

Abstract
Chondrocyte maturation during endochondral bone formation is regulated by a number of signals that either promote or inhibit maturation. Among these, two well-studied signaling pathways play crucial roles in modulating chondrocyte maturation: transforming growth factor-beta (TGF-β)/Smad3 signaling slows the rate of chondrocyte maturation, while Wingless/INT-1-related (Wnt)/β-catenin signaling enhances the rate of chondrocyte maturation. Axin1 and Axin2 are functionally equivalent and have been shown to inhibit Wnt/β-catenin signaling and stimulate TGF-β signaling. Here we show that while Wnt3a stimulates Axin2 in a negative feedback loop, TGF-β suppresses the expression of both Axin1 and Axin2 and stimulates β-catenin signaling. In Axin2 −/− chondrocytes, TGF-β treatment results in a sustained increase in β-catenin levels compared to wild-type chondrocytes. In contrast, overexpression of Axin enhanced TGF-β signaling while overexpression of β-catenin inhibited the ability of TGF-β to induce Smad3-sensitive reporters. Finally, the suppression of the Axins is Smad3-dependent since the effect is absent in Smad3 −/− chondrocytes. Altogether these findings show that the Axins act to integrate signals between the Wnt/β-catenin and TGF-β/Smad pathways. Since the suppression Axin1 and Axin2 expression by TGF-β reduces TGF-β signaling and enhances Wnt/β-catenin signaling, the overall effect is a shift from TGF-β toward Wnt/β-catenin signaling and an acceleration of chondrocyte maturation.
Keywords: axin, axin1, axin2, TGF-β, Wnt, β-catenin, chondrogenesis
INTRODUCTION
Chondrocyte development is regulated by a number of signaling pathways that either promote or inhibit maturation. While transforming growth factor-beta (TGF-β) and parathyroid hormone-related peptide (PTHrP) slow the rate of chondrocyte maturation, bone morphogenic proteins (BMPs), thyroid hormone, retinoic acid, and Wingless/INT-1-related (Wnt) proteins enhance the rate of chondrocyte maturation.1–6 Although these individual pathways have been extensively studied, less work has been completed to define signaling interactions that may be important for cartilage maturation. Understanding these interactions and their molecular targets is essential since chondrocytes in the growth plate simultaneously integrate these various complex signals. Recently, the TGF-β/BMP-related Smad pathways and the Wnt β-catenin pathways have emerged as important signals related to growth and development. Evidence suggests that the TGF-β/BMP Smad pathways and the Wnt/β-catenin pathways control both the proliferation and differentiation of chondrocytes in the growth plate and thus it is likely that these signals are highly interrelated.2,7–9
While Wnt/β-catenin signaling inhibits chondrogenesis in the developing skeleton,10,11 β-catenin signaling is reestablished in the growth plate where numerous Wnts are expressed once cartilage has formed and the skeletal elements have developed.9,12–14 Cell culture and chick limb bud models suggest that Wnt/β-catenin signaling stimulates chondrocyte maturation. Overexpression of Wnts 4, 8, and 9, β-catenin, and LEF-1 induce colX, alkaline phosphatase, and other genes associated with chondrocyte hypertrophy.9,14,15 Inhibition of β-catenin signaling by overexpression of Frzb-1, a secreted decoy receptor, dominant negative Wnt receptors, or dominant negative β-catenin results in delayed maturation.9,13,14 Thus, cell culture and chick limb bud models show that Wnt/β-catenin signaling is active during endochondral bone formation and suggest that β-catenin stimulates chondrocyte maturation.
Wnt/β-catenin has also been shown to regulate proliferation in numerous cell types and recent findings from our laboratory showed that the induction of proliferation by TGF-β was dependent upon β-catenin.7 In the absence of Wnt signaling molecules, β-catenin is part of a cytosolic protein complex that includes Axin, adenomatous polyposis coli (APC), and glycogen synthase kinase–3β (GSK).16,17 In this complex, β-catenin is phosphorylated by GSK,18 which results in ubiquitination by E3 ubiquitin ligases with subsequent degradation in the 26S proteasome.17,18 TGF-β signaling results in the phosphorylation of Smad3 and our prior work showed that Smad3 interacts with the E3 ligase, β-TCRP and prevents the ubiquitination of β-catenin.7 This indirect activation of β-catenin is necessary for the stimulation of proliferation by TGF-β in isolated murine sternal chondroctyes.7
Axin is another potential target for TGF-β and β-catenin interactions. Axin is a scaffold for the protein complex, described above, involved in β-catenin catabolism.17,19 It has interaction sites for numerous proteins involved in Wnt signal transduction, including β-catenin, GSK, casein kinase I, APC, Dishevelled (Dvl), and the LRP receptors.17,19 During Wnt signaling Axin is recruited to the LRP receptor complex where an interaction with Dv1 results in Axin degradation and release of active β-catenin.17 β-catenin subsequently translocates to the nucleus where it associates with TCF/LEF (T Cell Factor/Lymphoid Enhancer Factor) transcription factors, binds to DNA, and regulates gene transcription.17
In addition to its role in β-catenin signaling, Axin also has an important function in TGF-β/Smad signaling. The classic TGF-β–mediated signaling pathway involves Smad activation.20 Smads are a family of intracellular proteins that comprise three classes of signaling molecules: receptor-associated Smads (2 and 3 for TGF-β; 1, 5, and 8 for BMP signaling), the cofactor Smad4, and the inhibitory Smads (6 and 7).20,21 Receptor-associated Smads bind to type I receptors and upon ligand binding and activation, are phosphorylated and released into the cytoplasm. Recent data demonstrate that Axin1/Axin2 binds to Smad3 at the type I TGF receptor and is essential for its phosphorylation.22 The activated receptor-associated Smads then form a heterodimer with Smad4, translocate to the nucleus, and influence gene transcription.20,21 An additional mechanism through which Axin facilitates TGF-β signaling is by acting as a scaffold for the interaction between the E3 ubiquitin ligase, Arkadia and inhibitory Smad7.23 Smad7 degradation is accelerated in the presence of Axin, with a subsequent increase in TGF-β/Smad signaling.23
Axin has two functionally identical isoforms, Axin1 and Axin2; knockin of Axin2 into the deleted Axin1 gene rescues Axin1−/− mice.24 In vitro studies reveal that both Axin1 and Axin2 overexpression reduce β-catenin levels and signaling.16,25 However, Axin1 and Axin2 are differentially expressed and their gene deletions have different phenotypes. Axin1−/− mice die at embryonic day 9.5 (E9.5) with forebrain truncation, neural tube defects, and axis duplications.24 In contrast, Axin2−/− mice complete embryonic development but have craniofacial defects and premature closure of the cranial sutures due to increased β-catenin signaling.26 The focus of this article is to determine a potential role for the Axins in the coordination of TGF-β and Wnt/β-catenin signaling in cartilage.
