

Abstract
Although dementia is a clinical diagnosis, neuroimaging often is crucial for proper assessment. Magnetic resonance imaging (MRI) and computed tomography (CT) may identify nondegenerative and potentially treatable causes of dementia. Recent neuroimaging advances, such as the Pittsburgh Compound-B (PIB) ligand for positron emission tomography imaging in Alzheimer’s disease, will improve our ability to differentiate among the neurodegenerative dementias. High-resolution volumetric MRI has increased the capacity to identify the various forms of the frontotemporal lobar degeneration spectrum and some forms of parkinsonism or cerebellar neurodegenerative disorders, such as corticobasal degeneration, progressive supranuclear palsy, multiple system atrophy, and spinocerebellar ataxias. In many cases, the specific pattern of cortical and subcortical abnormalities on MRI has diagnostic utility. Finally, among the new MRI methods, diffusion-weighted MRI can help in the early diagnosis of Creutzfeldt-Jakob disease. Although only clinical assessment can lead to a diagnosis of dementia, neuroimaging is clearly an invaluable tool for the clinician in the differential diagnosis.
Keywords: MRI, PET, Pittsburgh Compound-B, dementia, neurodegenerative disease
Neuroimaging has dramatically changed our ability to accurately diagnose dementia. New neuroimaging methods facilitate diagnosis of most of the neurodegenerative conditions after symptom onset and show promise for diagnosis even in very early or pre-symptomatic phases with some diseases. The growing importance of neuroimaging in dementia is in part exemplified by the NIH’s support of the Alzheimer’s Disease Neuroimaging Initiative, a $60 million dollar effort to track changes in magnetic resonance imaging (MRI) and positron emission tomography (PET) scans over time in patients with Alzheimer’s disease (AD). Diagnostic certainty is very high with some conditions and techniques, including Creutzfeldt-Jakob disease (CJD) with diffusion-weighted MRI (DWI) and AD with Pittsburgh Compound-B (PIB) PET.
Despite advances in neuroimaging, the diagnosis of dementia is still largely a clinical diagnosis, based on history, disease course, and laboratory tests including neuroimaging. In evaluating a patient with dementia, it is critically important to eliminate obvious causes of cognitive decline, such as electrolyte imbalance, infections, thyroid dysfunction, and vitamin deficiency. As discussed in a separate article in this issue, brain tumors are usually identified by MRI with contrast (as well as magnetic resonance spectroscopy [MRS] or PET). Here we discuss neuroimaging of the more common dementias, including AD, frontotemporal dementia (FTD), vascular dementia (VaD), and atypical parkinsonian dementia. Also, we briefly discuss neuroimaging in the diagnosis of rapidly progressive dementias, including prion disease, and some neurogenetic disorders, such as cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) and spinocerebellar ataxias.
ALZHEIMER’S DISEASE
The diagnosis of AD continues to be based nearly exclusively on clinical criteria, although neuroimaging is beginning to be considered in many diagnostic algorithms. The neuropathological course of AD has been well described by the Braak and Braak staging, with damage beginning in the entorhinal cortex, spreading to the hippocampus, and subsequently to the rest of the cortex.1 Many AD patients with early age of onset (i.e., before 65 years) often present with complaints other than memory impairment, such as visuospatial problems, apraxia, or language deficits, reflecting a different pattern of cortical involvement in comparison with AD with late age of onset (i.e., after 65 years).2,3 Several structural MRI studies localize the pattern of the atrophy in early-onset AD to more posterior regions with prominent involvement of the precuneus and posterior cingulate.4–6 Several structural neuroimaging studies have shown the involvement of other regions: amygdala,7 anterior parahippocampal gyrus,7 and the occipital lobes.8 In particular, many structural studies have documented the involvement of the corpus callosum and prominent posterior cortical involvement.9–11
Usually, patients with AD show atrophy of the parietal lobes and the hippocampus on MRI (Fig. 1). In our dementia clinic, we find that the parietal atrophy is best seen on axial or coronal T1 or fluid-attenuated inversion recovery (FLAIR) with thinning of the posterior of the body of the corpus callosum on T1 sagittal sequences.12 Hippocampal atrophy is best seen with thin coronal T1 or FLAIR tomographic slices through the medial temporal lobes.13,14 In clinical practice, structural MRI can help support a clinical diagnosis, but unfortunately such techniques are not sufficient to establish a definitive diagnosis, as the overlap is great between atrophy associated with “healthy aging” versus AD.
Figure 1.

T1-weighted MRI scans in patients with pathologically-proven AD. (A) Coronal image showing bilateral hippocampal atrophy (arrows) in an 83-year-old woman (MMSE score 21). (B) Axial image showing biparietal and posterior cingulate atrophy (arrows) in a 62-year-old woman with early age of onset AD (MMSE 22). (C) Sagittal image showing thinning of the posterior body of the corpus callosum (arrow), associated with significant parietal and posterior frontal atrophy in a 59-year-old woman with early onset AD (MMSE 21). AD, Alzheimer’s disease, MMSE, Mini Mental Status Exam.
Functional neuroimaging (i.e., fluorodeoxy-glucose- [FDG-] PET and single photon emission computed tomography [SPECT]) may increase diagnostic confidence in the evaluation of dementia. In particular, FDG-PET reveals specific abnormalities in AD by showing reduced glucose metabolism in the parietal and superior/posterior temporal regions, posterior cingulate cortex, and precuneus.15–17 In advanced stages of AD, frontal lobe defects are also seen.18 FDG-PET has been reported to have a sensitivity of 93% and a specificity of 63% in predicting a pathological diagnosis of AD.19 In a recent study, the combination of clinical diagnosis and positive PET scan increased the probability of detecting AD pathology from 70 to 84%.20 In a clinical-pathological study, a positive SPECT scan raised the likelihood of AD to 92%, whereas a negative SPECT scan lowered the likelihood to 70%. SPECT was more useful when the clinical diagnosis was “possible” AD, with the likelihood of 84% with a positive SPECT, and 52% with a negative SPECT.21 Similar results were found with MR perfusion using a paramagnetic contrast agent.22,23
A very promising approach for improved diagnosis of neurodegenerative diseases is PET neuroimaging with protein-specific radiolabeled ligands such as 11C-labeled PIB (11C-PIB) for AD (Fig. 2). In a recent study, Aβ amyloid neuroimaging helped discriminate AD from frontotemporal lobar degeneration (FTLD) showing that all patients with AD had positive 11C-PIB scans by visual inspection, while 8 of 12 patients with FTLD and 7 of 8 controls had negative scans.24 As these patients were not pathology-confirmed, it is possible that the “false positive” patients had AD pathology. Although PIB holds promise for improved diagnostic accuracy in AD, its use in clinical practice is still under investigation.
Figure 2.
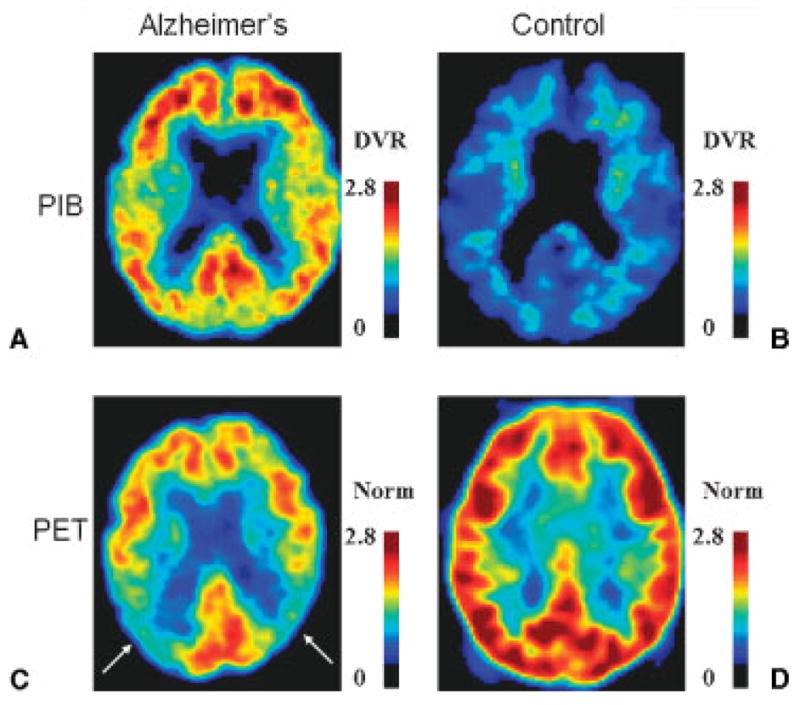
Molecular and functional PET in (A,C) a patient with AD and (B,D) a healthy control. PET neuroimaging with the Aβ amyloid ligand Pittsburgh Compound-B in (A) a 67-year-old man with moderate AD and (B) a 73-year-old cognitively normal woman. Axial DVR images referenced to the cerebellum are shown in neurologic orientation. (A) The patient with AD has increased tracer binding in the frontal, posterior cingulate, parietal and temporal cortices, and the striatum (not shown). (B) In contrast, the cognitively normal control does not demonstrate tracer uptake in the cortex. (C,D) Fluorodeoxyglucose PET in the same subjects. Axial images normalized to mean activity in the pons (Norm) are presented in neurologic orientation. (C) The patient with AD demonstrates prominent hypometabolism, particularly in parietal cortex (arrows), while (D) the normal control shows normal glucose metabolism. (Images courtesy of Gil Rabinovici, University of California, San Francisco, and William Jagust, University of California, Berkeley.) PET, positron emission tomography; AD, Alzheimer’s disease; DVR, distribution volume ratio.
Several studies have been conducted on patients with mild cognitive impairment (MCI), a clinical syndrome in which cognitive difficulties are reported by patients or their informants, but they are not severe enough for a diagnosis of dementia. Patients with amnestic MCI (a-MCI), who have objective impairments in memory on neuropsychological testing, have a rate of conversion to AD of ~12% per year and ~80% conversion in 6 years.25 Amnestic mild cognitive impairment patients who convert show more overall atrophy and focal atrophy involving the hippocampus, inferior and middle temporal gyrus, posterior cingulate, and precuneus, compared with nonconverters.26 In a recent MRI study conducted on 190 a-MCI subjects, the rates of progression of medial temporal atrophy (visual assessment-based) were associated with significantly increased risk of developing dementia within 3 years.27 In a-MCI progressing to AD, PET shows hypometabolism in the inferior parietal, posterior cingulate, and medial temporal region,28–30 while SPECT shows hypoperfusion in the inferior parietal lobule, angular gyrus, and precuneus.31 Finally, increased PIB uptake has been shown in frontal, increased PIB uptake has been shown in frontal, parietal, and lateral temporal cortices as well as in the posterior cingulate of patients with a-MCI, suggesting the presence of AD pathology.32 Although structural findings, particularly biparietal, posterior body of corpus callosum, and hippocampal atrophy, are supportive of AD, nuclear medicine techniques, in particular using 11C-PIB or other compounds labeling disease-specific proteins in vivo, will likely be the most useful for diagnosing AD in the future.
