

Abstract
Rationale: Bronchiolitis obliterans syndrome is the leading cause of chronic lung allograft dysfunction. We have demonstrated that respiratory viral infection is a bronchiolitis obliterans syndrome risk factor and virus-dependent injury induces expression of innate airway epithelial genes belonging to the interleukin (IL)-12 family. Thus, we hypothesized that epithelial cell IL-12 family members could mediate lung allograft dysfunction.
Objectives: We used mouse and human allograft specimens to evaluate the role of epithelial cell IL-12 family members in allograft dysfunction associated with and without viral infection.
Methods: Murine and human IL-12 family members were characterized and manipulated in allografts and then correlated with epithelial cell injury, immune cell accumulation, and collagen deposition.
Results: In a mouse model of lung transplantation, concurrent viral infection and allogeneic transplantation increased epithelial injury and this was followed by exaggerated accumulation of macrophages and collagen deposition. This virus-driven allograft dysfunction was associated with an epithelial innate response manifested by a synergistic increase in the production of the macrophage chemoattractant IL-12 p80 (p80), but not IL-12 or IL-23. Blockade or overexpression of donor epithelial p80 resulted in a corresponding abrogation or enhancement of macrophage accumulation and allograft dysfunction. We extended these findings to human recipients with viral infection and transplant bronchitis and again observed excessive epithelial p80 expression that correlated with increased macrophage accumulation.
Conclusions: These experiments support a role for an enhanced epithelial innate response as a central process in allograft dysfunction and identify the macrophage chemoattractant p80 as an innate epithelial effector of disease progression.
Keywords: graft rejection, innate immunity, lung transplantation, macrophage, virus
Lung transplantation is a life-saving treatment option for patients with end-stage respiratory disease. Despite improved surgical techniques, better allograft preservation, more potent immunosuppressants, and aggressive treatment of infections the overall survival rate after lung transplantation is the lowest among all solid organ transplants. A major contributor to this posttransplantation mortality is chronic lung allograft dysfunction. Although chronic allograft dysfunction is encountered after transplantation of all solid organs, the high incidence (50% at 5 yr) and case fatality rates (25–30%) as well as the relatively poor response to therapy make it the leading cause of late death after lung transplantation (1).
Although the biochemical mechanisms responsible for chronic lung allograft dysfunction remain unknown, identification of histologic features and risk factors has provided important insights into disease pathogenesis. Chronic lung allograft dysfunction is characterized histologically by airway epithelial cell injury, mononuclear cell influx, and fibrous obliteration of the small airways, and is referred to as bronchiolitis obliterans (BO) (2, 3). The clinical correlate of BO is bronchiolitis obliterans syndrome (BOS) and is defined functionally as a persistent worsening of airflow obstruction (4). Review of clinical conditions that are associated with the subsequent development of BO and BOS has identified acute vascular rejection, lymphocytic bronchitis (referred to as transplant bronchitis in this article), and in some studies cytomegalovirus (CMV) infection as disease risk factors (5, 6). More recently, we have identified antecedent community-acquired respiratory viral infection as a distinct risk factor for chronic allograft dysfunction (7). Because these risk factors result in epithelial cell injury and allograft inflammation, our laboratory and others have suggested that an exaggerated or abnormal epithelial injury response initiates allograft inflammation and chronic allograft dysfunction (3, 6, 8, 9).
To provide a molecular link between epithelial injury and chronic allograft dysfunction, we proposed that induction of innate epithelial injury response proteins initiates inflammatory cascades that lead to chronic allograft dysfunction. In that regard, we have previously demonstrated induction of the epithelial injury response gene encoding interleukin (IL)-12 p40 during paramyxoviral respiratory infection (10). The IL-12–related cytokines are encoded by five independently regulated genes: p40, p35, Epstein-Barr virus–induced gene-3 (EBI3), p19, and p28. Because of alternative heterodimeric partnering and monomer secretion, this family is composed of seven secreted proteins: IL-12 (a p40 and p35 heterodimer), IL-12 p80 (a p40 homodimer), p40 (a p40 monomer), IL-23 (a p40 and p19 heterodimer), EBI3 (an EBI3 monomer), IL-27 (a p28 and EBI3 heterodimer), and EBI3–p35 (an EBI3 and p35 heterodimer) (11–13). Although the factors that regulate the dimerization of p40 with other IL-12 family members are unknown, IL-12 p40 expression in mouse and human airway epithelial cells resulted in the selective expression of IL-12 p80 (p80) and p40 but not IL-12 (10). The IL-12–related cytokines have distinct biologic properties and p80, but not the other p40-containing IL-12 family members, is a macrophage chemoattractant that signals through IL-12 receptor β1 (IL-12Rβ1) (14, 15). Interestingly, exaggerated macrophage accumulation has been observed in animal and human models of lung allograft dysfunction (16–19), and the IL-12 family members have been implicated in kidney (20), liver (21), bone marrow (22–24), and cardiac allograft dysfunction (25, 26). Accordingly, we set out to examine whether innate epithelial injury response proteins belonging to the IL-12 family were critical mediators that linked epithelial cell injury with macrophage accumulation and chronic lung allograft dysfunction.
METHODS
See the online supplement for additional details.
Mice
C57BL/6J (H-2d), C57BL/6J IL-12 p40−/−, C57BL/6J IL-12 p35−/−, BALB/cJ (H-2b), and BALB/cJ 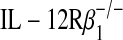 mice were obtained from Jackson Laboratory (Bar Harbor, ME). The Animal Studies Committee of the Washington University School of Medicine (St. Louis, MO) approved all animal studies.
mice were obtained from Jackson Laboratory (Bar Harbor, ME). The Animal Studies Committee of the Washington University School of Medicine (St. Louis, MO) approved all animal studies.
Murine Heterotopic Subcutaneous Tracheal Transplantation, Viral Infection, and Analysis of Tracheal Graft
Murine heterotopic subcutaneous tracheal transplantation was performed as originally described (27). To assess airway epithelial cell injury, immune cell accumulation, and collagen deposition grafts were harvested on Days 5, 14, and 28, respectively. Intranasal inoculation of Sendai virus (Fushimi strain, ATCC VR-105) or ultraviolet-inactivated Sendai virus was performed as previously described (10, 14, 28). To quantitate epithelial injury, the percentage of basement membrane (BM) covered by epithelium was calculated. The mean values from three representative sections of each graft were recorded as the percentage of denuded BM (length of denuded BM divided by total length of BM, multiplied by 100). To quantitate collagen deposition, duplicate measurement of allograft hydroxyproline was determined and compared with a cis-4-hydroxyl-l-proline standard curve (0–100 μg/ml; Sigma, St. Louis, MO). To immunolabel neutrophils, macrophages, lymphocytes, and IL-12 p40 we incubated tissue with rat anti-mouse neutrophil IgG (7/4 clone, 0.66 μg/ml; Serotec, Inc., Raleigh, NC), rat anti-mouse Mac-3 IgG (M3/84 clone, 2 μg/ml; BD Biosciences Pharmingen, San Diego, CA), rabbit anti-CD3 polyclonal antibody (1:50, vol/vol), or goat anti-mouse IL-12 p40 polyclonal antibody (2 μg/ml; Santa Cruz Biotechnology, Inc., Santa Cruz, CA). To quantitate total cell and immune cell accumulation in graft lumen, immunolabeled cells were counted in three high-power (×400) fields from three representative sections of each graft. To quantitate IL-12 family member concentration in grafts we used commercially available ELISA kits for mouse IL-12 p80/p40 and IL-12 (R&D Systems, Inc., Minneapolis, MN) and IL-23 (eBioscience, San Diego, CA).