METHODS
Chondrocyte Isolation
Primary chondrocytes were isolated from the sterna and ribs of 3-day-old mice as previously described.2 Tissues were placed in DMEM containing 2 mg/mL pronase for 45 min at 37°C on a 150 rpm shaking platform. Following a wash in HBSS, sterna and ribs were further digested in DMEM with 3mg/mL collagenase D (Sigma, St. Louis, MO) at 37°C and 5% CO2 for 1.5 h. Following a wash in HBSS, tissues were transferred into a Petri dish containing DMEM with 3 mg/mL collagenase D and incubated at 37°C and 5% CO2 in a cell incubator for 6 h. Isolated cells were then resuspended in 5 mL DMEM containing 10% NuSerum IV (Collaborative Biomedical Products, Bedford, MA) and supplemented with 50 U penicillin and streptomycin and 2 mM of L-glutamine. Cells were passed through a cell strainer, centrifuged at 2000 rpm for 3 min at 4°C, and after several washes, cells were plated, in 6-well plates at 5 × 105 cells per well (for protein isolation), or in 12-well plates at 5 × 104 cells per well (for RNA isolation). After 24 h in culture, the culture media were supplemented with 50 ng/mL of ascorbic acid, and, for treatment experiments, human TGF-β1 (Calbiochem, Darmstadt, Germany), Wnt3a (R&D Systems, Inc., Minneapolis, MN), or BMP-2 (Peprotech, Inc., Rockyhill, NJ) was added.
RNA Extraction and Quantitative Reverse-Transcriptase PCR
Total RNA was extracted from primary chondrocyte cultures using the Trizol protocol (Invitrogen, Carlsbad, CA) following the manufacturer’s recommendations. One microgram of total RNA was reverse-transcribed using the iscript cDNA synthesis kit (Bio-Rad, Hercules, CA) following the manufacturer’s recommended protocol. Two microliters of reverse-transcribed cDNA were used for quantitative PCR. cDNA levels were measured in real-time using the fluorescent dye SYBR Green I (SYBR Green PCR Master Mix, Applied Biosystems, Foster City, CA) using specific primers designed for mouse Axin1 (Forward: 5′-ACG GTA CAA CGA AG CAG AGA GCT -3′; Reverse: 5′-CGG ATC TCC TTT GGC ATT CGG TAA -3′), Axin2 (Forward: 5′-GAG TAG CGC CGT GTT AGT GAC T -3′; Reverse: 5′-CCA GGA AAG TCC GGA AGA GGT ATG -3′), and β-actin (Forward: 5′-TGT TAC CAA CTG GGA CGA CA -3′; Reverse: 5′-CTG GGT CAT CTT TTC ACG GT -3′). The PCR reaction used the RotorGene real-time DNA amplification system (Corbett Research, Sydney, Australia) and the following protocol was used: a 95°C denaturation step for 10 min followed by 45 cycles with denaturation for 30 sec at 95°C, annealing for 30 sec at 55°C, and extension for 30 sec at 72°C. Detection of the fluorescent product occurred after each extension period. PCR products were subjected to a melting curve analysis and the data were analyzed and quantified with the RotorGene analysis software. Gene expression was normalized to β-actin.
Western Blotting
Chondrocytes were treated with TGF-β, Wnt3a, or BMP-2, then at different time points, were washed with cold phosphate-buffered saline and lysed on ice using Golden Lysis Buffer supplemented with protease inhibitor cocktail tablets. The protein concentration of the soluble material was measured using Coomassie Plus Protein Assay kit (Pierce, Rockford, IL). Twenty micrograms of extracts were assayed by SDS-PAGE. After transfer to a nitrocellulose membrane, blots were probed with the following antibodies: mouse anti-total-β-catenin (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) at a concentration of 1:1000, rabbit anti-Axin (Zymed Laboratories, South San Francisco, CA) at a concentration of 1:250, and mouse anti-β-actin (Sigma) at a concentration of 1:5000. Horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse polyclonal antibodies (Bio-Rad Laboratories) were used as secondary antibodies. The immune complexes were detected using Pico or Femto chemiluminescent agents (Pierce).