FRONTOTEMPORAL LOBE DEGENERATION
Recent studies suggest that frontotemporal lobe degeneration (FTLD) is as common a cause of dementia as AD in individuals younger than 65 years33,34; yet, FTLD also occurs in older subjects, as nearly one quarter of patients with FTLD develop disease after the age of 65 years.35 Three distinct clinical presentations of FTLD are recognized: behavioral variant frontotemporal dementia (bvFTD), semantic dementia (SD), and progressive nonfluent aphasia (PNFA).36 Behavioral variant FTD is characterized by focal pathology in the frontal (dorsolateral, orbital, and medial frontal cortices) and anterior temporal regions. Semantic dementia is typified by changes in the anterior temporal lobe greater on the left than on the right side. Progressive nonfluent aphasia is characterized by mainly left-sided inferior frontal and insular atrophy.37–40
Structural neuroimaging, preferably with MRI, is an essential part of the evaluation of suspected FTLD (Fig. 3). In the early phase of disease, MRI may appear normal by routine visual assessment. However, as the disease progresses, focal atrophy of frontal lobes, hippocampus, and amygdala is often present.41,42 In SD, severe “knife-edge”–type atrophy is almost always present in the anterior temporal lobes.43 These changes are readily seen with coronal MRI (Fig. 3B) but can be missed by CT and even by MRI without coronal acquisitions.43 On T2 and proton density-weighted images, an increased signal intensity may be seen in the subcortical white matter of atrophied gyri, extending into the deep white matter.44 The corpus callosum can appear atrophic either anteriorly or diffusely, differentiating FTLD from AD, in which the posterior portion of the corpus callosum is more prominently affected (Fig. 1).12 Quantitative MRI, using either manual methods45–47 or the automated, computerized analysis known as voxel-based morphometry (VBM), have refined these observations.37,39,48 In agreement with pathological studies,41 both bvFTD and SD groups are mainly associated with gray matter loss in the ventromedial frontal regions, the bilateral insula, and the left anterior cingulate cortex.37,39 BvFTD patients also demonstrate atrophy throughout dorsolateral prefrontal cortex and medial premotor regions,37 whereas the SD subgroup shows involvement of the temporopolar and perirhinal cortices, and the anterior fusiform gyri.37,39,46–48 Patients with PNFA show selective atrophy in the left perisylvian region.39 Similar results have been found in autopsy-proven FTLD patients.49,50 Studies investigating the diagnostic utility of these findings show that frontal lobe volumes provided correct classification of 93% of bvFTD patients versus healthy controls,51 and smaller hippocampal and amygdala volumes significantly differentiate SD patients from patients with frontal variant bvFTD.52 More recently, automated cortical thickness measurement has also demonstrated differences in the regional distribution of brain atrophy between AD and bvFTD similar to those identified using VBM.53
Figure 3.

Brain coronal T1-weighted MRI from patients with different clinical presentations of frontotemporal lobar degeneration. (A) BvFTD in a 62-year-old man, MMSE score 24. (B) SD in a 66-year-old man, MMSE 26. (C) PNFA in a 66-year-old woman, MMSE 28. Note the bilateral gray matter loss in the inferior frontal gyrus (arrow), superior frontal gyrus (dashed arrow), the insula (dotted arrow), and the anterior cingulate (arrowhead) in (A) the patient with bvFTD; the atrophy of the left temporal lobe (arrow) in (B) the patient with SD; and the prominent atrophy in the left perisylvian region (arrow) in (C) the patient with PNFA. MRI, magnetic resonance imaging; bvFTD, behavioral variant frontotemporal dementia; MMSE, Mini Mental Status Exam; SD, semantic dementia; PNFA, progressive nonfluent aphasia.
Alzheimer’s disease can present in the early stages with disproportionate and prominent impairments on frontal lobe functions (“frontal variant” of AD),54 sufficient to prompt an initial clinical diagnosis of FTLD.55 Therefore, functional neuroimaging (i.e., PET and SPECT) has been enlisted to differentiate AD from FTLD in certain clinical situations.56,57 In pathologically confirmed AD and FTLD patients, FDG-PET significantly increased diagnostic accuracy with a sensitivity of 86% and a specificity of 97.6%, beyond clinical features alone.57 Specific patterns of hypometabolism are associated with specific clinical syndromes, with frontal hypometabolism being associated with bvFTD, temporal with SD, and left perisylvian with PNFA.16,58,59 SPECT has demonstrated that anterior temporal and frontal hypoperfusion are highly predictive of a diagnosis of FTLD as opposed to AD.56 PET neuroimaging, with protein-specific radiolabeled ligands, such as 11C-PIB, may be very useful for distinguishing “frontal variant” of AD from FTLD, but it is still experimental (Fig. 2).24 As discussed below, perfusion MRI using arterial spin labeling (ASL) technique also may be clinically helpful in the differential diagnosis of dementia, as one study showed that the combination of decreased frontal perfusion and preserved parietal perfusion provided a correct classification of bvFTD versus AD with 87% accuracy.60
VASCULAR DEMENTIA
Vascular dementia is caused by ischemic lesions, ranging from small vessel disease to large vessel strokes. Although it is well accepted that stroke can lead to dementia, lesions in white matter caused by small vessel disease are frequently found by MRI even in normal subjects, and therefore it is unclear how extensive these lesions need to be to cause cognitive impairment. Vascular etiologies are the third most common cause of dementia (~ 8 to 10%), following AD (60 to 70%), and dementia with Lewy bodies (DLB) (10 to 25%),61 but these numbers vary considerably according to the different criteria used for VaD.62 Furthermore, it is evident from autopsy studies that many patients have mixed dementias, often vascular disease with other conditions.61 Old criteria for VaD only included multi-infarct dementia,63 or dementia resulting from the cumulative effects of several clinically significant strokes, but the current criteria consider multi-infarct dementia as only one of several subtypes of VaD, including single-stroke dementia and small vessel disease.61 Most current criteria define the topography and severity of vascular lesions on MRI and require a temporal relationship (~3 months) between the clinical signs of stroke and dementia onset.62 This temporal relationship, however, is not always demonstrable when strokes are silent,64 such as small vessel incomplete strokes that affect cognition insidiously or strokes that affect the basal ganglia leading only to subtle extrapyramidal signs.65 Nevertheless, even in the absence of any focal neurological signs, the hemodynamic failure caused by severe carotid stenosis can lead to cognitive impairment.66 There is lack of consensus about the benefit of carotid endarterectomy on cognition, which in part may be due to the occurrence of microemboli from the procedure or the effect of general anesthesia.67 Recent studies have shown, however, improved cognitive function from carotid stenting when it is performed under local anesthesia with use of protective devices to avoid microemboli.68,69
The three main neuroimaging patterns in VaD are large vessels strokes (macroangiopathy, arteriosclerosis), small vessel disease (microangiopathy, arteriolosclerosis), and microhemorrhages (Fig. 4). Single large territorial strokes, especially in the middle cerebral artery (MCA) territory of the dominant hemisphere, or multiple smaller strokes in bilateral anterior cerebral artery (ACA) or posterior cerebral artery (PCA) territories, cause dementia in ~30% of stroke survivors.61,70,71 Single smaller strokes can also cause significant cognitive dysfunction when occurring in particular locations, such as the watershed territories, including the bilateral superior frontal gyrus or bilateral orbitofrontal (ACA/MCA), angular gyrus (ACA/MCA/PCA), temporo-occipital junction, and inferior temporal gyrus (MCA/PCA).61,71–73
Figure 4.

Axial T2 images in four patients with different types of VaD. (A) An 84-year-old woman with cognitive deficits (MMSE 26, which 2 years later declined to 15). T2-weighted MRI shows chronic right temporal pole infarction and only mild left hippocampal atrophy. (B) A 79-year-old man with behavioral, frontal-executive, and memory problems (MMSE 19). T2-weighted MRI shows chronic left thalamic lacunar stroke, bilateral caudate and frontal white matter small vessel disease, as well as bifrontal atrophy. (C) A 72-year-old woman with memory impairment (MMSE 24) diagnosed with mixed AD-VaD. T2-weighted MRI shows bilateral hippocampal atrophy and multiple microhemorrhages (focal hypointensities, arrows) suggestive of amyloid angiopathy. (D–F) A 49-year-old man with CADASIL and progressive behavioral and memory disturbances (MMSE 27). Axial T2-weighted MRI shows diffuse bilateral leukoencephalopathy involving (D) the centrum semiovale, (D,E) anterior and posterior deep white matter, (E) internal and external capsules (arrow), (F) subcortical white matter of temporal poles (arrows), and the pons, and (E) a lacunar stroke in the right thalamus and microhemorrhages primarily in the left thalamus. This MRI pattern, with the bilateral anterior temporal lobe white matter hyperintensity, is highly suggestive of CADASIL. VaD, vascular dementia; MMSE, Mini Mental Status Exam; MRI, magnetic resonance imaging; AD, Alzheimer’s disease; CADASIL, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy.