Generation and Characterization of Clara Cell Secretory Protein–p40 Transgenic C57BL/6J Mice
To achieve airway epithelial cell overexpression of mouse p40 we generated a C57BL/6J transgenic mouse strain, using 2.3 kb of the rat Clara cell secretory protein (CCSP) promoter (kindly provided by J. Whitsett, University of Cincinnati College of Medicine, Cincinnati, OH) (29). Expression of the CCSP-p40 transgene was quantitated in bronchoalveolar lavage (BAL) fluid by ELISA and we detected a selective increase in IL-12 p80/p40 but not IL-12 or IL-23.
Lung Transplant Recipient Care and Cohort Assembly
The Institutional Review Board of the Washington University School of Medicine approved all human studies. All recipients underwent pre- and post-transplantation care by the Lung Transplant Group at the Washington University School of Medicine as previously described (7, 30). To analyze IL-12 p40 expression in lung transplant recipients we performed three separate experiments. First, to analyze IL-12 p40 expression in allografts from recipients with a respiratory viral infection, we performed IL-12 p40 immunolabeling of 10 biopsies from recipients from whom transbronchial biopsies were obtained during respiratory viral infection (7). Second, to further evaluate IL-12 p40 expression in bronchitis we immunolabeled five open lung biopsy specimens for IL-12 p40. Third, to analyze and quantitate IL-12 family member expression and to determine whether specific family members were selectively increased in transplant recipients with a single type of allograft dysfunction, we prospectively collected BAL fluid and transbronchial biopsies. All BAL fluid specimens underwent microbiologic analysis for the presence of bacteria, fungus, mycobacteria, and virus according to standard microbiologic techniques and all transbronchial biopsies underwent pathologic analysis for acute rejection and transplant bronchitis according to the Lung Rejection Study Group (2). We prospectively collected BAL fluid and transbronchial biopsies from 86 recipients. Of these 86 specimens, we excluded 39 because infectious pathogens (n = 19) or two concurrent pathologic abnormalities (n = 20) were identified. The remaining 47 recipients with a single abnormality were segregated into four cohorts. Recipients without infection, acute rejection, or bronchitis constituted the Normal Transplant cohort (A0B0 without infection; n = 16). Recipients with only acute rejection constituted the Acute Rejection cohort (A1B0, n = 1; A2B0, n = 8; A3B0, n = 1 [total, n = 10]). Recipients with only CMV detected in the BAL fluid constituted the CMV cohort (A0B0 with CMV pneumonitis, n = 4; A0B0 with BAL positive for CMV by shell vial assay, n = 5 [total, n = 9]). Recipients with only transplant bronchitis constituted the Bronchitis cohort (A0B1, n = 7; A0B2, n = 5 [total, n = 12]). Normal nontransplantation subjects had no clinical history of lung disease, normal spirometry, and normal airway reactivity and constituted the Nontransplant Normal cohort (n = 10).
Human BAL Fluid Analysis
BAL fluid total cell count, differential, and IL-12 p80/p40 and IL-12 levels were determined as previously described (10). To measure human IL-23, we performed a receptor-binding assay with reagents according to the manufacturer's recommendations (R&D Systems, Inc.). We sequentially incubated 96-well plates with recombinant human IL-23 receptor–Fc chimera protein (1 μg/ml for 18 h at 25°C), blocking buffer (300 μl/well of 0.5% bovine serum albumin in phosphate-buffered saline for 1 h at 37°C), BAL fluid or recombinant human IL-23 (100 μl/well at 0–2,000 pg/ml; R&D Systems, Inc.), biotinylated anti-human IL-12 p40 (0.1 μg/ml for 2 h at 25°C, clone 8.6; Endogen, Woburn, MA), streptavidin-conjugated horseradish peroxidase, substrate solution, and stop solution. Values represent means of duplicate measurements of optical density at 450 nm (with wavelength correction set to 540 nm).
Statistical Analysis
Murine variables were compared by Kruskal-Wallis test. Continuous human variables were analyzed for statistical significance by one-way analysis of variance, which included a random subject effect and a false-discovery rate procedure to identify significant differences between specific groups. Categorical human variables were compared by χ2 test or Fisher exact test, the latter if the expected cell size was less than 5 in more than 20% of cells. Correlation between human BAL fluid levels of IL-12 p80 and immune cell accumulation was analyzed by Pearson test (two tailed). For all tests a p value less than 0.05 was considered significant. Data were analyzed with SPSS 10.0 (SPSS, Chicago, IL) and SAS 9.1.2 (SAS Institute, Cary, NC).
RESULTS
Transplantation and Respiratory Viral Infection Combine to Enhance Allograft Dysfunction
We have previously shown that respiratory viral infection of lung transplant recipients with parainfluenza, respiratory syncytial virus, influenza, and adenovirus is a distinct risk factor for developing all stages of BOS and death (7). To explore the role of airway epithelial cell injury and innate epithelial injury response proteins in mediating this association we used a mouse heterotopic tracheal transplantation model. This allogeneic tracheal transplant model results in the sequential injury of airway epithelial cells, accumulation of immune cells, and fibrous obliteration of the allograft lumen (27, 31–33). The role of respiratory viral infection in the progression of allograft dysfunction was assessed on the basis of a well-characterized murine parainfluenza virus-1 (Sendai virus) infection that results in viral replication in the airway epithelial cells and induction of innate epithelial injury response genes (10, 14, 28).
To examine the role of concurrent viral infection in the setting of allogeneic transplantation we first documented that viral protein expression peaked in tracheal airway epithelial cells 5 d after infection (data not shown). Compared with syngeneic or allogeneic transplantation conditions using uninfected donor tracheas, allogeneic transplantation using donor tracheas from infected mice resulted in enhanced epithelial cell injury characterized by incomplete reepithelialization of the basement membrane (Figures 1A and 1C; and see Figure E1 in the online supplement). Interestingly, this enhanced virus-driven epithelial injury was associated with a subsequent increase in the accumulation of inflammatory cells within the allograft lumen on Day 14 (Figures 1B and 1D) and with exaggerated collagen deposition on Day 28 posttransplantation (Figures 1E and 1F). This accelerated allograft dysfunction was not observed in syngeneic grafts from infected donors or allografts from donor mice that were inoculated with ultraviolet-inactivated virus, indicating that the virus-driven phenotype requires both an alloimmune mismatch and active viral replication (Figures 1A–1F). Characterization and quantitation of subsequent inflammatory cell accumulation in the allograft lumen indicated that this virus-driven response was associated with a significant accumulation of macrophages but not of neutrophils or lymphocytes (Figure 2).
Figure 1.