Transfection and Luciferase Assay
For transfection, primary sternal chondrocytes or chondrocyte cell lines (TMC-23 or C5.18) were plated at a density of 105 cells/well in 6-well plates. TMC-23 and C5.18 are well-described murine chondrocyte and rat chondrocyte cell lines, respectively.27,28 Cultures were transfected 12 h after plating with the TGF-β responsive reporters, p3TP-Luc (500 ng; a gift from Dr. Joan Massague) and 4×SBE or the Topflash reporter of Wnt/β-catenin signaling (500 ng; a gift from Dr. Jennifer Westendorf). In some experiments, cells were cotransfected with S33Y β-catenin (a gift from Dr. Kenneth Kinzler), Axin1 (a gift from Dr. Wei Hsu), or Smad3 (a gift from Dr. Rik Derynck) plasmids. Two hours after transfection, cultures were placed in normal culture media for 24 h, followed by treatment with BMP-2 (50 ng/mL), Wnt3a (100 ng/mL), or TGF-β (5 ng/mL) for the next 24 h. Cells were lysed using passive lysis buffer (Promega, Madison, WI) and luciferase activity in the cell lysate measured using the dual Luciferase assay system (Promega) with an Optocomp luminometer (MGM Instruments, Hamden, CT). Transfection efficiency was determined by cotransfection with pRL vector (Promega) and determining the Renilla uniformis luciferase activity. In all experiments, control vectors were used to keep the total amount of transfected DNA identical.
Statistical Analysis
Differences between various treatments or time points were compared using a two-way analysis of variance with Tukey–HSD post hoc analysis. Statistical significance was considered present when P < 0.05.
RESULTS
Wnt3a Induces Axin2 Gene and Protein Expression in Chondrocytes
Previous findings indicate that Wnt-signaling induces Axin2 expression.29,30 To determine if this regulation occurs in chondrocytes, we studied Axin2 mRNA and protein expression in Wnt3a-treated primary murine chondrocyte cultures. Axin2 gene expression was rapidly induced by Wnt3a treatment, with a 15-fold induction occurring at 6 h following treatment. Axin2 levels gradually decreased, but remained elevated at 24 h where a sevenfold induction was observed (Fig. 1A). To determine if the induction in gene expression was associated with increased protein levels, primary chondrocytes from Axin2LacZ/+ mice were cultured and treated with 100 ng/mL of Wnt3a and β-galactosidase activity was measured as a reporter of Axin2 protein expression at 0, 12, 24, 36, 48, and 72 h following treatment induction. Relative to baseline activity at time zero, β-galactosidase activity following Wnt3a treatment increased nearly eightfold after 12 h (Fig. 1B). This induction was not as pronounced at later time points, but remained elevated 72 h later. In contrast, Wnt3a did not induce the expression of Axin1 in chondrocytes (data not shown). Thus, regulation of Axin1 and Axin2 by Wnt3a in chondrocytes is similar to the pattern that is observed with other cell types.29,30
FIGURE 1.

Wnt3a induces Axin2 gene and protein expression. Primary murine sternal chondrocytes were isolated and placed in monolayer culture as described in “Methods.” After overnight culture they were treated with Wnt3a (100 ng/mL) or with a vehicle control for 0, 6, 12, and 24 h before total RNA was isolated. Quantitative Reverse-Transcriptase PCR was performed to assess Axin2 gene expression (normalized to β-actin) (A). Primary chondrocytes from Axin2LacZ/+ mice were placed in monolayer culture overnight and then treated with 100 ng/mL of Wnt3a. Cultures were harvested after 0, 12, 24, 36, 48, and 72 h and and β-galactosidase activity was measured as a reporter of Axin2 protein expression (B). The symbol * represents significance at P < 0.05 when compared to 0 h.
Axin Regulates β-Catenin Signaling in Chondrocytes
To study the reciprocal interaction of Axin expression on Wnt/β-catenin signaling in chondrocytes, TMC23 and C5.18 chondrocyte cell lines were transfected with the Topflash reporter construct and treated with Wnt3a (100 ng/mL) in the presence or absence of cotransfection with Axin1. Luciferase activity was measured 2 days after treatment as an indication of Wnt3a-induced β-catenin signaling activation. As expected, Wnt3a-treated cells had increased luciferase reporter activity compared to controls cells. Overexpression of Axin1 significantly inhibited Wnt3a-induced Topflash reporter activity in both cell lines (Fig. 2). Since Axin1 and Axin2 have been shown to be functionally equivalent in the regulation of Wnt/β-catenin signaling, the findings suggest that regulation of the Axin genes modulate β-catenin signaling.24
FIGURE 2.

Axin inhibits Wnt3a signaling in chondrocytes. TMC23 (A) and C5.18 (B) chondrocyte cell lines were transfected with the Topflash reporter construct and treated with Wnt3a (100 ng/mL) in the presence or absence of cotransfection with Axin1. Cell extracts were harvested after 48 h and luciferase activity measured. Transfection efficiency was determined by measurement of renilla luciferase activity. The symbol * represents significance at P < 0.05.
Axin Modulates Signaling Cross-talk between the β-Catenin and TGF-β Signaling Pathways
Wnt/β-catenin and TGF-β are two key signaling pathways involved in regulating chondrocyte differentiation.1–4 Prior work has suggested important interactions between these pathways and previously we showed that TGF-β/Smad3 induces β-catenin signaling in chondrocytes.7 However, a reciprocal effect of β-catenin on TGF-β/Smad3 signaling has not previously been examined. Initial experiments determined whether β-catenin alters the effect of TGF-β on activation of the Smad3-responsive reporter p3TP-Lux. C5.18 chondrocytes transfected with p3TP-Lux were treated with TGF-β in the presence or absence of cotransfection with Smad3 and/or β-catenin plasmids. Treatment with TGF-β for 48 h resulted in a 12-fold induction in luciferase activity, and this was increased to 20-fold in cells cotransfected with Smad3. Transfection of the β-catenin plasmid significantly inhibits TGF-β signaling in both the presence (P < 0.05) and absence (P < 0.01) of Smad3 (Fig. 3A). Combined with prior work, the experiment shows that TGF-β/Smad induces β-catenin, and β-catenin in turn downregulates TGF-β/Smad3 signaling.