Small vessel disease causes incomplete or complete infarcts in the white matter or in subcortical gray matter nuclei.74 On FLAIR images, incomplete infarcts present as hyperintensities, whereas complete infarcts present as lacunae, which are hypointense in relation to the brain and isointense to the cerebrospinal fluid. Lacunae must be differentiated from perivascular Virchow-Robin spaces. Lacunar strokes are small complete infarcts (2 to 15 mm). When located in the caudate head,75,76 anterior thalamus, or the mamillothalamic tract,73,77,78 lacunae can cause significant cognitive and/or behavioral dysfunction due to the extended functional deafferentation of the cortical areas, as has been shown by SPECT hypoperfusion in a series of left anterior thalamic lesions.78
Small vessel disease identified on MRI in the white matter is called leukoaraiosis.79 Leukoaraiosis presents as multiple punctuate or confluent lesions, but more often as incomplete infarcts, and is commonly seen in healthy elderly and in subjects with migraine. When small vessel disease causes subcortical VaD, this is associated with the pathology of Binswanger’s disease.80 Some studies have suggested that to assess in single cases how much the lesion load affects cognition, a threshold of 10 cm2 81 or 25% of total white matter82 is required before VaD is detectable clinically. The clinical diagnosis of dementia using standard criteria for VaD is usually based on the presence of memory and another cognitive dysfunction, but in VaD the memory deficit can be less pronounced than the frontal-executive dysfunction (e.g., problems organizing, planning, multitasking, etc.).83 Quantitative MRI studies in nondemented elderly subjects demonstrate that the total volume of subcortical lesions correlates with frontal-executive impairment.84–86 Interestingly, a correlation was not found between verbal memory and subcortical temporal lesions, but in the presence of co-occurring AD pathology, it is likely that the subcortical lesions play a synergistic effect, as shown in longitudinal studies.87
Because the pattern of white matter abnormality often is not pathognomonic for vascular lesions, non-vascular causes of white matter disease with cognitive impairment, such as multiple sclerosis, should be considered in the differential diagnosis of subcortical VaD. In subcortical VaD, the pattern of white matter abnormality can help to identify the underlying cause. Lesions in periventricular or deep white matter are usually associated with small vessel cerebrovascular disease (e.g., systemic hypertension, diabetes, hyperhomocysteinemia) or acute hypotensive states (e.g., orthostatic hypotension, congestive heart failure, arrhythmias).74 The involvement of subcortical white matter, often with small cortical infarcts, can be caused by cardiac, aortic, or carotid microemboli,88 vasculitis,89 and CADASIL.90 In vasculitis, MR angiography shows abnormalities in the minority of cases in which large vessels are involved, and paramagnetic contrast injection may show abnormal enhancement, but the diagnosis usually requires angiography or brain biopsy.89,91
CADASIL is a genetic small vessel disease that presents usually between ages 30 and 50, with migraine, strokes, cognitive impairment, and behavioral disorders, including depression. MRI shows confluent lesions in the periventricular white matter, characteristic white matter lesions in the temporal poles and in the external capsule, and striatocapsular lacunae and microhemorrhages (Fig. 4).90 Two recent studies found that only the number and total volume of lacunae, but not the volume of white matter FLAIR hyperintensity, significantly correlated with cognitive dysfunction in CADASIL.92,93
Microhemorrhages are the third major neuroimaging aspect of VaD, and in one study they were found in 65% of VaD cases.94 While macrohemorrhages associated with cognitive impairment (e.g., venous infarcts) can be seen on conventional T1- and T2-weighted spin echo images, microhemorrhages often cannot be seen in these sequences, but can be detected accurately using T2*-weighted gradient echo images. In many cases, it is likely that microhemorrhages and white matter ischemic disease are caused by systemic hypertension.95 One study found microhemorrhages in 18% of AD cases, in which they are often due to cerebral amyloid angiopathy (CAA).94 Microhemorrhages have been reported in both sporadic (16 to 38%) and hereditary (69%) forms of CAA. In CAA, unlike hypertension-related lesions, microhemorrhages are more frequently located at the corticosubcortical junctions in frontomesial, frontorbital, and parietal regions—areas that are crucial for behavior and cognition.96
Recognition of the different neuroimaging patterns of VaD is clinically relevant because specific patterns can help identify the underlying pathology and etiology, which may be treatable. Vascular-related MCI is likely the early, most treatable stage of VaD97; thus, it is an important health issue to detect and treat vascular MCI (e.g., reduction of vascular risk factors to prevent the conversion to VaD).98,99
ATYPICAL PARKINSONIAN DEMENTIAS
Dementia occurs in most parkinsonian syndromes, including idiopathic Parkinson’s disease (PD) and the atypical parkinsonian dementias. Dementia with Lewy bodies (also called Lewy body dementia) is a common cause of neurodegenerative dementia that is pathologically associated with α-synuclein-positive Lewy body neuronal inclusions in the cortex, brainstem, and substantia nigra. Progressive cognitive decline, mainly in memory, and visuospatial and executive functions can be associated with visual hallucinations, cognitive circadian fluctuations, and rapid eye movement sleep behavior disorder.100
The most common finding on MRI scanning in DLB is mild diffuse cortical atrophy. This feature unfortunately does not help distinguish DLB from other dementias that often have overlapping clinical findings, such as corticobasal degeneration or AD. In volumetric MRI studies, VBM analyses identified more insular and frontotemporal atrophy in DLB101–103 than in age-matched controls, but did not consistently detect areas more atrophic in DLB than in AD. Region of interest-based analyses showed more putaminal atrophy in one study104 and more dorsal midbrain atrophy in another of DLB compared with AD.103 In both studies, however, there was a large range of overlap, suggesting these methods will not help improve clinical diagnosis in an individual patient.
When cognitive symptoms are prevalent and parkinsonism is absent or subtle, as often occurs in the early phases, DLB is easily confused with AD. SPECT can help diagnose DLB through reduced striatal uptake of dopamine or its presymptomatic transporters. In a multicenter study, SPECT with the presynaptic dopamine transporter 123I-FP-CIT identified probable DLB with a sensitivity of 77.7% and specificity of 90.4%.105
Transcranial sonography has recently been shown to have excellent potential to detect iron deposits in deep nuclei106 and may help differentiate some atypical parkinsonian dementias from PD. Unfortunately, this technique is still not widely available and cannot be used in ~20% of subjects because of the thickness of the temporal bones; however, it may be an option for some patients, particularly when MRI cannot be performed. In one study with this technique, normal echogenic substantia nigra indicated multiple system atrophy (MSA) rather than PD (sensitivity, 90%; specificity, 98%), whereas third-ventricle dilation (>10 mm) with lenticular nucleus hyperechogenicity indicated progressive supranuclear palsy (PSP) rather than PD (sensitivity, 84%; specificity 98%).107
Corticobasal degeneration characteristically presents with progressive asymmetric cortical symptoms: language or speech disturbance, visuospatial deficit or hemineglect, motor apraxia or alien limb, and sometimes myoclonus.108 MRI demonstrates characteristically asymmetric frontal and/or parietal atrophy (Fig. 5A,B), with less frequent involvement of the temporal lobe. Visual assessment of the asymmetry has been reported to differentiate corticobasal degeneration from PSP with relatively high accuracy (sensitivity 87.5%, specificity 100%).109 A VBM analysis of our institution’s corticobasal degeneration cohort found a pattern of left frontoparietal atrophy (stronger than the contralateral areas); the most significant locus of atrophy was the junction between superior frontal and precentral sulci, which are functionally related to the frontal eye fields.110
Figure 5.

(A,B) T2-weighted images showing diffuse asymmetric (L > R) bilateral frontoparietal atrophy (arrows) in a 54-year-old woman with progressive nonfluent aphasia and mild parkinsonism (MMSE 29) due to pathology-proven corticobasal degeneration. (C,E) Sagittal and (D) coronal T1-weighted images from a high-resolution volumetric sequence showing (C, compared with the pons) reduced midbrain area and (D) thinned superior cerebellar peduncles (arrows), compared with (E) the middle cerebral peduncles (arrows) in a 61-year-old man with parkinsonism and frontal-executive dysfunction (MMSE 27) and autopsy-proven progressive supranuclear palsy. (F) Axial FLAIR and (G) coronal T2* images showing posterolateral putamen hypointensity with hyperintense rim (arrow and dotted arrow) in a 64-year-old man with multiple system atrophy (MMSE 26). MMSE, Mini Mental Status Examination; FLAIR, fluid attenuation inversion recovery.
Progressive supranuclear palsy is characterized by impairment of vertical eye movement and postural instability with falls.111 MRI usually shows third ventricle dilation and significant dorsal midbrain atrophy especially of the anteroposterior diameter (Fig. 5C); however, the visual assessment of these features is not sufficient to completely discriminate PSP from corticobasal degeneration.109 A study measured the anteroposterior midbrain diameter in the midsagittal slice and found it shorter than 15 mm in all 16 PSP cases, and longer than 17 mm in all 20 PD patients and 12 controls.112 The midbrain sizes of some MSA cases, however, overlapped with those of PSP. Using manually drawn regions of interest on the precise midsagittal slice (of 3-mm thick MRI tomographic slices), one study found that the ratio of the pons/midbrain area completely differentiated all 21 PSP subjects not only from all 23 PD subjects, but also from all 25 MSA subjects.113 A more recent study, however, on a larger cohort (33 PSP, 19 MSA, 108 PD) found some overlap even using this ratio.114 This methodology was improved upon by using a volumetric T1 scan to measure not only the pons/midbrain area ratio (P/M) on the midsagittal section, but also the middle/superior cerebellar peduncle width ratio (MCP/SCP) (Fig. 5D,E). Applying an index [(P/M) × (MCP/SCP)] showed 100% sensitivity and specificity in differentiating PSP from both PD and MSA.114
Multiple system atrophy presents characteristically with orthostatic hypotension, urinary dysfunction, and mild cerebellar symptoms. T2-weighted MRI often shows a posterolateral putaminal hypointensity, mainly due to iron deposition, with a hyperintense rim, mainly due to gliosis (Fig. 5F,G).115 Using T2* gradient echo sequences (see Emerging Neuroimaging Approaches section below) to detect this hypointensity and FLAIR to detect the hyperintense rim in a large sample of 52 MSA patients, fair accuracy was recently demonstrated (sensitivity 69%, specificity 97%) in differentiating MSA from PD.116 This study was performed with only a 1 Tesla (1T) MRI scanner, but the iron sensitivity in MSA is field strength-dependent117; therefore, this iron-related MRI finding is even more likely to be detected at the more commonly used 1.5T and 3T field strengths.
CEREBELLAR DISORDERS
Among the most frequent neurodegenerative cerebellar disorders in adulthood are the cerebellar variant of MSA (MSA-C), idiopathic cerebellar ataxia with (IDCA-P) or without parkinsonism (IDCA-C), and hereditary spinocerebellar ataxias (SCA), of which there are many genetically defined subtypes. MSA-C presents with more cerebellar symptoms than parkinsonism, and cognitive dysfunction may be subtle.118 MRI in MSA-C (but also in some cases of standard MSA) shows the degeneration of transverse pontine fibers as the characteristic “cross” sign (also called the “hot cross bun” sign). This sign is associated with middle cerebellar peduncle hyperintensity and with pontine atrophy.119 One study found this sign to have 97% sensitivity and 100% specificity in differentiating MSA-C from IDCA-P,120 but another study identified this sign in 3 of 14 (21%) patients with SCA Type 1 and in 7 of 11 (64%) of those with SCA Type 2.121 Subacute onset cerebellar disorders have many causes, including paraneoplastic conditions, often associated with anti-Yo antibodies (Fig. 7C,D)122 or other antibody-mediated conditions such as antiglutamic acid decarboxylase antibodies.123
Figure 7.

Two cases of non–prion-related rapidly progressive dementia or ataxia syndromes. Note that in these cases, the abnormalities are best seen in FLAIR images. (A,B) A 66-year-old man with intravascular lymphoma. (B) FLAIR multifocal abnormalities involving cerebral and cerebellar gray and white matter in a vascular distribution. These lesions, also involving the right hippocampus, showed patchy enhancement after contrast administration (not shown). (A) DWI shows a right periventricular focal region with diffusion restriction; DWI hyperintensity is common in lymphomas. (C,D) A 65-year-old woman with anti-Yo paraneoplastic cerebellitis. MRI shows mild diffuse hyperintensity of the cerebellar, compared with the cerebral, cortex with slight atrophy of the lateral folia. Note (D) the strong hyperintense FLAIR signal in superior, medial cerebellum (arrows), and (C) no major hyperintensity in the axial DWI scan. (E,F) A 60-year-old woman with paraneoplastic limbic encephalitis and FLAIR MRI showing hyperintensity of bilateral insula, medial (arrows) and inferior temporal cortex, hippocampus, amygdala (F) on FLAIR and only subtle hyperintensity (E) on DWI. FLAIR, fluid-attenuated inversion recovery; DWI, diffusion-weighted imaging; MRI, magnetic resonance imaging.