Transplantation and respiratory viral infection combine to enhance allograft dysfunction. In (A), C57BL/6J donor tracheas underwent heterotopic tracheal transplantation into C57BL/6J recipient mice (C57 → C57) or BALB/cJ recipient mice (C57 → BALB). C57BL/6J donor mice underwent intranasal inoculation with Sendai virus or ultraviolet-inactivated (UV) Sendai virus and 5 d later the tracheas were harvested for heterotopic tracheal transplantation into BALB/cJ recipient mice (Virus + C57 → BALB and UV Virus + C57 → BALB, respectively). Five days post-transplantation, the grafts were harvested and underwent hematoxylin and eosin (H&E) staining. Arrows in the Virus + C57 → BALB photomicrograph indicate denuded basement membrane (BM). In (B), 14 d posttransplantation grafts were harvested and underwent H&E staining. Scale bar: 20 μm. In (C), samples from conditions in (A) underwent quantification of epithelial injury, designated as percentage of denuded BM. In (D), samples from conditions in (B) underwent quantification of total lumenal cells per high-power field (HPF, ×400). In (E), grafts were harvested and underwent quantification of hydroxyproline on the indicated posttransplantation days. In (F), samples from (E) were used to calculate fold increase in hydroxyproline between Days 7 and 28. Representative photomicrographs are shown and quantitative values represent means ± SEM (n = 4 or 5). *p < 0.05 compared with C → C; **p < 0.05 compared with C → C and C → B.
Figure 2.

Transplantation and respiratory viral infection combine to selectively enhance macrophage accumulation. In (A–C), transplantation conditions were as described in Figure 1A. Fourteen days post-transplantation, grafts were harvested and representative sections underwent immunolabeling with rat anti-mouse neutrophil IgG to identify neutrophils (A), rat anti–Mac-3 to identify macrophages (B), or rat anti-CD3 to identify lymphocytes (C). For each condition nonimmune IgG control gave no signal above background (data not shown). Immunolabeled cells in the lumen of the graft from each condition underwent quantitation (cells per HPF, ×400). Values represent means ± SEM (n = 4 or 5) from representative slides. *p < 0.05 compared with C → C and C → B.
Transplantation and Respiratory Viral Infection Generate a Synergistic Increase in Allograft p80
To identify innate epithelial injury response mediators that may account for enhanced macrophage accumulation during virus-driven allograft dysfunction we focused on the IL-12 family members because they have been implicated in allograft dysfunction (20–26), and we have previously described virus-driven induction of the macrophage chemoattractant p80 (10, 14). Initially, we performed IL-12 p40 immunolabeling of the allografts to determine whether IL-12 family members were expressed after transplantation and, if present, to identify the cellular source of their production. We detected p40 immunolabeling in allografts with concurrent viral infection but not in syngeneic grafts, allografts without viral infection, or allografts from donors after inoculation with ultraviolet-inactivated virus. In addition, this p40 immunolabeling colocalized predominantly with airway epithelial cells (Figure 3A). More quantitative analysis of the individual IL-12 family members in the grafts revealed that allogeneic transplantation induced p80 expression and that allogeneic transplantation with concurrent viral infection resulted in a synergistic increase in p80, but not IL-12 or IL-23 (Figures 3B–3D).
Figure 3.

Transplantation and respiratory viral infection generate a synergistic increase in allograft p80. In (A), C57BL/6J donor mice underwent intranasal inoculation with Sendai virus and 5 d later underwent tracheal harvest for heterotopic tracheal transplantation into BALB/cJ recipient mice. Five days post-transplantation, allografts were harvested and underwent immunolabeling with rat anti–IL-12 p40 and detection with red chromogenic substrate. Samples incubated with rat IgG control gave no signal above background (data not shown). Scale bar: 20 μm. In (B–D), tracheas from control C57BL/6J mice (column 1) and from mice after viral inoculation only (Virus only, columns 2–5) were obtained on the indicated day after viral inoculation. Transplantation (TX) conditions were as described in Figure 1A (columns 6–9) and harvested 5 d post-transplantation (equivalent to 10 d after viral inoculation). All tracheas underwent tissue sonication, determination of protein concentration, and duplicate measurements for p80 (B), IL-12 (C), and IL-23 (D). Values represent means ± SEM (n = 4–6). For comparison of transplant cohorts, *p < 0.05 compared with C → C; **p < 0.05 compared with C → C and C → B.
Blockade of p80 Function Attenuates Virus-driven Allograft Dysfunction
Given that we had demonstrated a synergistic increase in p80 expression during virus-driven allograft dysfunction, we next reasoned that this phenotype would be attenuated in recipient mice that could not respond to p80 (i.e., IL-12 receptor β1–deficient mice). Compared with a wild-type recipient, virus-driven allogeneic transplantation in an IL-12 receptor 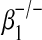 recipient resulted in less epithelial cell injury on Day 5, macrophage accumulation on Day 14, and collagen deposition on Day 28 (see Figure E2). These results further support a critical role for p80 in mediating virus-driven allograft dysfunction.
recipient resulted in less epithelial cell injury on Day 5, macrophage accumulation on Day 14, and collagen deposition on Day 28 (see Figure E2). These results further support a critical role for p80 in mediating virus-driven allograft dysfunction.
Inhibition or Blockade of p80 Function Attenuates Allograft Dysfunction
To test the possibility that epithelial cell p80 may be critical in mediating allograft dysfunction in the absence of viral infection, we initially employed a loss-of-function approach using donor tracheas from IL-12 p40−/− mice that are unable to generate p80. In the IL-12 p40−/− allografts we observed preserved epithelial integrity, decreased macrophage accumulation, minimal collagen deposition, and abrogated lumen obliteration (Figures 4B–4D, second columns; and see Figure E3). Similar to the results with IL-12 p40−/− donor allografts, allogeneic transplantation into an 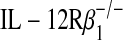 recipient mouse also attenuated this dysfunction (Figures 4B–4D, third columns). To exclude the possibility that developmental or compensatory alterations in the IL-12 p40−/− or
recipient mouse also attenuated this dysfunction (Figures 4B–4D, third columns). To exclude the possibility that developmental or compensatory alterations in the IL-12 p40−/− or 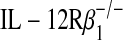 mouse strain may have contributed to this phenotype, we transiently blocked p80 by administering anti–IL-12 p40 neutralizing antibody to the recipient mouse. Relative to administration of control IgG antibody, we observed that IL-12 p40 neutralization after allogeneic transplantation attenuated allograft dysfunction (Figures 4B–4D, fifth columns). Taken together, blockade of p80 or inhibition of its function during allogeneic transplantation attenuated macrophage accumulation as well as allograft dysfunction.
mouse strain may have contributed to this phenotype, we transiently blocked p80 by administering anti–IL-12 p40 neutralizing antibody to the recipient mouse. Relative to administration of control IgG antibody, we observed that IL-12 p40 neutralization after allogeneic transplantation attenuated allograft dysfunction (Figures 4B–4D, fifth columns). Taken together, blockade of p80 or inhibition of its function during allogeneic transplantation attenuated macrophage accumulation as well as allograft dysfunction.
Figure 4.

