:6384–6392. doi: 10.1128/MCB.22.18.6384-6392.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davis AP, Symington LS. The Rad52-Rad59 complex interacts with Rad51 and replication protein A. DNA Repair (Amst) 2003;2:1127–1134. doi: 10.1016/s1568-7864(03)00121-6. [DOI] [PubMed] [Google Scholar]
- 38.Peterson SE, Stellwagen AE, Diede SJ, Singer MS, Haimberger ZW, Johnson CO, Tzoneva M, Gottschling DE. The function of a stem-loop in telomerase RNA is linked to the DNA repair protein Ku. Nat Genet. 2001;27:64–67. doi: 10.1038/83778. [DOI] [PubMed] [Google Scholar]
- 39.Martin SG, Laroche T, Suka N, Grunstein M, Gasser SM. Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell. 1999;97:621–633. doi: 10.1016/s0092-8674(00)80773-4. [DOI] [PubMed] [Google Scholar]
- 40.Bressan DA, Baxter BK, Petrini JH. The Mre11-Rad50-Xrs2 protein complex facilitates homologous recombination-based double-strand break repair in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:7681–7687. doi: 10.1128/mcb.19.11.7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tomita K, Matsuura A, Caspari T, Carr AM, Akamatsu Y, Iwasaki H, Mizuno K, Ohta K, Uritani M, Ueno M. Competition between the Rad50 complex and the Ku heterodimer reveals a role for Exo1 in processing double-strand breaks but not telomeres. Mol Cell Biol. 2003;23:5186–5197. doi: 10.1128/MCB.23.15.5186-5197.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Milne GT, Jin S, Shannon KB, Weaver DT. Mutations in two Ku homologs define a DNA end-joining repair pathway in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:4189–4198. doi: 10.1128/mcb.16.8.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clikeman JA, Khalsa GJ, Barton SL, Nickoloff JA. Homologous recombinational repair of double-strand breaks in yeast is enhanced by MAT heterozygosity through yKU-dependent and -independent mechanisms. Genetics. 2001;157:579–589. doi: 10.1093/genetics/157.2.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD, Haber JE. Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell. 1998;94:399–409. doi: 10.1016/s0092-8674(00)81482-8. [DOI] [PubMed] [Google Scholar]
- 45.Fisher TS, Zakian VA. Ku: a multifunctional protein involved in telomere maintenance. DNA Repair (Amst) 2005;4:1215–1226. doi: 10.1016/j.dnarep.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 46.Banerjee S, Smith S, Myung K. Suppression of gross chromosomal rearrangements by yKu70-yKu80 heterodimer through DNA damage checkpoints. Proc Natl Acad Sci U S A. 2006;103:1816–1821. doi: 10.1073/pnas.0504063102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song K, Jung D, Jung Y, Lee SG, Lee I. Interaction of human Ku70 with TRF2. FEBS Lett. 2000;481:81–85. doi: 10.1016/s0014-5793(00)01958-x. [DOI] [PubMed] [Google Scholar]
- 48.Chai W, Ford LP, Lenertz L, Wright WE, Shay JW. Human Ku70/80 associates physically with telomerase through interaction with hTERT. J Biol Chem. 2002;277:47242–47247. doi: 10.1074/jbc.M208542200. [DOI] [PubMed] [Google Scholar]
- 49.Ting NS, Yu Y, Pohorelic B, Lees-Miller SP, Beattie TL. Human Ku70/80 interacts directly with hTR, the RNA component of human telomerase. Nucleic Acids Res. 2005;33:2090–2098. doi: 10.1093/nar/gki342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ahmed A, Tollefsbol TO. Telomerase, telomerase inhibition, and cancer. J Anti Aging Med. 2003;6:315–325. doi: 10.1089/109454503323028911. [DOI] [PubMed] [Google Scholar]
- 51.Sharma GG, Gupta A, Wang H, Scherthan H, Dhar S, Gandhi V, Iliakis G, Shay JW, Young CS, Pandita TK. hTERT associates with human telomeres and enhances genomic stability and DNA repair. Oncogene. 2003;22:131–46. doi: 10.1038/sj.onc.1206063. [DOI] [PubMed] [Google Scholar]
- 52.Chung HK, Cheong C, Song J, Lee HW. Extratelomeric functions of telomerase. Curr Mol Med. 2005;5:233–241. doi: 10.2174/1566524053586635. [DOI] [PubMed] [Google Scholar]