Cognitive impairment is being increasingly identified and studied in the SCAs. Among the 28 SCA genotypes identified to date, cognitive deficits vary both within and between mutations and may be more related to the cerebral than to cerebellar involvement.124 Cognitive deficits are very mild or absent in SCA6, which has pure cerebellar degeneration on MRI and involves executive dysfunction in SCA1 and SCA3 variants that show putaminal and caudate atrophy on MRI.125 In SCA2, multiple cognitive domains are impaired in ~25% of cases, and these show significant cerebellar, but also cerebral, atrophy on structural MRI scans.126 SCA17 presents frequently with dystonia, parkinsonism, and cognitive dysfunction in up to 80% of cases; virtually all patients have cerebral atrophy on MRI.127 SCA17 can present similarly to MSA and PSP128 and have the putaminal T2 signal changes usually associated with MSA.129
PRION DISEASES AND OTHER RAPIDLY PROGRESSIVE DEMENTIAS
Currently, MRI is more useful for the diagnosis of prion disease than for any other dementia. Hyperintensity of the cortical gyri (cortical ribboning), striatum (caudate and putamen), and/or thalamus on trace DWI scans has high sensitivity and specificity for the diagnosis of sporadic CJD (sCJD) (Fig. 6) and some forms of genetic prion diseases. These DWI findings have at least 91 to 92% sensitivity and 94 to 95% specificity for sCJD,130,131 whereas the pulvinar sign (greater hyperintensity of the posterior thalamus relative to the anterior putamen) on T2-weighted MRI has high diagnostic utility for variant CJD132,133 (vCJD) (Fig. 6). In sCJD, the most common pattern of MRI FLAIR and DWI hyperintensity is cortical and subcortical (68%), followed by the cortex alone (24%) and subcortical alone (5%).130 DWI hyper-intensity in CJD is much easier to see than T2 or FLAIR hyperintensity. In our experience, the DWI hyperintensity in subcortical regions almost invariably corresponds to apparent diffusion coefficient (ADC) map hypointensity, confirming restricted diffusion in these regions. Unfortunately, without extensive post image processing, ADC maps of the cortex often can be difficult to interpret. Cortical areas most commonly involved on DWI in sCJD include the cingulate gyrus, superior and middle frontal gyrus, insula, precuneus, angular gyrus, and parahippocampal gyri (P. Vitali and M. Geschwind, unpublished data). Interestingly, the rolandic cortex is usually spared on MRI FLAIR and DWI in CJD, as is the case with most neurodegenerative diseases that cause dementia.130,134 In prion disorders associated with a high degree of amyloid deposition, such as Gerstmann-Sträussler-Scheinker disease and other genetic variants, PET ligands that bind to prion amyloid deposits, such as 2-(1-{6-[(2-[F-18]fluoroethyl) (methyl)amino]-2-naphthyl} ethylidene) malononitrile (FDDNP), may offer additional diagnostic opportunities.135
Figure 6.

MRI findings in CJD. (A,B) A patient with probable variant CJD and three common MRI patterns in sporadic CJD: (C,D) predominantly subcortical, (E,F) both cortical and subcortical, and (G,H) predominantly cortical. Note that in sporadic CJD the abnormalities are always (C,E,G) more evident on DWI than (D,F,H) on FLAIR images. (A,B) A 21-year-old woman with probable variant CJD with MRI showing bilateral thalamic hyperintensity in the mesial pars (mainly dorsomedian nucleus) and posterior pars (pulvinar) of the thalamus, the so-called “double hockey stick sign.” Also note the “pulvinar sign,” with the posterior thalamus (pulvinar) being more hyperintense than the anterior putamen. The three sporadic CJD cases are pathology-proven. (C,D) A 52-year-old woman with MRI showing strong hyperintensity in bilateral striatum (solid arrows, both caudate and putamen) and slight hyperintensity in mesial and posterior thalamus (dotted arrow). (E,F) A 68-year-old man with MRI showing hyperintensity in bilateral striatum (note anteroposterior gradient in the putamen, which is commonly seen in CJD), thalamus, right insula (dotted arrow), anterior and posterior cingulate gyrus (arrow, L > R), and left temporal-parietal-occipital junction (arrow). (G,H) A 76-year-old woman with MRI showing diffuse hyperintense signal mainly in bilateral parietal and occipital cortex, right posterior insula (dashed arrow) and left inferior frontal cortex (arrow), but no significant subcortical abnormalities. CJD, Creutzfeldt-Jakob disease; MRI, magnetic resonance imaging; DWI, diffusion-weighted imaging; FLAIR, fluid-attenuated inversion recovery.
Other rapidly progressive dementias can have MRI findings similar to CJD. Bartonella encephalopathy, Wilson’s disease, and Wernicke’s encephalopathy can show DWI hyperintensity in the deep gray nuclei,136–138 while antibody-mediated encephalopathies and neurofilament inclusion body dementia can have FLAIR hyperintensity in the cortex and/or deep nuclei. In these conditions, unlike prion disease, the underlying white matter is also often involved (M. Geschwind, unpublished data).139,140 Seizures, including nonconvulsive status epilepticus, can result in cortical ribboning on DWI; however, unlike CJD, these findings resolve within days after cessation of seizures.141,142 Limbic encephalopathies often have T2-weighted, particularly FLAIR, medial temporal lobe and/or patchy white matter hyperintensity (Fig. 7E,F).143,144 Many rapidly progressive dementias can cause leukoencephalopathy, including progressive multifocal leukoencephalopathy and leukodystrophies, or mixed gray and white matter involvement, such as mitochondrial diseases and lymphoma (Fig. 7A,B).145,146 Infections and toxic conditions can also be a cause of dementia and are discussed in other articles in this issue. MRI with gadolinium contrast should be used in the evaluation of most rapidly progressive dementias.
EMERGING NEUROIMAGING APPROACHES IN DEMENTIA
Nuclear Medicine Methods
PET and SPECT are the most promising neuroimaging techniques for the future of dementia diagnosis. These methods will be even more useful when ligands specific for the different proteins (e.g., tau, TDP-43, α-synuclein, PrP) involved in neurodegenerative disorders are developed.135
Diffusion-Weighted Magnetic Resonance Imaging
With increasingly sophisticated techniques for analysis, the utility of DWI is becoming more apparent. Restricted diffusion resulting in hyperintensity on trace DWI scans, such as that seen in CJD or acute stroke, can easily be appreciated by visual assessment. Hyper-intensity on DWI scans, however, can be caused by T2 shine-through in addition to restricted diffusion, a problem that can be addressed with ADC maps.147 Diffusion-weighted imaging can also be helpful in the diagnosis of other, more common forms of dementia because enlargement of extracellular spaces caused by neuronal loss can be detected as an increase in ADC (i.e., increased diffusion).147,148 Increased diffusion occurring in settings such as neuronal loss or vasogenic edema causes DWI hypointensity, which is more difficult to see by visual assessment than DWI hyperintensity because of masking from concomitant hyperintensity due to T2 shine-through effect. To circumvent this problem, a region of interest-based approach to analysis of ADC maps where the T2 effect has already been subtracted may be effective. This approach has been studied for diagnostic utility in subcortical dementias. For example, one study found that ADC values less than 0.733 × 10−3 mm2/s in the middle cerebellar peduncle differentiated MSA from PSP with 91% sensitivity and 84% specificity.149 While this approach is feasible in white matter and in subcortical gray matter, further postprocessing is required to measure ADC in the cortex.150 One study using precise segmentation on coronal DWI slices in MCI patients found that hippocampal ADC values greater than the median predicted conversion to AD better than did hippocampal volume.151
T2-T2′-T2* Relaxometry and Susceptibility-Weighted Magnetic Resonance Imaging
It is well known that iron deposition increases in certain deep brain structures, such as the putamen, in aging, but this also occurs to an even greater extent in several neurodegenerative disorders. T2 time is shortened by iron deposition, causing T2 hypointensity, but T2 time is lengthened (T2 hyperintensity) by increased water content due to the neuronal loss; these opposing effects cancel out T2 signal, making it difficult to detect iron deposition in neurodegenerative disease with spin echo techniques.152 A recent study, however, used a more sensitive technique by looking at histograms of T2 signal and detected increased iron deposition in AD patients compared with the controls.153 Furthermore, today, with higher field strength MRI, such as 3T or greater, and new technologies such as T2*, T2′ (derived from T2 and T2*), and T2 rho, cerebral iron can be better assessed.152
Susceptibility-weighted MRI is an emerging technique that uses the information not only from the magnitude, but also from the phase, of the signal from a gradient echo MRI sequence. The phase shift is strongly dependent on the susceptibility of the tissues to the local inhomogeneity of the magnetic field and therefore is differentially sensitive to iron (and hemorrhagic products) and calcium. The capacity of susceptibility-weighted MRI to differentiate calcium from iron and from hemorrhagic products (all hyperdense in CT and hypointense in T2 and T2*) make this sequence especially promising in the aging brain, where increases in calcium and iron deposition are commonly observed.154
Arterial Spin Labeling
Although cerebral blood perfusion now is routinely assessed by dynamic susceptibility imaging acquired during paramagnetic contrast injection, ASL can measure absolute cerebral blood flow without the use of any contrast agent. This technique uses magnetic labeling of inflowing arterial blood. Because the signal-to-noise ratio is low at 1.5T, intensive image postprocessing is necessary, and this technique is not yet used in the clinical setting. With the advent of 3T scanners, the improved signal-to-noise ratio is expected to increase the clinical application of this technique. In cortical dementias, ASL may offer an alternative to PET/SPECT in detecting hypoperfused areas, as ASL is less expensive and avoids exposure to radioactivity. Similar to PET/SPECT, hypoperfusion assessed by ASL in AD can be detected even after correcting for atrophy.155 In one study, frontal-parietal perfusion gradients detected by ASL differentiated clinical AD from clinical bvFTD with 90% sensitivity and 83% specificity.60
Functional Magnetic Resonance Imaging
Functional MRI (fMRI) indirectly assesses neural activity through blood oxygen level-dependent (BOLD) changes.156 Although the neurovascular mechanism underlying BOLD changes is still poorly understood and is an area of study, fMRI increasingly is being used in neurological research. Furthermore, studying cognitive-behavioral functions in early neurodegenerative disorders should identify the neuroanatomical networks affected by these diseases.157 Currently, the intrinsic inter- and intrasubject variability of the BOLD signal and the intrinsic dependence of hemodynamic factors prevent the use of this technique for differential diagnostic purposes. Although fMRI usually requires active participation from subjects, which can be difficult in patients with certain cognitive disorders, this is not necessary for resting state network studies, in which variation in fMRI signal is measured over time while subjects are resting quietly. This technique can be employed, in a research context, to evaluate the functional connectivity in the different brain areas, even in cognitively impaired patients. It may soon have diagnostic applicability as well.158
Acknowledgments
This work was supported by NIA K23AG021989 and NIH-NIA 1P01 AG021601. Dr. Vitali is supported by a research grant from Fondazione Tronchetti-Provera, Milan, Italy.
Footnotes
Neuroimaging Essentials for the Clinician; Guest Editor, Rohit Bakshi, M.D.