FIGURE 3.

β-catenin inhibits TGF-β signaling in chondrocytes while the induction of β-catenin by TGF-β is enhanced in Axin2-deficient chondrocytes. (A) Chondrocyte C5.18 cells were transfected with TGF-β responsive reporter, p3TP-Lux in the presence or absence of cotransfections with Smad3 and/or β-catenin and treated with or without TGF-β (5 ng/mL) for 48 h. Transfection of β-catenin significantly inhibited TGF-β or TGF-β+Smad3-induced p3TP-Lux reporter activity (*P < 0.05, **P < 0.01). (B) Protein lysates from TGF-β-treated primary chondrocytes from WT and Axin2−/− mice were assayed for β-catenin by Western blot. TGF-β treatment enhances β-catenin accumulation in both WT and Axin2−/− cells. However, the elevated levels of β-catenin persist in Axin2 −/− cells.
To examine if Axin2 participates in this cross-talk, primary chondrocytes isolated from wild-type and Axin2 −/− mice were treated with TGF-β and total β-catenin levels determined by Western blot (Fig. 3B). While the magnitude of TGF-β stimulation of β-catenin in both the wild-type and Axin2 −/− cultures was similar, the pattern of accumulation varied. In wild-type cultures, maximal levels of β-catenin were observed at 2 h and then levels declined progressively with a return to basal levels by 36–48 h following treatment. In contrast, peak β-catenin levels were sustained for 12 h in Axin2 −/− cultures and remained elevated after 48 h after treatment. The findings confirm that TGF-β induces β-catenin accumulation and establish a role for Axin2 in mediating the degradation of β-catenin in chondrocytes.
Axin Enhances TGF-β Signaling
Previous studies have established that Axin binds to type I TGF-β receptor and facilitates the phosphorylation of Smad3.22 To examine if this interaction is functionally important in chondrocytes, C5.18 chondrocyte cultures were transfected with 4×SBE, a reporter for Smad3 activity, and were treated with 0.02, 0.2, or 2.0 ng/mL of TGF-β in the presence of absence of cotransfection with an Axin1 plasmid. For each concentration of TGF-β, Axin1 cotransfection consistently enhanced reporter activity by approximately 20% (Fig. 4). The findings establish that Axin regulates TGF-β signaling in chondrocytes consistent with results in other cell types.22
FIGURE 4.

Axin enhances TGF-β signaling in chondrocytes. C5.18 chondrocyte cultures were transfected with 4×SBE and treated with 0.02, 0.2, or 2.0 ng/mL of TGF-β in the presence of absence of cotransfection with Axin plasmid. Cultures were harvested after 24 h and show that Axin cotransfection consistently enhanced reporter activity approximately 20% at each TGF-β concentration.
TGF-β Inhibits Axin1 and Axin2 Expression in Chondrocytes
Since Axin expression modulates TGF-β effects on both β-catenin and Smad3 signals, we examined if TGF-β in turn regulates expression of the Axins. Primary murine chondrocytes were treated with either 1 or 5 ng/mL of TGF-β and total RNA isolated from the cultures after 48 h. TGF-β resulted in a marked dose-dependent suppression of Axin1, with approximately 70% and 90% inhibitions observed in cultures treated with 1 and 5 ng/mL of TGF-β, respectively(Fig. 5A).
FIGURE 5.

TGF-β inhibits Axin1 and Axin2 expression in chondrocytes. Primary murine chondrocytes were isolated and placed in overnight culture. The cultures were then treated with TGF-β (1 or 5 ng/mL) and total RNA was harvested at 48 h. Real-time RT PCR was used to determine the effect on Axin1 gene expression (A). Chondrocyte cultures with treated with TGF-β (5 ng/mL) and total RNA harvested at various times between 0 and 72 h and real-time RT-PCR used to determine the effect on expressions of Axin1 (B) and Axin2 (C) gene expression. β-galactosidase activity was measured in primary chondrocytes from Axin2LacZ/+ knockin mice and revealed that Axin2 protein expression is also negatively regulated by TGF-β progressively over time (D). The symbol *represents significance at P < 0.05 when compared to 0 ng/mL TGF-β or 0 h.
Using a concentration of TGF-β of 5 ng/mL, Axin1 and Axin2 mRNA expression was measured following 0, 12, 24, 36, 48, and 72 h of exposure to TGF-β. TGF-β resulted in a time-dependent inhibition of both Axin1 and Axin2 levels, with increasing inhibition observed over time. Initial Axin1 inhibition was observed within 12 h (30% inhibition) and was maximal by 36 h (80% inhibition). In contrast, Axin2 inhibition was delayed until 24 h (50% inhibition) and was maximal at 72 h (90% inhibition) (Fig. 5B, C).
To determine if TGF-β also regulates Axin2 protein expression, β-galactosidase activity was measured in Axin2LacZ/+ knockin mice following treatment with TGF-β. Similar to the observed effect on mRNA expression, TGF-β resulted in a progressive and sustained decrease in β-galactosidase activity that paralleled the decrease in gene expression. Maximal suppression of protein synthesis also occurred at 72 h (80% inhibition) (Fig. 5D).