Inhibition or blockade of p80 function attenuates allograft dysfunction. In (A–D), wild-type C57BL/6J (C) or C57BL/6J IL-12 p40−/− (p40−/− C) donor tracheas underwent transplantation into wild-type BALB/cJ (B) or BALB/cJ IL-12 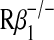 (Rβ1−/− B) mice. Additional cohorts of wild-type BALB/cJ recipients received IgG or anti–IL-12 p40 neutralizing antibody (α-p40 Ab, 1.0 mg administered intraperitoneally) every 4 d beginning 1 d before transplantation. At 14 or 28 d post-transplantation, grafts underwent H&E staining (A), quantification of the percentage of denuded BM (B), immune cell accumulation (C), and hydroxyproline (D) as described in Figure 1. Values represent means ± SEM (n = 4 or 5). *p < 0.05 compared with C → B.
(Rβ1−/− B) mice. Additional cohorts of wild-type BALB/cJ recipients received IgG or anti–IL-12 p40 neutralizing antibody (α-p40 Ab, 1.0 mg administered intraperitoneally) every 4 d beginning 1 d before transplantation. At 14 or 28 d post-transplantation, grafts underwent H&E staining (A), quantification of the percentage of denuded BM (B), immune cell accumulation (C), and hydroxyproline (D) as described in Figure 1. Values represent means ± SEM (n = 4 or 5). *p < 0.05 compared with C → B.
Overexpression of p80 Accelerates Allograft Dysfunction
To further implicate p80 as an innate effector of allograft dysfunction, we next examined transplantation conditions with allografts that overexpressed p80 in donor airway epithelial cells. To achieve this, we generated a C57BL/6J transgenic mouse strain that used the rat Clara cell secretory protein promoter to selectively drive murine IL-12 p40 expression in airway epithelial cells (29). Allogeneic transplantation using donor tracheas from these transgenic mice that overexpressed p80 increased macrophage accumulation and tended to increase the degree of epithelial cell injury and collagen deposition (Figures 5B–5D, second columns). A second approach to examine the role of p80 overexpression used donor tracheas from the IL-12 p35−/− mouse. Our previous results have demonstrated that IL-12 p35 deficiency results in airway epithelial cell p80 overexpression under inflammatory conditions such as respiratory viral infection (10). In agreement with these results, allogeneic transplantation with donor tracheas from IL-12 p35−/− mice resulted in enhanced p80 expression (64.4 ± 4.6 pg/ml, mean ± SEM) and accelerated allograft dysfunction (Figures 5B–5D, third columns). To determine whether this accelerated phenotype relied on recipient expression of IL-12Rβ1, we performed allogeneic transplantation with IL-12 p35−/− donor tracheas and 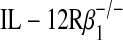 recipient mice. Loss of IL-12Rβ1 expression in the recipient mice attenuated allograft dysfunction, indicating that the phenotype in IL-12 p35−/− allografts was mediated, at least in part, by expression of IL-12 family members (Figures 5B–5D, fourth columns).
recipient mice. Loss of IL-12Rβ1 expression in the recipient mice attenuated allograft dysfunction, indicating that the phenotype in IL-12 p35−/− allografts was mediated, at least in part, by expression of IL-12 family members (Figures 5B–5D, fourth columns).
Figure 5.

Overexpression of p80 accelerates allograft dysfunction. In (A–D), wild-type C57BL/6J (C), Clara cell secretory protein (CCSP)–p40 transgenic C57BL/6J (Tg-p40 C), or IL-12 p35−/− C57 (p35−/− C) donor tracheas underwent transplantation into wild-type BALB/cJ (B) or BALB/cJ 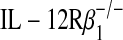 (Rβ1−/− B) mice. At 14 or 28 d post-transplantation, grafts underwent H&E staining (A), quantification of the percentage of denuded basement membrane (B), immune cell accumulation (C), and hydroxyproline (D) as described in Figure 1. Values represent means ± SEM (n = 4 or 5). *p < 0.05 compared with C → B.
(Rβ1−/− B) mice. At 14 or 28 d post-transplantation, grafts underwent H&E staining (A), quantification of the percentage of denuded basement membrane (B), immune cell accumulation (C), and hydroxyproline (D) as described in Figure 1. Values represent means ± SEM (n = 4 or 5). *p < 0.05 compared with C → B.
Recipient Alloreactivity Is Unchanged under Transplantation Conditions that Overexpress or Block p80
To determine whether T-cell alloreactivity was altered under transplantation conditions that either enhanced p80 expression (viral infection, p40 transgenic, and IL-12 p35−/− donors) or blocked p80 function (IL-12 Rβ1−/− recipient) we performed a mixed lymphocyte reaction with recipient splenocytes harvested after transplantation. Compared with alloreactivity measured in naive spleen cells, we observed a significant increase in alloreactivity from splenocytes in wild-type recipients that had undergone allogeneic heterotopic tracheal transplantation. However, this allogeneic response was not different under transplantation conditions that either enhanced p80 expression or blocked its function (see Figure E4).
Enhanced Expression of Epithelial p80 in Human Transplant Recipients Correlates with Transplant Bronchitis and Accumulation of Macrophages
To test the possibility that p80 may also mediate virus-driven allograft dysfunction in human lung transplant recipients, we performed p40 immunohistochemical analysis of allograft tissue obtained at the time of a concurrent respiratory viral infection (n = 10). In contrast to absent p40 immunolabeling in normal nontransplanted individuals (10), seven of these transplant allografts demonstrated p40 immunolabeling that colocalized to airway epithelial cells (Figure 6A, left column). Interestingly, all five of the specimens with a histologic diagnosis of transplant bronchitis demonstrated epithelial cell p40 expression. To further evaluate p40 expression in biopsy specimens with a larger number of airways containing preserved epithelium we immunolabeled open lung biopsies from five recipients with transplant bronchitis, and again demonstrated an increase in epithelial cell p40 expression in all of these specimens (Figure 6A, right column).
Figure 6.

Enhanced expression of epithelial p80 in human recipients with transplant bronchitis. In (A), lung tissue from recipients with allograft dysfunction due to respiratory viral infection (n = 10, left) and transplant bronchitis (n = 5, right) underwent detection of IL-12 p40 as described in Figure 3 (bottom row). IgG gave no signal above background (top row), arrows indicate immune cells, and representative photomicrographs are shown. Scale bar: 20 μm. In (B–D), BAL fluid from nontransplanted normal individuals (NL, n = 10), normal transplanted recipients (Transplant NL, n = 16), transplant recipients with acute rejection only (n = 10), transplant recipients with cytomegalovirus only (CMV, n = 9), and transplant recipients with bronchitis only (n = 12) was concentrated 10-fold and duplicate samples were used to measure p80/p40 (B), IL-12 (C), and IL-23 (D) by ELISA. Values represent means ± SEM. *p < 0.05 compared with normal transplanted cohort.