References
- 1.Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 2.Maurer K, Volk S, Gerbaldo H. Auguste D and Alzheimer’s disease. Lancet. 1997;349:1546–1549. doi: 10.1016/S0140-6736(96)10203-8. [DOI] [PubMed] [Google Scholar]
- 3.Klunemann HH, Fronhofer W, Wurster H, Fischer W, Ibach B, Klein HE. Alzheimer’s second patient: Johann F and his family. Ann Neurol. 2002;52:520–523. doi: 10.1002/ana.10309. [DOI] [PubMed] [Google Scholar]
- 4.Ishii K, Kawachi T, Sasaki H, et al. Voxel-based morphometric comparison between early- and late-onset mild Alzheimer’s disease and assessment of diagnostic performance of z score images. AJNR Am J Neuroradiol. 2005;26:333–340. [PMC free article] [PubMed] [Google Scholar]
- 5.Karas G, Scheltens P, Rombouts S, et al. Precuneus atrophy in early-onset Alzheimer’s disease: a morphometric structural MRI study. Neuroradiology. 2007;49:967–976. doi: 10.1007/s00234-007-0269-2. [DOI] [PubMed] [Google Scholar]
- 6.Frisoni GB, Pievani M, Testa C, et al. The topography of grey matter involvement in early and late onset Alzheimer’s disease. Brain. 2007;130(Pt 3):720–730. doi: 10.1093/brain/awl377. [DOI] [PubMed] [Google Scholar]
- 7.Krasuski JS, Alexander GE, Horwitz B, et al. Volumes of medial temporal lobe structures in patients with Alzheimer’s disease and mild cognitive impairment (and in healthy controls) Biol Psychiatry. 1998;43:60–68. doi: 10.1016/s0006-3223(97)00013-9. [DOI] [PubMed] [Google Scholar]
- 8.Rusinek H, de Leon MJ, George AE, et al. Alzheimer disease: measuring loss of cerebral gray matter with MR imaging. Radiology. 1991;178:109–114. doi: 10.1148/radiology.178.1.1984287. [DOI] [PubMed] [Google Scholar]
- 9.Teipel SJ, Bayer W, Alexander GE, et al. Regional pattern of hippocampus and corpus callosum atrophy in Alzheimer’s disease in relation to dementia severity: evidence for early neocortical degeneration. Neurobiol Aging. 2003;24:85–94. doi: 10.1016/s0197-4580(02)00044-1. [DOI] [PubMed] [Google Scholar]
- 10.Tomaiuolo F, Scapin M, Di Paola M, et al. Gross anatomy of the corpus callosum in Alzheimer’s disease: regions of degeneration and their neuropsychological correlates. Dement Geriatr Cogn Disord. 2007;23:96–103. doi: 10.1159/000097371. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, Schuff N, Jahng GH, et al. Diffusion tensor imaging of cingulum fibers in mild cognitive impairment and Alzheimer disease. Neurology. 2007;68:13–19. doi: 10.1212/01.wnl.0000250326.77323.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamauchi H, Fukuyama H, Nagahama Y, et al. Comparison of the pattern of atrophy of the corpus callosum in frontotemporal dementia, progressive supranuclear palsy, and Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2000;69:623–629. doi: 10.1136/jnnp.69.5.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Likeman M, Anderson VM, Stevens JM, et al. Visual assessment of atrophy on magnetic resonance imaging in the diagnosis of pathologically confirmed young-onset dementias. Arch Neurol. 2005;62:1410–1415. doi: 10.1001/archneur.62.9.1410. [DOI] [PubMed] [Google Scholar]
- 14.Scheltens P, Fox N, Barkhof F, De Carli C. Structural magnetic resonance imaging in the practical assessment of dementia: beyond exclusion. Lancet Neurol. 2002;1:13–21. doi: 10.1016/s1474-4422(02)00002-9. [DOI] [PubMed] [Google Scholar]
- 15.Minoshima S, Giordani B, Berent S, Frey KA, Foster NL, Kuhl DE. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer’s disease. Ann Neurol. 1997;42:85–94. doi: 10.1002/ana.410420114. [DOI] [PubMed] [Google Scholar]
- 16.Ishii K, Sakamoto S, Sasaki M, et al. Cerebral glucose metabolism in patients with frontotemporal dementia. J Nucl Med. 1998;39:1875–1878. [PubMed] [Google Scholar]
- 17.Herholz K. PET studies in dementia. Ann Nucl Med. 2003;17:79–89. doi: 10.1007/BF02988444. [DOI] [PubMed] [Google Scholar]
- 18.Choo IH, Lee DY, Youn JC, et al. Topographic patterns of brain functional impairment progression according to clinical severity staging in 116 Alzheimer disease patients: FDG-PET study. Alzheimer Dis Assoc Disord. 2007;21:77–84. doi: 10.1097/WAD.0b013e3180687418. [DOI] [PubMed] [Google Scholar]
- 19.Hoffman JM, Welsh-Bohmer KA, Hanson M, et al. FDG-PET imaging in patients with pathologically verified dementia. J Nucl Med. 2000;41:1920–1928. [PubMed] [Google Scholar]
- 20.Jagust W, Reed B, Mungas D, Ellis W, Decarli C. What does fluorodeoxyglucose PET imaging add to a clinical diagnosis of dementia? Neurology. 2007;69:871–877. doi: 10.1212/01.wnl.0000269790.05105.16. [DOI] [PubMed] [Google Scholar]
- 21.Jagust W, Thisted R, Devous MD, Sr, et al. SPECT perfusion imaging in the diagnosis of Alzheimer’s disease: a clinical-pathologic study. Neurology. 2001;56:950–956. doi: 10.1212/wnl.56.7.950. [DOI] [PubMed] [Google Scholar]
- 22.Sandson TA, O’Connor M, Sperling RA, Edelman RR, Warach S. Noninvasive perfusion MRI in Alzheimer’s disease: a preliminary report. Neurology. 1996;47:1339–1342. doi: 10.1212/wnl.47.5.1339. [DOI] [PubMed] [Google Scholar]
- 23.Bozzao A, Floris R, Baviera ME, Apruzzese A, Simonetti G. Diffusion and perfusion MR imaging in cases of Alzheimer’s disease: correlations with cortical atrophy and lesion load. AJNR Am J Neuroradiol. 2001;22:1030–1036. [PMC free article] [PubMed] [Google Scholar]
- 24.Rabinovici GD, Furst AJ, O’Neil JP, et al. 11C-PIB PET imaging in Alzheimer disease and frontotemporal lobar degeneration. Neurology. 2007;68:1205–1212. doi: 10.1212/01.wnl.0000259035.98480.ed. [DOI] [PubMed] [Google Scholar]
- 25.Petersen RC, Doody R, Kurz A, et al. Current concepts in mild cognitive impairment. Arch Neurol. 2001;58:1985–1992. doi: 10.1001/archneur.58.12.1985. [DOI] [PubMed] [Google Scholar]
- 26.Chetelat G, Landeau B, Eustache F, et al. Using voxel-based morphometry to map the structural changes associated with rapid conversion in MCI: a longitudinal MRI study. Neuroimage. 2005;27:934–946. doi: 10.1016/j.neuroimage.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 27.DeCarli C, Frisoni GB, Clark CM, et al. Qualitative estimates of medial temporal atrophy as a predictor of progression from mild cognitive impairment to dementia. Arch Neurol. 2007;64:108–115. doi: 10.1001/archneur.64.1.108. [DOI] [PubMed] [Google Scholar]
- 28.Chetelat G, Desgranges B, de la Sayette V, Viader F, Eustache F, Baron JC. Mild cognitive impairment: can FDG-PET predict who is to rapidly convert to Alzheimer’s disease? Neurology. 2003;60:1374–1377. doi: 10.1212/01.wnl.0000055847.17752.e6. [DOI] [PubMed] [Google Scholar]
- 29.Mosconi L, Perani D, Sorbi S, et al. MCI conversion to dementia and the APOE genotype: a prediction study with FDG-PET. Neurology. 2004;63:2332–2340. doi: 10.1212/01.wnl.0000147469.18313.3b. [DOI] [PubMed] [Google Scholar]
- 30.Anchisi D, Borroni B, Franceschi M, et al. Heterogeneity of brain glucose metabolism in mild cognitive impairment and clinical progression to Alzheimer disease. Arch Neurol. 2005;62:1728–1733. doi: 10.1001/archneur.62.11.1728. [DOI] [PubMed] [Google Scholar]
- 31.Hirao K, Ohnishi T, Hirata Y, et al. The prediction of rapid conversion to Alzheimer’s disease in mild cognitive impairment using regional cerebral blood flow SPECT. Neuroimage. 2005;28:1014–1021. doi: 10.1016/j.neuroimage.2005.06.066. [DOI] [PubMed] [Google Scholar]
- 32.Kemppainen NM, Aalto S, Wilson IA, et al. PET amyloid ligand [11C]PIB uptake is increased in mild cognitive impairment. Neurology. 2007;68:1603–1606. doi: 10.1212/01.wnl.0000260969.94695.56. [DOI] [PubMed] [Google Scholar]
- 33.Johnson JK, Diehl J, Mendez MF, et al. Frontotemporal lobar degeneration: demographic characteristics of 353 patients. Arch Neurol. 2005;62:925–930. doi: 10.1001/archneur.62.6.925. [DOI] [PubMed] [Google Scholar]
- 34.Ratnavalli E, Brayne C, Dawson K, Hodges JR. The prevalence of frontotemporal dementia. Neurology. 2002;58:1615–1621. doi: 10.1212/wnl.58.11.1615. [DOI] [PubMed] [Google Scholar]
- 35.Rosso SM, Donker Kaat L, Baks T, et al. Frontotemporal dementia in The Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain. 2003;126(Pt 9):2016–2022. doi: 10.1093/brain/awg204. [DOI] [PubMed] [Google Scholar]
- 36.Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 37.Rosen HJ, Gorno-Tempini ML, Goldman WP, et al. Patterns of brain atrophy in frontotemporal dementia and semantic dementia. Neurology. 2002;58:198–208. doi: 10.1212/wnl.58.2.198. [DOI] [PubMed] [Google Scholar]
- 38.Rosen HJ, Kramer JH, Gorno-Tempini ML, Schuff N, Weiner M, Miller BL. Patterns of cerebral atrophy in primary progressive aphasia. Am J Geriatr Psychiatry. 2002;10:89–97. [PubMed] [Google Scholar]
- 39.Gorno-Tempini ML, Dronkers NF, Rankin KP, et al. Cognition and anatomy in three variants of primary progressive aphasia. Ann Neurol. 2004;55:335–346. doi: 10.1002/ana.10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neary D, Snowden JS, Mann DM. Classification and description of frontotemporal dementias. Ann N Y Acad Sci. 2000;920(51–52):46–51. doi: 10.1111/j.1749-6632.2000.tb06904.x. [DOI] [PubMed] [Google Scholar]
- 41.Kril JJ, Halliday GM. Clinicopathological staging of frontotemporal dementia severity: correlation with regional atrophy. Dement Geriatr Cogn Disord. 2004;17:311–315. doi: 10.1159/000077161. [DOI] [PubMed] [Google Scholar]