BMP Does Not Alter Axin Expression or Regulate β-Catenin Signaling
The delayed inhibition of Axin2 by TGF-β suggests that the effect may be related to alterations in the maturational state of the cells, rather than to a direct signaling effect. For this reason, we examined whether BMP-2, which has effects opposite TGF-β and promotes chondrocyte maturation, regulates the expression of Axin2.2,3 In contrast to the observations made with TGF-β, primary chondrocyte cells from Axin2LacZ/+ knockin mice treated with BMP-2 (100 ng/mL) had no elevation of β-galactosidase reporter activity (Fig. 6A).
FIGURE 6.

BMP does not alter Axin expression or Wnt/β-catenin signaling. Primary chondrocyte cells from Axin2LacZ/+ knockin mice were treated with BMP-2 (100 ng/mL) and cell extracts harvested between 0 and 72 h. β-galactosidase reporter activity was measured (A). C5.18 cells were transfected with the Wnt Topflash reporter and treated with BMP-2 (100 ng/mL) in the presence or absence of Wnt3a. Cell extracts were harvested after 24 h and luciferase activity was measured. The luciferase activity was normalized to renilla luciferase (B).
To further examine BMP/β-catenin interactions, C5.18 cells transfected with the Wnt signaling reporter, Topflash, were treated with BMP-2 in the presence or absence of Wnt3a. Unlike effects observed with TGF-β, BMP-2-treatment did not induce luciferase activity, indicating an absence of stimulation of β-catenin signaling.7 Furthermore, BMP did not alter the stimulatory effects of β-catenin (Fig. 6B).
Altogether, the findings demonstrate unique crosstalk between the TGF-β/Smad3 and the Wnt/β-catenin pathways and suggest that the regulation of Axin2 gene and protein levels by TGF-β is due to a pathway-specific signaling event.
TGF-β Inhibits Axin1 and Axin2 through Smad3 Signaling
Since Smad3 has been shown to be a key downstream effector of TGF-β signaling in chondrocytes, we examined whether or not Smad3 plays a role in TGF-β-mediated Axin inhibition.2,7,31 For these experiments, primary chondrocytes were isolated from Smad3 −/− mice. In contrast to the marked inhibitions of Axin1 and Axin2 observed in wild-type cultures treated with TGF-β (Fig. 2), no decrease in the expression of either Axin1 or Axin2 occurred in Smad3 −/− chondrocyte cultures at either 48 or 72 h following exposure to TGF-β (Fig. 7A).
FIGURE 7.

Smad3 −/− cells do not exhibit TGF-β-mediated inhibition of Axin expression. Primary chondrocytes from Smad 3−/− mice were isolated and cultured with 5 ng/mL TGF-β treatment for 24 and 48 h. Total RNA was harvested and RT-PCR performed to measure the relative suppression of Axin1 or Axin2 gene expression in TGF-β-treated cultures compared to control cultures after 48 h of treatment (A). Total cell protein extracts were obtained from wild-type and Smad3 −/− chondrocyte cultures treated with TGF-β or control medium and Axin1 protein levels measured by Western blot. β-actin was used as a loading control (B).
To confirm these findings, a Western blot for Axin1 was performed on protein extracts obtained from wild-type and Smad3 −/− chondrocyte cultures treated with and without TGF-β. As previously observed TGF-β stimulated a marked decrease in Axin1 protein expression following 60 h of treatment. In contrast, Axin1 protein levels in Smad3−/− cells remained unchanged when compared to untreated controls (Fig. 7B). These data indicate that the regulation of Axin1 and Axin2 by TGF-β is dependent upon Smad3 signaling.
DISCUSSION
This study further supports the presence of crosstalk between the TGF-β/Smad3 and Wnt/β-catenin signaling pathways. While we previously demonstrated that TGF-β/Smad3 signaling induced β-catenin signaling, the current findings show that β-catenin, in turn, inhibits TGF-β signaling in chondrocytes.7 Furthermore, we show that Axin1 and Axin2, two functionally equivalent proteins that modulate both signaling pathways, are involved in the interaction.24
TGF-β was found to be a potent inhibitor of both Axin1 and Axin2. In contrast, BMP treatment had no effect on expression of the Axins, suggesting that the effect was not related to alterations in the maturational state of the cells, but was related to a specific signaling event downstream of TGF-β. Prior work in our laboratory established that Smad3 induced β-catenin signaling by interfering with the ability of the E3 ubiquitin ligase, β-TCRP, to stimulate degradation of β-catenin.7 A genetic model using Smad3-deficient chondrocytes was used to definitively demonstrate a role for Smad3 signaling in the suppression of expression of both Axin1 and Axin2. This is the first model that we are aware of establishing TGF-β/Smad3 in the regulation of Axin expression.