To determine which IL-12 family members were generated from this epithelial cell p40 expression we simultaneously assayed BAL fluid for the IL-12 family members in prospectively collected BAL fluid. In addition, to determine whether specific alterations in IL-12 family member expression correlated with certain types of allograft dysfunction we segregated subjects into five cohorts: normal individuals (Nontransplant Normal), transplant recipients without acute rejection, infection, or bronchitis (Transplant Normal), recipients with only acute rejection (Acute Rejection), recipients with only CMV infection (CMV), and recipients with only transplant bronchitis (Bronchitis). Demographic characteristics were indistinguishable in all cohorts (Table 1). Interestingly, BAL concentrations of p80/p40 were selectively increased in the cohort with transplant bronchitis. As seen in the mouse transplant model, the increase in epithelial cell p40 expression resulted in a specific increase in BAL p80/p40 because the levels of IL-12 and IL-23 were not increased (Figures 6B–6D). Consistent with the observation that p80 is a macrophage chemoattractant, in the subjects with transplant bronchitis we observed a significant correlation between BAL fluid concentrations of p80 and accumulation of macrophages, but not of neutrophils or lymphocytes (Figure 7).
TABLE 1.
CHARACTERISTICS OF STUDY POPULATION
| Nontransplant Normal (n = 10) | Transplant Normal (n = 16) | Acute Rejection (n = 10) | CMV (n = 9) | Bronchitis (n = 12) | p Value | |
|---|---|---|---|---|---|---|
| Female, n (%) | 8 (80) | 6 (38) | 4 (40) | 4 (44) | 7 (58) | 0.25 |
| Age at transplantation, yr (mean ± SD) | 34.3 ± 8.7 | 48.6 ± 13.6 | 45.3 ± 17.5 | 55.0 ± 14.5 | 45.3 ± 17.5 | 0.07 |
| Pretransplantation disease, n (%) | 0.15 | |||||
| COPD/α1-AT | 8 (50) | 7 (70) | 4 (44) | 3 (25) | ||
| CF/bronchiectasis | 5 (31) | 3 (30) | 2 (22) | 8 (66) | ||
| Other | 3 (19) | 3 (33) | 1 (8) | |||
| Type of transplant, n (%) | 0.56 | |||||
| Single | 1 (6) | 1 (11) | 1 (8) | |||
| Bilateral | 15 (94) | 10 (100) | 8 (89) | 11 (92) | ||
| FEV1 at bronchoscopy, L (mean ± SD) | 3.37 ± 1.13 | 2.80 ± 0.87 | 2.43 ± 0.96 | 2.39 ± 0.98 | 2.16 ± 0.78 | 0.07 |
| Time from transplantation to bronchoscopy, d, median (interquartile range) | 182 (55–502) | 76 (46–109) | 88 (57–255) | 226 (54–723) | 0.21 |
Definition of abbreviations: α1-AT = α1-antitrypsin deficiency; CF = cystic fibrosis; CMV = cytomegalovirus; COPD = chronic obstructive pulmonary disease.
Figure 7.

p80 expression in human recipients with transplantation bronchitis correlates with BAL fluid macrophage accumulation. In (A–C), BAL p80 concentrations from transplant recipients with bronchitis were correlated with BAL neutrophils (A), macrophages (B), and lymphocytes (C). r = correlation coefficient.
DISCUSSION
Our studies in mice and humans demonstrate that an enhanced innate epithelial cell response occurs during conditions associated with subsequent chronic allograft dysfunction. We observed that concurrent viral infection and allogeneic transplantation resulted in an exaggerated innate epithelial response characterized by a synergistic increase in epithelial cell injury and expression of IL-12 p40. The induction of epithelial IL-12 p40 resulted in a selective increase in p80 and these levels correlated with macrophage accumulation. In addition, modification of the epithelial response using genetically modified mouse strains that blocked or enhanced p80 function abrogated or accelerated chronic allograft dysfunction, suggesting this epithelial cell response is an initial event that may drive the chronic phenotype. Extension of these findings to human subjects demonstrated an enhanced epithelial cell response in recipients with community-acquired respiratory viral infection and transplant bronchitis, specific conditions that are also associated with subsequent allograft dysfunction. Taken together, we propose that the innate epithelial cell protein p80 is an effector protein that serves to initiate biologic processes that culminate in chronic allograft dysfunction. Identification of a role for epithelial p80 expression in the development of chronic allograft dysfunction provides novel insight into disease pathogenesis and provides a potential target for studies aimed at modifying disease progression.
We have previously identified respiratory viral infection as a distinct risk factor for chronic allograft dysfunction and death in lung transplant recipients. The current studies identify epithelial cell expression of p40 during respiratory viral infection, and also suggest that p80 may mediate virus-driven allograft dysfunction. Identification of the virus-driven mediators of subsequent allograft dysfunction is critical because evidence indicates that the true burden of viral infections on chronic allograft dysfunction is still underappreciated. Previous studies have captured recipients with viral infections retrospectively and therefore may have underestimated the true incidence of viral infection (7, 34). Prospective protocols to track viral symptoms and identify virus with polymerase chain reaction–based assays have demonstrated a much higher incidence of respiratory viral infection (0.66–1.56 infections per patient-year of follow-up) (35–37). Likewise, the detection of viral genomes in human cardiac and renal allografts has been increased using polymerase chain reaction–based technology, and the detection of viral genomes in these allografts is also associated with chronic allograft dysfunction (38–41). Accordingly, viral infection of lung and other solid organ allografts has been increasingly recognized as a risk factor for chronic dysfunction, and identification of the proteins, such as p80, that mediate this postviral allograft dysfunction provides novel insight into the mediators of disease progression.
We have modeled postviral allograft dysfunction in humans using an animal model and demonstrated that respiratory viral infection in the setting of allogeneic transplantation accelerated chronic allograft dysfunction. Although this lung transplant model is limited by the subcutaneous placement of the allograft, it does provide a reductionist model to examine airway epithelial cell injury response gene induction in an allogeneic setting. We demonstrated a synergistic increase in p80 during allogeneic transplant and viral infection, thus identifying p80 as a potential mediator of this phenotype. This synergistic increase in p80 expression may result from a direct effect of the virus on the epithelial cells or indirectly through the virus-driven induction of additional cytokines that may then induce p80. For example, viral infection may directly increase p80 secretion by enhancing known transcriptional activators of the IL-12 p40 gene (i.e., nuclear factor-κB, Ets-2, CCAAT enhancer–binding protein-β, PU.1, ICSBP, AP-1, IRF-1, and p300) or by inhibiting transcriptional repressors (i.e., histone deacetylase-1 and erythroid Kruppel-like factor). Alternatively, the viral infection may directly enhance p80 secretion by increasing IL-12 p40 mRNA stability as has been demonstrated for CCL5 (RANTES [regulated upon activation, T-cell expressed and secreted]), an additional chemokine that is induced by paramyxoviral infection (42). Whether the synergistic increase in p80 results from viral induction of additional cytokines and or alterations in mRNA stability is currently being investigated.
The selective expression of epithelial cell p80 in virus-driven allograft dysfunction and in human recipients with transplant bronchitis indicates that specific mechanisms exist which regulate the secretion of various IL-12 family members. Simultaneous measurement of the IL-12 family members in BAL fluid specimens allowed for direct comparisons between the family members, and demonstrated a selective increase in p80 but not in IL-12 or IL-23. In addition to allograft transplantation, we have also noted this same pattern of expression in mouse viral bronchiolitis and human subjects with asthma (10). Accordingly, we now postulate that epithelial cells may be intrinsically programmed for p40 homodimerization. One possibility that may favor p40 homodimerization is a relatively high level of p40 monomer with or without a low abundance of additional p40-binding partners, such as p35 and p19. Although the biochemical mechanism resulting in p80 secretion remains unknown, the selective expression of epithelial cell p80 in these disease states suggests that p80 possesses unique biologic properties, and this is in agreement with our previous observations that demonstrated p80, but not other IL-12 family members, is a macrophage chemoattractant (14).