- 42.Miller BL, Gearhart R. Neuroimaging in the diagnosis of frontotemporal dementia. Dement Geriatr Cogn Disord. 1999;10(S1):71–74. doi: 10.1159/000051217. [DOI] [PubMed] [Google Scholar]
- 43.Kipps CM, Davies RR, Mitchell J, Kril JJ, Halliday GM, Hodges JR. Clinical significance of lobar atrophy in frontotemporal dementia: application of an MRI visual rating scale. Dement Geriatr Cogn Disord. 2007;23:334–342. doi: 10.1159/000100973. [DOI] [PubMed] [Google Scholar]
- 44.Kitagaki H, Mori E, Yamaji S, et al. Frontotemporal dementia and Alzheimer disease: evaluation of cortical atrophy with automated hemispheric surface display generated with MR images. Radiology. 1998;208:431–439. doi: 10.1148/radiology.208.2.9680572. [DOI] [PubMed] [Google Scholar]
- 45.Frisoni GB, Beltramello A, Geroldi C, Weiss C, Bianchetti A, Trabucchi M. Brain atrophy in frontotemporal dementia. J Neurol Neurosurg Psychiatry. 1996;61:157–165. doi: 10.1136/jnnp.61.2.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chan D, Fox NC, Scahill RI, et al. Patterns of temporal lobe atrophy in semantic dementia and Alzheimer’s disease. Ann Neurol. 2001;49:433–442. [PubMed] [Google Scholar]
- 47.Galton CJ, Gomez-Anson B, Antoun N, et al. Temporal lobe rating scale: application to Alzheimer’s disease and frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2001;70:165–173. doi: 10.1136/jnnp.70.2.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mummery CJ, Patterson K, Price CJ, Ashburner J, Frackowiak RS, Hodges J. A voxel-based morphometry study of semantic dementia: relationship between temporal lobe atrophy and semantic memory. Ann Neurol. 2000;47:36–45. [PubMed] [Google Scholar]
- 49.Whitwell JL, Josephs KA, Rossor MN, et al. Magnetic resonance imaging signatures of tissue pathology in frontotemporal dementia. Arch Neurol. 2005;62:1402–1408. doi: 10.1001/archneur.62.9.1402. [DOI] [PubMed] [Google Scholar]
- 50.Rabinovici GD, Seeley WW, Kim EJ, et al. Distinct MRI atrophy patterns in autopsy-proven Alzheimer’s disease and frontotemporal lobar degeneration. Am J Alzheimers Dis Other Demen. 2007;22:474–488. doi: 10.1177/1533317507308779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fukui T, Kertesz A. Volumetric study of lobar atrophy in Pick complex and Alzheimer’s disease. J Neurol Sci. 2000;174:111–121. doi: 10.1016/s0022-510x(00)00261-6. [DOI] [PubMed] [Google Scholar]
- 52.Barnes J, Whitwell JL, Frost C, Josephs KA, Rossor M, Fox NC. Measurements of the amygdala and hippocampus in pathologically confirmed Alzheimer disease and frontotemporal lobar degeneration. Arch Neurol. 2006;63:1434–1439. doi: 10.1001/archneur.63.10.1434. [DOI] [PubMed] [Google Scholar]
- 53.Du AT, Schuff N, Kramer JH, et al. Different regional patterns of cortical thinning in Alzheimer’s disease and frontotemporal dementia. Brain. 2007;130(Pt 4):1159–1166. doi: 10.1093/brain/awm016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Johnson JK, Head E, Kim R, Starr A, Cotman CW. Clinical and pathological evidence for a frontal variant of Alzheimer disease. Arch Neurol. 1999;56:1233–1239. doi: 10.1001/archneur.56.10.1233. [DOI] [PubMed] [Google Scholar]
- 55.Varma AR, Snowden JS, Lloyd JJ, Talbot PR, Mann DM, Neary D. Evaluation of the NINCDS-ADRDA criteria in the differentiation of Alzheimer’s disease and frontotemporal dementia. J Neurol Neurosurg Psychiatry. 1999;66:184–188. doi: 10.1136/jnnp.66.2.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Talbot PR, Lloyd JJ, Snowden JS, Neary D, Testa HJ. A clinical role for 99mTc-HMPAO SPECT in the investigation of dementia? J Neurol Neurosurg Psychiatry. 1998;64:306–313. doi: 10.1136/jnnp.64.3.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Foster NL, Heidebrink JL, Clark CM, et al. FDG-PET improves accuracy in distinguishing frontotemporal dementia and Alzheimer’s disease. Brain. 2007;130(Pt 10):2616–2635. doi: 10.1093/brain/awm177. [DOI] [PubMed] [Google Scholar]
- 58.Nestor PJ, Graham NL, Fryer TD, Williams GB, Patterson K, Hodges JR. Progressive non-fluent aphasia is associated with hypometabolism centred on the left anterior insula. Brain. 2003;126(Pt 11):2406–2418. doi: 10.1093/brain/awg240. [DOI] [PubMed] [Google Scholar]
- 59.Diehl-Schmid J, Grimmer T, Drzezga A, et al. Decline of cerebral glucose metabolism in frontotemporal dementia: a longitudinal 18F-FDG-PET-study. Neurobiol Aging. 2007;28:42–50. doi: 10.1016/j.neurobiolaging.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 60.Du AT, Jahng GH, Hayasaka S, et al. Hypoperfusion in frontotemporal dementia and Alzheimer disease by arterial spin labeling MRI. Neurology. 2006;67:1215–1220. doi: 10.1212/01.wnl.0000238163.71349.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jellinger KA. The enigma of vascular cognitive disorder and vascular dementia. Acta Neuropathol. 2007;113:349–388. doi: 10.1007/s00401-006-0185-2. [DOI] [PubMed] [Google Scholar]
- 62.Pohjasvaara T, Mantyla R, Ylikoski R, Kaste M, Erkinjuntti T. Comparison of different clinical criteria (DSM-III, ADDTC, ICD-10, NINDS-AIREN, DSM-IV) for the diagnosis of vascular dementia. National Institute of Neurological Disorders and Stroke—Association Internationale pour la Recherche et l’Enseignement en Neurosciences. Stroke. 2000;31:2952–2957. doi: 10.1161/01.str.31.12.2952. [DOI] [PubMed] [Google Scholar]
- 63.Hachinski VC, Illiff LD, Zilhka E, du Boulay GH, McAllister VL, Marchall J. Cerebral blood flow in dementia. Arch Neurol. 1975;32:632–637. doi: 10.1001/archneur.1975.00490510088009. [DOI] [PubMed] [Google Scholar]
- 64.Vermeer SE, Longstreth WT, Jr, Koudstaal PJ. Silent brain infarcts: a systematic review. Lancet Neurol. 2007;6:611–619. doi: 10.1016/S1474-4422(07)70170-9. [DOI] [PubMed] [Google Scholar]
- 65.Staekenborg SS, van der Flier WM, van Straaten EC, Lane R, Barkhof F, Scheltens P. Neurological signs in relation to type of cerebrovascular disease in vascular dementia. Stroke. 2008;39:317–322. doi: 10.1161/STROKEAHA.107.493353. [DOI] [PubMed] [Google Scholar]
- 66.Johnston SC, O’Meara ES, Manolio TA, et al. Cognitive impairment and decline are associated with carotid artery disease in patients without clinically evident cerebrovascular disease. Ann Intern Med. 2004;140:237–247. doi: 10.7326/0003-4819-140-4-200402170-00005. [DOI] [PubMed] [Google Scholar]
- 67.Berman L, Pietrzak RH, Mayes L. Neurocognitive changes after carotid revascularization: a review of the current literature. J Psychosom Res. 2007;63:599–612. doi: 10.1016/j.jpsychores.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 68.Mlekusch W, Mlekusch I, Haumer M, et al. Improvement of neurocognitive function after protected carotid artery stenting. Catheter Cardiovasc Interv. 2008;71:114–119. doi: 10.1002/ccd.21407. [DOI] [PubMed] [Google Scholar]
- 69.Turk AS, Chaudry I, Haughton VM, et al. Effect of carotid artery stenting on cognitive function in patients with carotid artery stenosis: preliminary results. AJNR Am J Neuroradiol. 2008;29:265–268. doi: 10.3174/ajnr.A0828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leys D, Henon H, Mackowiak-Cordoliani MA, Pasquier F. Poststroke dementia. Lancet Neurol. 2005;4:752–759. doi: 10.1016/S1474-4422(05)70221-0. [DOI] [PubMed] [Google Scholar]
- 71.Guermazi A, Miaux Y, Rovira-Canellas A, et al. Neuroradiological findings in vascular dementia. Neuroradiology. 2007;49:1–22. doi: 10.1007/s00234-006-0156-2. [DOI] [PubMed] [Google Scholar]
- 72.Pohjasvaara T, Mantyla R, Salonen O, et al. MRI correlates of dementia after first clinical ischemic stroke. J Neurol Sci. 2000;181(1–2):111–117. doi: 10.1016/s0022-510x(00)00437-8. [DOI] [PubMed] [Google Scholar]
- 73.Auchus AP, Chen CP, Sodagar SN, Thong M, Sng EC. Single stroke dementia: insights from 12 cases in Singapore. J Neurol Sci. 2002;203–204:85–89. doi: 10.1016/s0022-510x(02)00272-1. [DOI] [PubMed] [Google Scholar]
- 74.Roman GC, Erkinjuntti T, Wallin A, Pantoni L, Chui HC. Subcortical ischaemic vascular dementia. Lancet Neurol. 2002;1:426–436. doi: 10.1016/s1474-4422(02)00190-4. [DOI] [PubMed] [Google Scholar]
- 75.Kumral E, Evyapan D, Balkir K. Acute caudate vascular lesions. Stroke. 1999;30:100–108. doi: 10.1161/01.str.30.1.100. [DOI] [PubMed] [Google Scholar]
- 76.Mizuta H, Motomura N. Memory dysfunction in caudate infarction caused by Heubner’s recurring artery occlusion. Brain Cogn. 2006;61:133–138. doi: 10.1016/j.bandc.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 77.Carrera E, Bogousslavsky J. The thalamus and behavior: effects of anatomically distinct strokes. Neurology. 2006;66:1817–1823. doi: 10.1212/01.wnl.0000219679.95223.4c. [DOI] [PubMed] [Google Scholar]
- 78.Shim YS, Kim JS, Shon YM, Chung YA, Ahn KJ, Yang DW. A serial study of regional cerebral blood flow deficits in patients with left anterior thalamic infarction: anatomical and neuropsychological correlates. J Neurol Sci. 2008;266(1–2):84–91. doi: 10.1016/j.jns.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 79.Hachinski VC, Potter P, Merskey H. Leuko-araiosis. Arch Neurol. 1987;44:21–23. doi: 10.1001/archneur.1987.00520130013009. [DOI] [PubMed] [Google Scholar]
- 80.Roman GC. Senile dementia of the Binswanger type. A vascular form of dementia in the elderly. JAMA. 1987;258:1782–1788. doi: 10.1001/jama.1987.03400130096040. [DOI] [PubMed] [Google Scholar]