Prior work suggests a complex role for β-catenin signaling as a modulator of chondrogenesis and chondrocyte maturation.32 β-catenin signaling is a strong inhibitor of chondrogenesis, but appears to be necessary for the normal function of the growth plate.10,11 Thus, although this signaling pathway prevents the formation of cartilage, its reestablishment in chondrocytes is necessary.2–4 In the growth plate β-catenin appears to have a role in the regulation of both proliferation and differentiation.7,8 In vivo models of β-catenin loss of function show reduced chondrocyte proliferation compared to controls.8 Our recent findings show that the induction of β-catenin signaling is necessary for the stimulation of proliferation by TGF-β.2 In this model, β-catenin was necessary for the induction of cyclin D1 gene expression in chondrocytes downstream of TGF-β.7 A role for β-catenin in cell proliferation through induction of cyclin D1 has been established in numerous cell types and has particular importance in cancer models.8,33–35
In addition to its effects as a stimulator of proliferation, in chondrocytes a role for β-catenin as a signal involved in acceleration of maturation has also been established.9,14,36,37 While development models have suggested a role in maturation, in vitro studies have clearly demonstrated that gain of β-catenin function results in accelerated chondrocyte maturation while loss of function has resulted in delayed maturation.9,14,36,37 This has been demonstrated in various models including isolated chick sternal chondrocytes, developing chick limbs in vivo, and ectopic ossification models.9,14,36,37
Since maturation typically occurs in cells that exit the cell cycle, the dual stimulation of both proliferation and maturation are events that tend to oppose and are not typically dually mediated by a single signaling pathway. However, in the growth plate cell fate decisions are determined by the various growth factors and signaling molecules present in the local environment. Prior work in our laboratory and by others demonstrates that the closely related TGF-β and BMP signaling pathways have opposite effects on chondrocyte maturation.2,3 Thus, while the TGF-β–Smad pathways inhibit maturation, the BMP/Smad pathways enhance chondrocyte maturation.1,3,38 Moreover, our work shows that the absence of Smad3 results in an increase in basal BMP signaling and that the BMP signal is necessary for maturation.2 Thus, it is clear that the overall effect of the various signals is dependent on the presence or absence of complementary or competing signaling pathways.
β-catenin is a signaling pathway that potentially links and integrates both proliferation and differentiation and may have disparate effects on these two events dependent upon the presence or absence of associated pathways.32 In this context, TGF-β and the Axins appear to have a particularly important role. In association with prior work, the current studies show that TGF-β stimulates β-catenin signaling while in turn β-catenin signaling inhibits TGF-β signaling.7 Moreover, TGF-β reduces the expression of the Axins. The cumulative effect of decreased Axin expression makes chondrocytes more sensitive to Wnt/β-catenin signaling and less sensitive to TGF-β Smad signaling. The overall impact of these signaling events is a negative feedback loop on TGF-β signaling and a positive feedback loop on Wnt/β-catenin signaling.
Figure 8 summarizes the Axin-mediated interaction between TGF-β/Smad3 and Wnt/β-catenin signaling suggested by our findings. TGF-β signaling activates Smad3. Smad3 was previously shown to be essential for the stimulation of β-catenin expression through an inhibitory role on the E3 ubiquitin ligase, β-TRCP.7 While Smad3 activation results in dual stimulation of both TGF-β and β-catenin signaling, β-catenin signaling acts to downregulate TGF-β signaling. The Smad3-mediated suppression of Axin expression further inhibits TGF-β/Smad3 signaling, but makes cells more sensitive to Wnt/β-catenin. Thus, TGF-β/Smad3 regulates elements of the β-catenin signaling pathway that act to stimulate β-catenin and result in enhanced responsiveness to Wnt signals. At the same time, these β-catenin pathway molecules suppress TGF-β signaling. The release of cells from TGF-β signaling may be one mechanism leading to maturation and provide a mechanism through which β-catenin shifts toward stimulating the maturation process.
FIGURE 8.
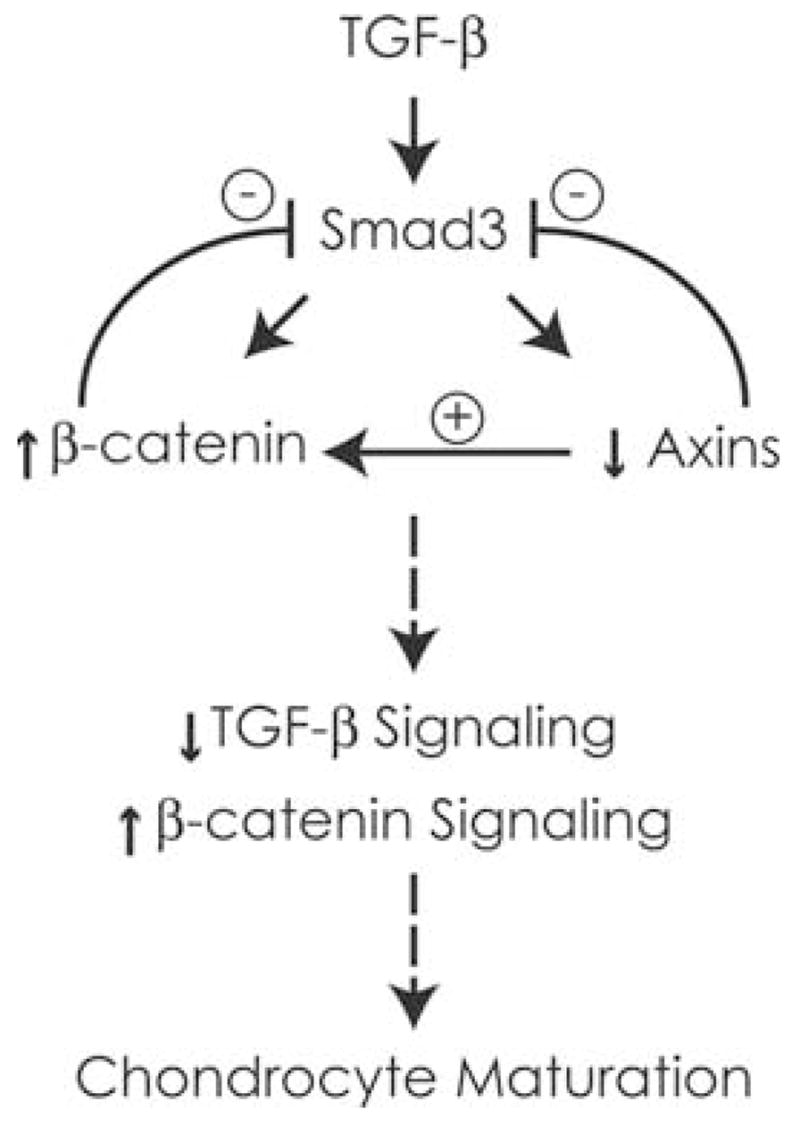
Summary of TGF-β/Smad and β-catenin signaling interactions in chondrocytes. TGF-β signaling inhibits chondrocyte maturation and induces Wnt/β-catenin signaling by directly activating β-catenin and reducing expression of Axin1 and Axin2. While β-catenin is necessary for the induction of cyclin D1 and chondrocyte proliferation downstream of TGF-β/Smad3, the induction of β-catenin and suppression of axins result in a decrease in TGF-β signaling. The overall effect of enhanced β-catenin signaling and inhibited Smad3 signaling is a release of chondrocytes from the inhibitory effects of TGF-β signaling on maturation. β-catenin in the presence of other stimulators of maturation, including BMP signaling molecules and Runx2, enhances chondrocyte maturation.