Although our results suggest that p80 is the IL-12 family member that mediates chronic allograft dysfunction, it is possible that additional IL-12 p40–containing family members may contribute to the observed phenotypes. Because the p40 protein is common to multiple IL-12 family members (e.g., p80, IL-12, IL-23, and p40 monomer), the IL-12 p40–deficient mouse strain is deficient in all these proteins. Likewise, IL-12Rβ1 is required for appropriate IL-12 family member signaling, so that the IL-12Rβ1–deficient stain is incapable of responding to p80 as well as the remaining family members. Accordingly, phenotypes observed in experiments with these mice may also be attributed to manipulation of IL-12 and IL-23 as well as p80. However, the experiment using the p40 transgenic strain with selective overexpression of p80, but not of IL-12 or IL-23, suggests these cytokines are not necessary to mediate chronic allograft dysfunction. Furthermore, for IL-12 or IL-23 to contribute to the observed phenotype the levels of these cytokines would have to increase or decrease. Because we observed no significant changes in IL-12 or IL-23 levels during allograft dysfunction (in mice or humans), we believe it is unlikely that IL-12 or IL-23 is sufficient to mediate the observed phenotype.
We observed a relationship between p80 expression, macrophage accumulation in the allograft, and subsequent allograft dysfunction. This cascade of events places macrophage recruitment as a central process in chronic lung allograft dysfunction and is in agreement with previous studies (16–19). Because we have previously demonstrated that p80 does not activate macrophage phagocytosis or oxidative burst (14), we suggest the macrophage may promote allograft dysfunction by amplifying allograft inflammation. In that regard, p80-dependent induction of inducible nitric oxide synthetase and tumor necrosis factor-α in macrophages has been reported previously (43, 44). Additional studies in our laboratory indicate that p80 can induce expression of the chemokines CCL5 (RANTES) and IL-8 in primary culture murine macrophages (L. Yan and M.J. Walter, unpublished results). Of note, all four of these immunomodulatory proteins have also been implicated in chronic lung allograft dysfunction. Taken together, p80 may amplify allograft inflammation through its dual capacity to recruit macrophages and to induce their secretion of additional proinflammatory proteins.
Our data support a critical role for p80-dependent macrophage accumulation in mediating allograft dysfunction. To examine whether p80 also altered additional components of the alloimmune response, we measured T-cell alloimmunity under conditions without and with altered p80 expression. We did not observe an association between degree of alloimmunity, p80 expression, and allograft dysfunction. For example, we failed to demonstrate increased alloimmunity under conditions of enhanced p80 expression and accelerated allograft dysfunction. Likewise, we did not document decreased alloimmunity under conditions with blockade of p80 function and attenuated allograft dysfunction. Although this suggests p80 expression did not alter alloimmunity, we cannot exclude the possibility that p80-dependent effects on additional lymphocyte functions, such as cytokine secretion, may have partially contributed to the observed phenotypes.
Our findings support the proposal that an enhanced innate epithelial cell response is an initial event in chronic lung allograft dysfunction (8). Consistent with our findings, others have also demonstrated epithelial cell expression of inflammatory mediators during allograft dysfunction. For example, epithelial cell production of monocyte chemoattractant protein-1, IL-8, and inducible nitric oxide synthase has been demonstrated in recipients with acute vascular rejection and/or allograft dysfunction (16, 45–47). In addition, epithelial cell upregulation of intercellular adhesion molecule-1 as well as class I and II major histocompatibility complex antigens has been observed in some (48–50), but not all, human studies (51, 52). To our knowledge, the current study is the first to identify a specific pattern of an epithelial cell inflammatory protein in post–lung transplant bronchitis and the first to demonstrate a transplant-specific induction of p80, relative to other IL-12 family members. This epithelial response was unique to viral infection (in mice) and transplant bronchitis (in humans) but was absent in other conditions such as acute rejection and CMV infection. These results, along with a study that identified different gene transcript profiles between subjects with transplant bronchitis and acute rejection (53), support the concept that distinct pathophysiologic mechanisms (and mediators) are likely responsible for mediating these various conditions.
In summary, we have identified epithelial cell expression of the macrophage chemoattractant p80 in conditions that are associated with chronic lung allograft dysfunction (i.e., viral infection and bronchitis). These findings support a role for p80 as an innate epithelial cell effector that mediates chronic lung allograft dysfunction, and because epithelial cell injury, viral infection, and macrophage accumulation are observed in renal, liver, and cardiac allograft dysfunction, we propose that p80 may also mediate allograft dysfunction after transplantation of these organs.
Supplementary Material
Acknowledgments
The authors thank G. Fan, Q. Yan, L. Rikimaru, H. Chen, and R. McCarthy for technical assistance; W. Black for statistical assistance; J. Whitsett for providing the rat CCSP promoter; S. Brody and D. Schuster for helpful discussion and review of the manuscript; and J. Fassler, T. Francescon, C. Miller, L. Roldan, and G. Richardson for assistance in chart reviews.
Supported by National Institutes of Health grants R01-HL71947 and R01-HL083894 (M.J.W.), P50-56,419 and R01-69149 (M.C.), and 5M01RR00036 to the general clinical research center.
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1164/rccm.200512-1886OC on May 25, 2006
Conflict of Interest Statement: None of the authors has a financial relationship with a commercial entity that has an interest in the subject of the manuscript.