- 81.Boone KB, Miller BL, Lesser IM, et al. Neuropsychological correlates of white-matter lesions in healthy elderly subjects. A threshold effect. Arch Neurol. 1992;49:549–554. doi: 10.1001/archneur.1992.00530290141024. [DOI] [PubMed] [Google Scholar]
- 82.van Straaten EC, Scheltens P, Knol DL, et al. Operational definitions for the NINDS-AIREN criteria for vascular dementia: an interobserver study. Stroke. 2003;34:1907–1912. doi: 10.1161/01.STR.0000083050.44441.10. [DOI] [PubMed] [Google Scholar]
- 83.Kramer JH, Mungas D, Reed BR, et al. Forgetting in dementia with and without subcortical lacunes. Clin Neuropsychol. 2004;18:32–40. doi: 10.1080/13854040490507136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mungas D, Harvey D, Reed BR, et al. Longitudinal volumetric MRI change and rate of cognitive decline. Neurology. 2005;65:565–571. doi: 10.1212/01.wnl.0000172913.88973.0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Au R, Massaro JM, Wolf PA, et al. Association of white matter hyperintensity volume with decreased cognitive functioning: the Framingham Heart Study. Arch Neurol. 2006;63:246–250. doi: 10.1001/archneur.63.2.246. [DOI] [PubMed] [Google Scholar]
- 86.Carey CL, Woods SP, Damon J, et al. Discriminant validity and neuroanatomical correlates of rule monitoring in frontotemporal dementia and Alzheimer’s disease. Neuropsychologia. 2008;46:1081–1087. doi: 10.1016/j.neuropsychologia.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 87.Debette S, Bombois S, Bruandet A, et al. Subcortical hyperintensities are associated with cognitive decline in patients with mild cognitive impairment. Stroke. 2007;38:2924–2930. doi: 10.1161/STROKEAHA.107.488403. [DOI] [PubMed] [Google Scholar]
- 88.Yong SW, Bang OY, Lee PH, Li WY. Internal and cortical border-zone infarction: clinical and diffusion-weighted imaging features. Stroke. 2006;37:841–846. doi: 10.1161/01.STR.0000202590.75972.39. [DOI] [PubMed] [Google Scholar]
- 89.Kuker W. Cerebral vasculitis: imaging signs revisited. Neuroradiology. 2007;49:471–479. doi: 10.1007/s00234-007-0223-3. [DOI] [PubMed] [Google Scholar]
- 90.Chabriat H, Levy C, Taillia H, et al. Patterns of MRI lesions in CADASIL. Neurology. 1998;51:452–457. doi: 10.1212/wnl.51.2.452. [DOI] [PubMed] [Google Scholar]
- 91.Josephson SA, Papanastassiou AM, Berger MS, et al. The diagnostic utility of brain biopsy procedures in patients with rapidly deteriorating neurological conditions or dementia. J Neurosurg. 2007;106:72–75. doi: 10.3171/jns.2007.106.1.72. [DOI] [PubMed] [Google Scholar]
- 92.Liem MK, van der Grond J, Haan J, et al. Lacunar infarcts are the main correlate with cognitive dysfunction in CADASIL. Stroke. 2007;38:923–928. doi: 10.1161/01.STR.0000257968.24015.bf. [DOI] [PubMed] [Google Scholar]
- 93.Viswanathan A, Gschwendtner A, Guichard JP, et al. Lacunar lesions are independently associated with disability and cognitive impairment in CADASIL. Neurology. 2007;69:172–179. doi: 10.1212/01.wnl.0000265221.05610.70. [DOI] [PubMed] [Google Scholar]
- 94.Cordonnier C, van der Flier WM, Sluimer JD, Leys D, Barkhof F, Scheltens P. Prevalence and severity of micro-bleeds in a memory clinic setting. Neurology. 2006;66:1356–1360. doi: 10.1212/01.wnl.0000210535.20297.ae. [DOI] [PubMed] [Google Scholar]
- 95.Koennecke HC. Cerebral microbleeds on MRI: prevalence, associations, and potential clinical implications. Neurology. 2006;66:165–171. doi: 10.1212/01.wnl.0000194266.55694.1e. [DOI] [PubMed] [Google Scholar]
- 96.Lee SH, Kim SM, Kim N, Yoon BW, Roh JK. Cortico-subcortical distribution of microbleeds is different between hypertension and cerebral amyloid angiopathy. J Neurol Sci. 2007;258(1–2):111–114. doi: 10.1016/j.jns.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 97.Hachinski V, Iadecola C, Petersen RC, et al. National Institute of Neurological Disorders and Stroke–Canadian Stroke Network vascular cognitive impairment harmonization standards. Stroke. 2006;37:2220–2241. doi: 10.1161/01.STR.0000237236.88823.47. [DOI] [PubMed] [Google Scholar]
- 98.Tzourio C, Anderson C, Chapman N, et al. Effects of blood pressure lowering with perindopril and indapamide therapy on dementia and cognitive decline in patients with cerebrovascular disease. Arch Intern Med. 2003;163:1069–1075. doi: 10.1001/archinte.163.9.1069. [DOI] [PubMed] [Google Scholar]
- 99.Shlyakhto E. Observational Study on Cognitive Function and Systolic Blood Pressure Reduction (OSCAR): preliminary analysis of 6-month data from >10,000 patients and review of the literature. Curr Med Res Opin. 2007;23(S5):S13–S18. doi: 10.1185/030079907X260719. [DOI] [PubMed] [Google Scholar]
- 100.Weisman D, McKeith I. Dementia with Lewy bodies. Semin Neurol. 2007;27:42–47. doi: 10.1055/s-2006-956754. [DOI] [PubMed] [Google Scholar]
- 101.Burton EJ, Karas G, Paling SM, et al. Patterns of cerebral atrophy in dementia with Lewy bodies using voxel-based morphometry. Neuroimage. 2002;17:618–630. [PubMed] [Google Scholar]
- 102.Brenneis C, Wenning GK, Egger KE, et al. Basal forebrain atrophy is a distinctive pattern in dementia with Lewy bodies. Neuroreport. 2004;15:1711–1714. doi: 10.1097/01.wnr.0000136736.73895.03. [DOI] [PubMed] [Google Scholar]
- 103.Whitwell JL, Weigand SD, Shiung MM, et al. Focal atrophy in dementia with Lewy bodies on MRI: a distinct pattern from Alzheimer’s disease. Brain. 2007;130(Pt 3):708–719. doi: 10.1093/brain/awl388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cousins DA, Burton EJ, Burn D, Gholkar A, McKeith IG, O’Brien JT. Atrophy of the putamen in dementia with Lewy bodies but not Alzheimer’s disease: an MRI study. Neurology. 2003;61:1191–1195. doi: 10.1212/01.wnl.0000091889.20347.30. [DOI] [PubMed] [Google Scholar]
- 105.McKeith I, O’Brien J, Walker Z, et al. Sensitivity and specificity of dopamine transporter imaging with 123I-FP-CIT SPECT in dementia with Lewy bodies: a phase III, multicentre study. Lancet Neurol. 2007;6:305–313. doi: 10.1016/S1474-4422(07)70057-1. [DOI] [PubMed] [Google Scholar]
- 106.Berg D. Disturbance of iron metabolism as a contributing factor to SN hyperechogenicity in Parkinson’s disease: implications for idiopathic and monogenetic forms. Neurochem Res. 2007;32:1646–1654. doi: 10.1007/s11064-007-9346-5. [DOI] [PubMed] [Google Scholar]
- 107.Walter U, Dressler D, Probst T, et al. Transcranial brain sonography findings in discriminating between parkinsonism and idiopathic Parkinson disease. Arch Neurol. 2007;64:1635–1640. doi: 10.1001/archneur.64.11.1635. [DOI] [PubMed] [Google Scholar]
- 108.Litvan I, Agid Y, Goetz C, et al. Accuracy of the clinical diagnosis of corticobasal degeneration: a clinicopathologic study. Neurology. 1997;48:119–125. doi: 10.1212/wnl.48.1.119. [DOI] [PubMed] [Google Scholar]
- 109.Soliveri P, Monza D, Paridi D, et al. Cognitive and magnetic resonance imaging aspects of corticobasal degeneration and progressive supranuclear palsy. Neurology. 1999;53:502–507. doi: 10.1212/wnl.53.3.502. [DOI] [PubMed] [Google Scholar]
- 110.Boxer AL, Geschwind MD, Belfor N, et al. Patterns of brain atrophy that differentiate corticobasal degeneration syndrome from progressive supranuclear palsy. Arch Neurol. 2006;63:81–86. doi: 10.1001/archneur.63.1.81. [DOI] [PubMed] [Google Scholar]
- 111.Litvan I, Agid Y, Jankovic J, et al. Accuracy of clinical criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) Neurology. 1996;46:922–930. doi: 10.1212/wnl.46.4.922. [DOI] [PubMed] [Google Scholar]
- 112.Warmuth-Metz M, Naumann M, Csoti I, Solymosi L. Measurement of the midbrain diameter on routine magnetic resonance imaging: a simple and accurate method of differentiating between Parkinson disease and progressive supranuclear palsy. Arch Neurol. 2001;58:1076–1079. doi: 10.1001/archneur.58.7.1076. [DOI] [PubMed] [Google Scholar]
- 113.Oba H, Yagishita A, Terada H, et al. New and reliable MRI diagnosis for progressive supranuclear palsy. Neurology. 2005;64:2050–2055. doi: 10.1212/01.WNL.0000165960.04422.D0. [DOI] [PubMed] [Google Scholar]
- 114.Quattrone A, Nicoletti G, Messina D, et al. MR imaging index for differentiation of progressive supranuclear palsy from Parkinson disease and the Parkinson variant of multiple system atrophy. Radiology. 2008;246:214–221. doi: 10.1148/radiol.2453061703. [DOI] [PubMed] [Google Scholar]
- 115.Schwarz J, Weis S, Kraft E, et al. Signal changes on MRI and increases in reactive microgliosis, astrogliosis, and iron in the putamen of two patients with multiple system atrophy. J Neurol Neurosurg Psychiatry. 1996;60:98–101. doi: 10.1136/jnnp.60.1.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.von Lewinski F, Werner C, Jorn T, Mohr A, Sixel-Doring F, Trenkwalder C. T2*-weighted MRI in diagnosis of multiple system atrophy. A practical approach for clinicians. J Neurol. 2007;254:1184–1188. doi: 10.1007/s00415-006-0496-1. [DOI] [PubMed] [Google Scholar]
- 117.Savoiardo M. Differential diagnosis of Parkinson’s disease and atypical parkinsonian disorders by magnetic resonance imaging. Neurol Sci. 2003;24(S1):S35–S37. doi: 10.1007/s100720300036. [DOI] [PubMed] [Google Scholar]
- 118.Burk K, Daum I, Rub U. Cognitive function in multiple system atrophy of the cerebellar type. Mov Disord. 2006;21:772–776. doi: 10.1002/mds.20802. [DOI] [PubMed] [Google Scholar]