The ability of β-catenin to stimulate maturation probably is dependent upon the presence of other positive regulators of maturation.32 Prior work suggests that the induction of maturational markers by β-catenin occurs in association with other signals that stimulate maturation, including BMP signals and Runx2.14,37 Runx2 has been shown to be essential for chondrocyte maturation.39 Recent work in a chick chondrocyte model has shown a consensus TCF/Lef-binding site in the proximal promoter of Runx2 that is required for induction of Runx2 by β-catenin signaling.37 Furthermore, it was shown that the activation of the colX promoter by β-catenin was dependent upon the Runx2-binding site and that induction of both colX and Runx2 by BMP2 is enhanced by simultaneous β-catenin signaling.37
The current findings support the concept that the process of chondrocyte maturation during endochondral ossification is controlled by complex interactions between key signaling pathways. The experiments confirm an important role for the Wnt/β-catenin signaling pathway in the integration of TGF-β and BMP signals. The TGF-β/Smad3-mediated induction of β-catenin and inhibition of the Axins result in a feedback inhibition of TGF-β signaling and a stimulation of β-catenin signaling. Further study will define the potential importance of this event in permitting chondrocytes to transition to hypertrophy and progress toward terminal maturation.
Acknowledgments
The work was supported by National Health Service Awards AR048681 (R.J.O.) and AR053717 (R.J.O.).
References
- 1.Ferguson CM, et al. Smad 2 and 3 mediate TGF-β1-induced inhibition of chondrocyte maturation. Endocrinology. 2000;141:4728–4735. doi: 10.1210/endo.141.12.7848. [DOI] [PubMed] [Google Scholar]
- 2.Li TF, et al. Smad3-deficient chondrocytes have enhanced BMP signaling and accelerated differentiation. J Bone Miner Res. 2006;21:4–16. doi: 10.1359/JBMR.050911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kobayashi T, et al. BMP signaling stimulates cellular differentiation at multiple steps during cartilage development. Proc Natl Acad Sci USA. 2005;102:18023–18027. doi: 10.1073/pnas.0503617102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iwamoto M, et al. Runx2 expression and action in chondrocytes are regulated by retinoid signaling and parathyroid hormone-related peptide (PTHrP) Osteoarthr Cart. 2003;11:6–15. doi: 10.1053/joca.2002.0860. [DOI] [PubMed] [Google Scholar]
- 5.Ionescu AM, et al. PTHrP modulates chondrocyte differentiation through AP-1 and CREB signaling. J Biol Chem. 2001;276:11639–11647. doi: 10.1074/jbc.M006564200. [DOI] [PubMed] [Google Scholar]
- 6.Ballock RT, et al. Thyroid hormone regulates terminal differentiation of growth plate chondrocytes through local induction of bone morphogenetic proteins. Trans Orthop Res Soc. 2000;25:160. [Google Scholar]
- 7.Li TF, et al. Transforming growth factor-beta stimulates cyclin D1 expression through activation of beta-catenin signaling in chondrocytes. J Biol Chem. 2006;281:21296–21304. doi: 10.1074/jbc.M600514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akiyama H, et al. Interactions between Sox9 and beta-catenin control chondrocyte differentiation. Genes Dev. 2004;18:1072–1087. doi: 10.1101/gad.1171104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Enomoto-Iwamoto M, et al. The Wnt antagonist Frzb-1 regulates chondrocyte maturation and long bone development during limb skeletogenesis. Dev Biol. 2002;251:142–156. doi: 10.1006/dbio.2002.0802. [DOI] [PubMed] [Google Scholar]
- 10.Day TF, et al. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005;8:739–750. doi: 10.1016/j.devcel.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 11.Hill TP, et al. Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell. 2005;8:727–738. doi: 10.1016/j.devcel.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 12.Church V, et al. Wnt regulation of chondrocyte differentiation. J Cell Sci. 2002;115:4809–4818. doi: 10.1242/jcs.00152. [DOI] [PubMed] [Google Scholar]
- 13.Hartmann C, Tabin CJ. Dual roles of Wnt signaling during chondro-genesis in the chicken limb. Development. 2000;127:3141–3159. doi: 10.1242/dev.127.14.3141. [DOI] [PubMed] [Google Scholar]
- 14.Dong Y, et al. Wnt-mediated regulation of chondrocyte maturation: modulation by TGF-beta. J Cell Biochem. 2005;95:1057–1068. doi: 10.1002/jcb.20466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Topol L, et al. Wnt-5a inhibits the canonical Wnt pathway by promoting GSK-3-independent beta-catenin degradation. J Cell Biol. 2003;162:899–908. doi: 10.1083/jcb.200303158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kikuchi A. Roles of Axin in the Wnt signalling pathway. Cell Signal. 1999;11:777–788. doi: 10.1016/s0898-6568(99)00054-6. [DOI] [PubMed] [Google Scholar]