References
- 1.Trulock EP, Edwards LB, Taylor DO, Boucek MM, Keck BM, Hertz MI. Registry of the International Society for Heart and Lung Transplantation: twenty-second official adult lung and heart-lung transplant report—2005. J Heart Lung Transplant 2005;24:956–967. [DOI] [PubMed] [Google Scholar]
- 2.Yousem SA, Berry GJ, Cagle PT, Chamberlain D, Husain AN, Hruban RH, Marchevsky A, Ohori NP, Ritter J, Stewart S, et al.; Lung Rejection Study Group. Revision of the 1990 working formulation for the classification of pulmonary allograft rejection. J Heart Lung Transplant 1996;15:1–15. [PubMed] [Google Scholar]
- 3.Neuringer IP, Chalermskulrat W, Aris R. Obliterative bronchiolitis or chronic lung allograft rejection: a basic science review. J Heart Lung Transplant 2005;24:3–19. [DOI] [PubMed] [Google Scholar]
- 4.Estenne M, Maurer JR, Boehler A, Egan JJ, Frost A, Hertz M, Mallory GB, Snell GI, Yousem S. Bronchiolitis obliterans syndrome 2001: an update of the diagnostic criteria. J Heart Lung Transplant 2002;21:297–310. [DOI] [PubMed] [Google Scholar]
- 5.Sharples LD, McNeil K, Stewart S, Wallwork J. Risk factors for bronchiolitis obliterans: a systemic review of recent publications. J Heart Lung Transplant 2002;21:271–281. [DOI] [PubMed] [Google Scholar]
- 6.Estenne M, Hertz MI. Bronchiolitis obliterans after human lung transplantation. Am J Respir Crit Care Med 2002;166:440–444. [DOI] [PubMed] [Google Scholar]
- 7.Khalifah AP, Hachem RR, Chakinala MM, Schechtman KB, Patterson GA, Schuster DP, Mohanakumar T, Trulock EP, Walter MJ. Respiratory viral infections are a distinct risk for bronchiolitis obliterans syndrome and death. Am J Respir Crit Care Med 2004;170:181–187. [DOI] [PubMed] [Google Scholar]
- 8.Chakinala MM, Walter MJ. Community acquired respiratory viral infections after lung transplantation: clinical features and long-term consequences. Semin Thorac Cardiovasc Surg 2004;16:342–349. [DOI] [PubMed] [Google Scholar]
- 9.Strieter RM, Belperio JA, Keane MP. Host innate defenses in the lung: the role of cytokines. Curr Opin Infect Dis 2003;16:193–198. [DOI] [PubMed] [Google Scholar]
- 10.Walter MJ, Kajiwara N, Karanja P, Castro M, Holtzman MJ. IL-12 p40 production by barrier epithelial cells during airway inflammation. J Exp Med 2001;193:339–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walter M, Holtzman M. Epithelial production of IL-12 p80 during viral infection and asthma supports an altered paradigm for airway inflammation. In: Eissa N, Huston DP, editors. Lung biology in health and disease: therapeutic targets in airway inflammation. New York: Marcel Dekker; 2003. pp. 535–561.
- 12.Trinchieri G, Pflanz S, Kastelein RA. The IL-12 family of heterodimeric cytokines: new players in the regulation of T cell responses. Immunity 2003;19:641–644. [DOI] [PubMed] [Google Scholar]
- 13.Langrish CL, McKenzie BS, Wilson NJ, de Waal Malefyt R, Kastelein RA, Cua DJ. IL-12 and IL-23: master regulators of innate and adaptive immunity. Immunol Rev 2004;202:96–105. [DOI] [PubMed] [Google Scholar]
- 14.Russell TD, Yan Q, Fan G, Khalifah AP, Bishop DK, Brody SL, Walter MJ. IL-12 p40 homodimer–dependent macrophage chemotaxis and respiratory viral inflammation are mediated through IL-12 receptor β1. J Immunol 2003;171:6866–6874. [DOI] [PubMed] [Google Scholar]
- 15.Ha SJ, Lee CH, Lee SB, Kim CM, Jang KL, Shin HS, Sung YC. A novel function of IL-12p40 as a chemotactic molecule for macrophages. J Immunol 1999;163:2902–2908. [PubMed] [Google Scholar]
- 16.Belperio JA, Keane MP, Burdick MD, Lynch JP III, Xue YY, Berlin A, Ross DJ, Kunkel SL, Charo IF, Strieter RM. Critical role for the chemokine MCP-1/CCR2 in the pathogenesis of bronchiolitis obliterans syndrome. J Clin Invest 2001;108:547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Magnan A, Mege JL, Reynaud M, Thomas P, Capo C, Garbe L, Meric B, Badier M, Bongrand P, Viard L, et al.; Marseille and Montreal Lung Transplantation Group. Monitoring of alveolar macrophage production of tumor necrosis factor-α and interleukin-6 in lung transplant recipients. Am J Respir Crit Care Med 1994;150:684–689. [DOI] [PubMed] [Google Scholar]
- 18.Ward C, Snell GI, Zheng L, Orsida B, Whitford H, Williams TJ, Walters EH. Endobronchial biopsy and bronchoalveolar lavage in stable lung transplant recipients and chronic rejection. Am J Respir Crit Care Med 1998;158:84–91. [DOI] [PubMed] [Google Scholar]
- 19.Oyaizu T, Okada Y, Shoji W, Matsumura Y, Shimada K, Sado T, Sato M, Kondo T. Reduction of recipient macrophages by gadolinium chloride prevents development of obliterative airway disease in a rat model of heterotopic tracheal transplantation. Transplantation 2003;76:1214–1220. [DOI] [PubMed] [Google Scholar]
- 20.de Mattos AM, Meyer MM, Norman DJ, Bennett WM, Sprague J, Bakke AC. Interleukin-12 p40 m-RNA expression in human kidney allograft biopsies. Transpl Immunol 1997;5:199–203. [DOI] [PubMed] [Google Scholar]
- 21.Thai NL, Li Y, Fu F, Qian S, Demetris AJ, Duquesnoy RJ, Fung JJ. Interleukin-2 and interleukin-12 mediate distinct effector mechanisms of liver allograft rejection. Liver Transpl Surg 1997;3:118–129. [DOI] [PubMed] [Google Scholar]
- 22.Nakamura H, Komatsu K, Ayaki M, Kawamoto S, Murakami M, Uoshima N, Yagi T, Hasegawa T, Yasumi M, Karasuno T, et al. Serum levels of soluble IL-2 receptor, IL-12, IL-18, and IFN-γ in patients with acute graft-versus-host disease after allogeneic bone marrow transplantation. J Allergy Clin Immunol 2000;106:S45–S50. [DOI] [PubMed] [Google Scholar]
- 23.Yang YG, Dey B, Sergio JJ, Sykes M. Interleukin-12 prevents severe acute graft-versus-host disease (GVHD) and GVHD-associated immune dysfunction in a fully major histocompatibility complex haplotype–mismatched murine bone marrow transplantation model. Transplantation 1997;64:1343–1352. [DOI] [PubMed] [Google Scholar]
- 24.Sykes M, Pearson DA, Taylor PA, Szot GL, Goldman SJ, Blazar BR. Dose and timing of interleukin (IL)-12 and timing and type of total-body irradiation: effects on graft-vs.-host disease inhibition and toxicity of exogenous IL-12 in murine bone marrow transplant recipients. Biol Blood Marrow Transplant 1999;5:277–284. [DOI] [PubMed] [Google Scholar]
- 25.Piccotti JR, Chan SY, Li K, Eichwald EJ, Bishop DK. Differential effects of IL-12 receptor blockade with IL-12 p40 homodimer on the induction of CD4+ and CD8+ IFN-γ–producing cells. J Immunol 1997;158:643–648. [PubMed] [Google Scholar]
- 26.Piccotti KL Jr, Chan SY, Ferrante J, Magram J, Eichwald EJ, Bishop DK. Alloantigen-reactive Th1 development in IL-12–deficient mice. J Immunol 1998;160:1132–1138. [PubMed] [Google Scholar]