- 119.Abe K, Hikita T, Yokoe M, Mihara M, Sakoda S. The “cross” signs in patients with multiple system atrophy: a quantitative study. J Neuroimaging. 2006;16:73–77. doi: 10.1177/1051228405279988. [DOI] [PubMed] [Google Scholar]
- 120.Burk K, Buhring U, Schulz JB, Zuhlke C, Hellenbroich Y, Dichgans J. Clinical and magnetic resonance imaging characteristics of sporadic cerebellar ataxia. Arch Neurol. 2005;62:981–985. doi: 10.1001/archneur.62.6.981. [DOI] [PubMed] [Google Scholar]
- 121.Mandelli ML, De Simone T, Minati L, et al. Diffusion tensor imaging of spinocerebellar ataxias types 1 and 2. AJNR Am J Neuroradiol. 2007;28:1996–2000. doi: 10.3174/ajnr.A0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Peterson K, Rosenblum M, Kotanides H, Posner J. Paraneoplastic cerebellar degeneration. I A clinical analysis of 55 anti-Yo antibody-positive patients. Neurology. 1992;42:1931–1937. doi: 10.1212/wnl.42.10.1931. [DOI] [PubMed] [Google Scholar]
- 123.Chang CC, Eggers SD, Johnson JK, Haman A, Miller BL, Geschwind MD. Anti-GAD antibody cerebellar ataxia mimicking Creutzfeldt-Jakob disease. Clin Neurol Neurosurg. 2007;109:54–57. doi: 10.1016/j.clineuro.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 124.Burk K. Cognition in hereditary ataxia. Cerebellum. 2007;6:280–286. doi: 10.1080/14734220601115924. [DOI] [PubMed] [Google Scholar]
- 125.Klockgether T, Skalej M, Wedekind D, et al. Autosomal dominant cerebellar ataxia type I. MRI-based volumetry of posterior fossa structures and basal ganglia in spinocerebellar ataxia types 1, 2 and 3. Brain. 1998;121(Pt 9):1687–1693. doi: 10.1093/brain/121.9.1687. [DOI] [PubMed] [Google Scholar]
- 126.Brenneis C, Bosch SM, Schocke M, Wenning GK, Poewe W. Atrophy pattern in SCA2 determined by voxel-based morphometry. Neuroreport. 2003;14:1799–1802. doi: 10.1097/00001756-200310060-00008. [DOI] [PubMed] [Google Scholar]
- 127.Mariotti C, Alpini D, Fancellu R, et al. Spinocerebellar ataxia type 17 (SCA17): oculomotor phenotype and clinical characterization of 15 Italian patients. J Neurol. 2007;254:1538–1546. doi: 10.1007/s00415-007-0579-7. [DOI] [PubMed] [Google Scholar]
- 128.Lin IS, Wu RM, Lee-Chen GJ, Shan DE, Gwinn-Hardy K. The SCA17 phenotype can include features of MSA-C, PSP and cognitive impairment. Parkinsonism Relat Disord. 2007;13:246–249. doi: 10.1016/j.parkreldis.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 129.Loy CT, Sweeney MG, Davis MB, et al. Spinocerebellar ataxia type 17: extension of phenotype with putaminal rim hyperintensity on magnetic resonance imaging. Mov Disord. 2005;20:1521–1523. doi: 10.1002/mds.20529. [DOI] [PubMed] [Google Scholar]
- 130.Young GS, Geschwind MD, Fischbein NJ, et al. Diffusion-weighted and fluid-attenuated inversion recovery imaging in Creutzfeldt-Jakob disease: high sensitivity and specificity for diagnosis. AJNR Am J Neuroradiol. 2005;26:1551–1562. [PMC free article] [PubMed] [Google Scholar]
- 131.Shiga Y, Miyazawa K, Sato S, et al. Diffusion-weighted MRI abnormalities as an early diagnostic marker for Creutzfeldt-Jakob disease. Neurology. 2004;63:443–449. doi: 10.1212/01.wnl.0000134555.59460.5d. [DOI] [PubMed] [Google Scholar]
- 132.Collie DA, Sellar RJ, Zeidler M, Colchester CF, Knight R, Will RG. MRI of Creutzfeldt-Jakob disease: imaging features and recommended MRI protocol. Clin Radiol. 2001;56:726–739. doi: 10.1053/crad.2001.0771. [DOI] [PubMed] [Google Scholar]
- 133.Zeidler M, Sellar RJ, Collie DA, et al. The pulvinar sign on magnetic resonance imaging in variant Creutzfeldt-Jakob disease. Lancet. 2000;355:1412–1418. doi: 10.1016/s0140-6736(00)02140-1. [DOI] [PubMed] [Google Scholar]
- 134.Lin YR, Young GS, Chen NK, Dillon WP, Wong S. Creutzfeldt-Jakob disease involvement of rolandic cortex: a quantitative apparent diffusion coefficient evaluation. AJNR Am J Neuroradiol. 2006;27:1755–1759. [PMC free article] [PubMed] [Google Scholar]
- 135.Boxer AL, Rabinovici GD, Kepe V, et al. Amyloid imaging in distinguishing atypical prion disease from Alzheimer disease. Neurology. 2007;69:283–290. doi: 10.1212/01.wnl.0000265815.38958.b6. [DOI] [PubMed] [Google Scholar]
- 136.Chu K, Kang DW, Kim HJ, Lee YS, Park SH. Diffusion-weighted imaging abnormalities in Wernicke’s encephalopathy: reversible cytotoxic edema? Arch Neurol. 2002;59:123–127. doi: 10.1001/archneur.59.1.123. [DOI] [PubMed] [Google Scholar]
- 137.Sener RN. Diffusion MRI findings in Wilson’s disease. Comput Med Imaging Graph. 2003;27:17–21. doi: 10.1016/s0895-6111(02)00047-2. [DOI] [PubMed] [Google Scholar]
- 138.Singhal AB, Newstein MC, Budzik R, et al. Diffusion-weighted magnetic resonance imaging abnormalities in Bartonella encephalopathy. J Neuroimaging. 2003;13:79–82. [PubMed] [Google Scholar]
- 139.Josephs KA, Holton JL, Rossor MN, et al. Neurofilament inclusion body disease: a new proteinopathy? Brain. 2003;126(Pt 10):2291–2303. doi: 10.1093/brain/awg231. [DOI] [PubMed] [Google Scholar]
- 140.Mihara M, Sugase S, Konaka K, et al. The “pulvinar sign” in a case of paraneoplastic limbic encephalitis associated with non-Hodgkin’s lymphoma. J Neurol Neurosurg Psychiatry. 2005;76:882–884. doi: 10.1136/jnnp.2004.049783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chu K, Kang DW, Kim JY, Chang KH, Lee SK. Diffusion-weighted magnetic resonance imaging in nonconvulsive status epilepticus. Arch Neurol. 2001;58:993–998. doi: 10.1001/archneur.58.6.993. [DOI] [PubMed] [Google Scholar]
- 142.Hufnagel A, Weber J, Marks S, et al. Brain diffusion after single seizures. Epilepsia. 2003;44:54–63. doi: 10.1046/j.1528-1157.2003.07802.x. [DOI] [PubMed] [Google Scholar]
- 143.Vernino S, Geschwind MD, Boeve B. Autoimmune encephalopathies. Neurologist. 2007;13:140–147. doi: 10.1097/01.nrl.0000259483.70041.55. [DOI] [PubMed] [Google Scholar]
- 144.Vincent A, Buckley C, Schott JM, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain. 2004;127(Pt 3):701–712. doi: 10.1093/brain/awh077. [DOI] [PubMed] [Google Scholar]
- 145.Geschwind MD, Yoon G, Goldman J. Adult onset genetic disorders involving the frontal lobes. In: Miller BL, editor. The Human Frontal Lobes: Functions and Disorders. New York, NY: The Guilford Press; 2007. pp. 552–575. [Google Scholar]
- 146.Geschwind MD, Haman A, Miller BL. Rapidly progressive dementia. Neurol Clin. 2007;25:783–807. doi: 10.1016/j.ncl.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Burdette JH, Elster AD, Ricci PE. Acute cerebral infarction: quantification of spin-density and T2 shine-through phenomena on diffusion-weighted MR images. Radiology. 1999;212:333–339. doi: 10.1148/radiology.212.2.r99au36333. [DOI] [PubMed] [Google Scholar]
- 148.Minati L, Grisoli M, Bruzzone MG. MR spectroscopy, functional MRI, and diffusion-tensor imaging in the aging brain: a conceptual review. J Geriatr Psychiatry Neurol. 2007;20:3–21. doi: 10.1177/0891988706297089. [DOI] [PubMed] [Google Scholar]
- 149.Paviour DC, Thornton JS, Lees AJ, Jager HR. Diffusion-weighted magnetic resonance imaging differentiates Parkinsonian variant of multiple-system atrophy from progressive supranuclear palsy. Mov Disord. 2007;22:68–74. doi: 10.1002/mds.21204. [DOI] [PubMed] [Google Scholar]
- 150.Vitali P, Henry RG, Chung S, et al. ADC measurements at the gyral level and of deep nuclei in sporadic Jakob-Creutzfeldt disease. LP03:18:6. Presented at 32nd Congress of the European Society of Neuroradiology; Genoa, Italy Neuroradiology. 2007. p. S149. [Google Scholar]
- 151.Kantarci K, Petersen RC, Boeve BF, et al. DWI predicts future progression to Alzheimer disease in amnestic mild cognitive impairment. Neurology. 2005;64:902–904. doi: 10.1212/01.WNL.0000153076.46126.E9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Stankiewicz J, Panter SS, Neema M, Arora A, Batt CE, Bakshi R. Iron in chronic brain disorders: imaging and neurotherapeutic implications. Neurotherapeutics. 2007;4:371–386. doi: 10.1016/j.nurt.2007.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Schenck JF, Zimmerman EA, Li Z, et al. High-field magnetic resonance imaging of brain iron in Alzheimer disease. Top Magn Reson Imaging. 2006;17:41–50. doi: 10.1097/01.rmr.0000245455.59912.40. [DOI] [PubMed] [Google Scholar]
- 154.Thomas B, Somasundaram S, Thamburaj K, et al. Clinical applications of susceptibility weighted MR imaging of the brain—a pictorial review. Neuroradiology. 2008;50:105–116. doi: 10.1007/s00234-007-0316-z. [DOI] [PubMed] [Google Scholar]
- 155.Hayasaka S, Du AT, Duarte A, et al. A non-parametric approach for co-analysis of multi-modal brain imaging data: application to Alzheimer’s disease. Neuroimage. 2006;30:768–779. doi: 10.1016/j.neuroimage.2005.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Logothetis NK, Pfeuffer J. On the nature of the BOLD fMRI contrast mechanism. Magn Reson Imaging. 2004;22:1517–1531. doi: 10.1016/j.mri.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 157.Sperling R. Functional MRI studies of associative encoding in normal aging, mild cognitive impairment, and Alzheimer’s disease. Ann N Y Acad Sci. 2007;1097:146–155. doi: 10.1196/annals.1379.009. [DOI] [PubMed] [Google Scholar]
- 158.Greicius MD, Srivastava G, Reiss AL, Menon V. Default-mode network activity distinguishes Alzheimer’s disease from healthy aging: evidence from functional MRI. Proc Natl Acad Sci U S A. 2004;101:4637–4642. doi: 10.1073/pnas.0308627101. [DOI] [PMC free article] [PubMed] [Google Scholar]