- 17.Cadigan KM, Liu YI. Wnt signaling: complexity at the surface. J Cell Sci. 2006;119:395–402. doi: 10.1242/jcs.02826. [DOI] [PubMed] [Google Scholar]
- 18.Ha NC, et al. Mechanism of phosphorylation-dependent binding of APC to beta-catenin and its role in beta-catenin degradation. Mol Cell. 2004;15:511–521. doi: 10.1016/j.molcel.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 19.Bhasin N, et al. Differential regulation of chondrogenic differentiation by the serotonin2B receptor and retinoic acid in the embryonic mouse hindlimb. Dev Dyn. 2004;230:201–209. doi: 10.1002/dvdy.20038. [DOI] [PubMed] [Google Scholar]
- 20.Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily. Science. 2002;296:1646–1647. doi: 10.1126/science.1071809. [DOI] [PubMed] [Google Scholar]
- 21.Massague J, Chen Y. Controlling TGF-beta signaling. Genes Dev. 2000;14:627–640. [PubMed] [Google Scholar]
- 22.Furuhashi M, et al. Axin facilitates Smad3 activation in the transforming growth factor beta signaling pathway. Mol Cell Biol. 2001;21:5132–5141. doi: 10.1128/MCB.21.15.5132-5141.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu W, et al. Axin is a scaffold protein in TGF-beta signaling that promotes degradation of Smad7 by Arkadia. EMBO J. 2006;25:1646–1658. doi: 10.1038/sj.emboj.7601057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chia IV, Costantini F. Mouse axin and axin2/conductin proteins are functionally equivalent in vivo. Mol Cell Biol. 2005;25:4371–4376. doi: 10.1128/MCB.25.11.4371-4376.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kishida M, et al. Axin prevents Wnt-3a-induced accumulation of beta-catenin. Oncogene. 1999;18:979–985. doi: 10.1038/sj.onc.1202388. [DOI] [PubMed] [Google Scholar]
- 26.Yu HM, et al. The role of Axin2 in calvarial morphogenesis and craniosynostosis. Development. 2005;132:1995–2005. doi: 10.1242/dev.01786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feng JQ, et al. NF-kappaB specifically activates BMP-2 gene expression in growth plate chondrocytes in vivo and in a chondrocyte cell line in vitro. J Biol Chem. 2003;278:29130–29135. doi: 10.1074/jbc.M212296200. [DOI] [PubMed] [Google Scholar]
- 28.Harada S, et al. Osteogenic protein-1 up-regulation of the collagen X promoter activity is mediated by a MEF-2-like sequence and requires an adjacent AP-1 sequence. Mol Endocrinol. 1997;11:1832–1845. doi: 10.1210/mend.11.12.0022. [DOI] [PubMed] [Google Scholar]
- 29.Lustig B, et al. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol Cell Biol. 2002;22:1184–1193. doi: 10.1128/MCB.22.4.1184-1193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jho EH, et al. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22:1172–1183. doi: 10.1128/MCB.22.4.1172-1183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang X, et al. TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J Cell Biol. 2001;153:35–46. doi: 10.1083/jcb.153.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tamamura Y, et al. Developmental regulation of Wnt/beta-catenin signals is required for growth plate assembly, cartilage integrity, and endochondral ossification. J Biol Chem. 2005;280:19185–19195. doi: 10.1074/jbc.M414275200. [DOI] [PubMed] [Google Scholar]
- 33.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 34.Saegusa M, et al. Beta-catenin simultaneously induces activation of the p53-p21WAF1 pathway and overexpression of cyclin D1 during squamous differentiation of endometrial carcinoma cells. Am J Pathol. 2004;164:1739–1749. doi: 10.1016/s0002-9440(10)63732-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rowlands TM, et al. Dissecting the roles of beta-catenin and cyclin D1 during mammary development and neoplasia. Proc Natl Acad Sci USA. 2003;100:11400–11405. doi: 10.1073/pnas.1534601100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kitagaki J, et al. Activation of beta-catenin-LEF/TCF signal pathway in chondrocytes stimulates ectopic endochondral ossification. Osteoarthr Cart. 2003;11:36–43. doi: 10.1053/joca.2002.0863. [DOI] [PubMed] [Google Scholar]
- 37.Dong YF, et al. Wnt induction of chondrocyte hypertrophy through the Runx2 transcription factor. J Cell Physiol. 2006;208:77–86. doi: 10.1002/jcp.20656. [DOI] [PubMed] [Google Scholar]
- 38.Chen P, et al. Osteogenic protein-1 promotes growth and maturation of chick sternal chondrocytes in serum-free cultures. J Cell Sci. 1995;108:105–114. doi: 10.1242/jcs.108.1.105. [DOI] [PubMed] [Google Scholar]
- 39.Yoshida CA, et al. Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. 2004;18:952–963. doi: 10.1101/gad.1174704. [DOI] [PMC free article] [PubMed] [Google Scholar]