- 27.Hertz M, Jessurun J, King MB, Savik SK, Murray JJ. Reproduction of the obliterative bronchiolitis lesion after heterotopic transplantation of mouse airways. Am J Pathol 1993;142:1945–1951. [PMC free article] [PubMed] [Google Scholar]
- 28.Walter MJ, Morton JD, Kajiwara N, Agapov E, Holtzman MJ. Viral induction of a chronic asthma phenotype and genetic segregation from the acute response. J Clin Invest 2002;110:165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stripp BR, Sawaya PL, Luse DS, Wikenheiser KA, Wert SE, Huffman JA, Lattier DL, Singh G, Katyal SL, Whitsett JA. Cis-acting elements that confer lung epithelial cell expression of the CC10 gene. J Biol Chem 1992;267:14703–14712. [PubMed] [Google Scholar]
- 30.Khalifah AP, Hachem RR, Chakinala MM, Yusen RD, Aloush A, Patterson GA, Mohanakumar T, Trulock EP, Walter MJ. Minimal acute rejection after lung transplantation: a risk for bronchiolitis obliterans syndrome. Am J Transplant 2005;5:2022–2030. [DOI] [PubMed] [Google Scholar]
- 31.Hele DJ, Yacoub MH, Belvisi MG. The heterotopic tracheal allograft as an animal model of obliterative bronchiolitis. Respir Res 2001;2:169–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neuringer IP, Aris RM, Burns KA, Bartolotta TL, Chalermskulrat W, Randell SH. Epithelial kinetics in mouse heterotopic tracheal allografts. Am J Transplant 2002;2:410–419. [DOI] [PubMed] [Google Scholar]
- 33.Neuringer IP, Mannon RB, Coffman TM, Parsons M, Burns K, Yankaskas JR, Aris RM. Immune cells in a mouse airway model of obliterative bronchiolitis. Am J Respir Cell Mol Biol 1998;19:379–386. [DOI] [PubMed] [Google Scholar]
- 34.Billings JL, Hertz MI, Savik K, Wendt CH. Respiratory viruses and chronic rejection in lung transplant recipients. J Heart Lung Transplant 2002;21:559–566. [DOI] [PubMed] [Google Scholar]
- 35.Bridges ND, Spray TL, Collins MH, Bowles NE, Towbin JA. Adenovirus infection in the lung results in graft failure after lung transplantation. J Thorac Cardiovasc Surg 1998;116:617–623. [DOI] [PubMed] [Google Scholar]
- 36.Weinberg A, Zamora MR, Li S, Torres F, Hodges TN. The value of polymerase chain reaction for the diagnosis of viral respiratory tract infections in lung transplant recipients. J Clin Virol 2002;25:171–175. [DOI] [PubMed] [Google Scholar]
- 37.Kumar D, Erdman D, Keshavjee S, Peret T, Tellier R, Hadjiliadis D, Johnson G, Ayers M, Siegal D, Humar A. Clinical impact of community-acquired respiratory viruses on bronchiolitis obliterans after lung transplant. Am J Transplant 2005;5:2031–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shirali GS, Ni J, Chinnock RE, Johnston JK, Rosenthal GL, Bowles NE, Towbin JA. Association of viral genome with graft loss in children after cardiac transplantation. N Engl J Med 2001;344:1498–1503. [DOI] [PubMed] [Google Scholar]
- 39.Douche-Aourik F, Bourlet T, Mosnier JF, Jacques J, Decoene C, Stankowiak C, Pozzetto B, Andreoletti L. Association between enterovirus endomyocardial infection and late severe cardiac events in some adult patients receiving heart transplants. J Med Virol 2005;75:47–53. [DOI] [PubMed] [Google Scholar]
- 40.Aiello FB, Calabrese F, Rigotti P, Furian L, Marino S, Cusinato R, Valente M. Acute rejection and graft survival in renal transplanted patients with viral diseases. Mod Pathol 2004;17:189–196. [DOI] [PubMed] [Google Scholar]
- 41.Hirsch HH, Steiger J. Polyomavirus BK. Lancet Infect Dis 2003;3:611–623. [DOI] [PubMed] [Google Scholar]
- 42.Koga T, Look DC, Taguchi M, Holtzman MJ. Virus-inducible expression of a host chemokine gene relies on replication-linked mRNA stabilization. Proc Natl Acad Sci USA 1999;96:5680–5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pahan K, Sheikh FG, Liu X, Hilger S, McKinney M, Petro TM. Induction of nitric-oxide synthase and activation of NF-κB by interleukin-12 p40 in microglial cells. J Biol Chem 2001;276:7899–7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jana M, Dasgupta S, Saha RN, Liu X, Pahan K. Induction of tumor necrosis factor-α (TNF-α) by interleukin-12 p40 monomer and homodimer in microglia and macrophages. J Neurochem 2003;86:519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elssner A, Jaumann F, Dobmann S, Behr J, Schwaiblmair M, Reichenspurner H, Furst H, Briegel J, Vogelmeier C; Munich Lung Transplant Group. Elevated levels of interleukin-8 and transforming growth factor-β in bronchoalveolar lavage fluid from patients with bronchiolitis obliterans syndrome: proinflammatory role of bronchial epithelial cells. Transplantation 2000;70:362–367. [DOI] [PubMed] [Google Scholar]
- 46.Gabbay E, Walters EH, Orsida B, Whitford H, Ward C, Kotsimbos TC, Snell GI, Williams TJ. Post-lung transplant bronchiolitis obliterans syndrome (BOS) is characterized by increased exhaled nitric oxide levels and epithelial inducible nitric oxide synthase. Am J Respir Crit Care Med 2000;162:2182–2187. [DOI] [PubMed] [Google Scholar]
- 47.Mason NA, Springall DR, Pomerance A, Evans TJ, Yacoub MH, Polak JM. Expression of inducible nitric oxide synthase and formation of peroxynitrite in posttransplant obliterative bronchiolitis. J Heart Lung Transplant 1998;17:710–714. [PubMed] [Google Scholar]
- 48.Milne DS, Gascoigne AD, Wilkes J, Sviland L, Ashcroft T, Malcolm AJ, Corris PA. MHC class II and ICAM-1 expression and lymphocyte subsets in transbronchial biopsies from lung transplant recipients. Transplantation 1994;57:1762–1766. [PubMed] [Google Scholar]
- 49.Taylor PM, Rose ML, Yacoub MH. Expression of MHC antigens in normal human lungs and transplanted lungs with obliterative bronchiolitis. Transplantation 1989;48:506–510. [DOI] [PubMed] [Google Scholar]
- 50.Devouassoux G, Pison C, Drouet C, Pin I, Brambilla C, Brambilla E. Early lung leukocyte infiltration, HLA and adhesion molecule expression predict chronic rejection. Transpl Immunol 2001;8:229–236. [DOI] [PubMed] [Google Scholar]
- 51.Shreeniwas R, Schulman LL, Narasimhan M, McGregor CC, Marboe CC. Adhesion molecules (E-selectin and ICAM-1) in pulmonary allograft rejection. Chest 1996;110:1143–1149. [DOI] [PubMed] [Google Scholar]
- 52.Hasegawa S, Ritter JH, Patterson A, Ockner DM, Sawa H, Mohanakumar T, Cooper JD, Wick MR. Expression of intercellular and vascular cell adhesion molecules and class II major histocompatibility antigens in human lungs: lack of influence by conditions of organ preservation. J Heart Lung Transplant 1995;14:897–905. [PubMed] [Google Scholar]
- 53.Xu X, Golden JA, Dolganov G, Jones KD, Donnelly S, Weaver T, Caughey GH. Transcript signatures of lymphocytic bronchitis in lung allograft biopsy specimens. J Heart Lung Transplant 2005;24:1055–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.