

Abstract
Many antigens recognized by tumor-reactive cytotoxic CD8+ T cells are self-antigens. Tyrosinase-related protein 2 (TRP-2) is a melanogenic enzyme expressed by both melanocytes and melanomas that is reported to be a candidate melanoma rejection antigen. To study the role of self-reactive CD8+ T cells in tumor immunity and autoimmunity, we generated mice which bear a T cell receptor transgene (TCR Tg) specific for the TRP-2(180-188) epitope. TRP-2 TCR Tg mice did not spontaneously develop depigmentation despite systemic expression of TRP-2 in the skin. Peripheral T cells from these TCR Tg mice exhibited a naïve phenotype and proliferated in response to TRP-2 in vitro. In addition, transfer of in vitro-activated Tg T cells reduced B16 pulmonary tumor burden, but not subcutaneous tumors. We next sought to determine the in vivo responses of the Tg T cells to endogenous and tumor-derived TRP-2. Adoptive transfer of naïve TCR Tg T cells into wild-type C57BL/6 mice, in combination with a TRP-2-pulsed dendritic cell vaccine, induced proliferation of the Tg T cells and resulted in migration of the Tg T cells into a subcutaneous B16 melanoma tumor. Although these tumor-infiltrating Tg T cells remained reactive against TRP-2, they did not reduce growth of the primary subcutaneous tumor; similarly, these in vivo-primed effector cells had no significant effect on growth of pulmonary nodules. These data demonstrate that despite in vivo priming, tumor-infiltrating T cells may fail to reduce tumor burden. Determining the basis for the inability of the tumor micro-environment to sustain effective anti-tumor responses will be critical for designing newer, more potent anti-tumor immunotherapies.
Keywords: melanoma, T cells, adoptive immunotherapy, T cell receptor transgene
Introduction
Identification of tumor-associated antigens (TAAs) that can be recognized by cytotoxic T cells (CTLs) has been a major advancement in understanding tumor immunity. CTLs that recognize TAAs like Melan-A (1), gp-100 (2), tyrosinase (3), tyrosinase-related protein-1 (TRP-1) (4), and tyrosinase-related protein-2 (TRP-2) (5) have been isolated from melanoma patients. These antigens are expressed by both the melanoma as well as the normal melanocytes and are referred to as melanocyte differentiation antigens (6).
Because melanocyte differentiation antigens are self-antigens, immune responses against them are regulated by central and peripheral tolerance mechanisms. Generally, highly self-reactive CD8+ T cells are deleted during thymic development (7-10). However, deletion of self-reactive CD8+ T cells is incomplete and self reactive CD8+ T cells can escape thymic selection (11, 12) and persist in the periphery under the control of several regulatory mechanisms, including anergy, ignorance, suppression and deletion, so as to prevent autoimmunity (13-17).
TRP-2 is a melanogenic enzyme (dopachrome tautomerase, dct) that is expressed by both melanocytes and melanomas. (18, 19). Studies using a variety of vaccination protocols, including DNA encoding TRP-2 or dendritic cells pulsed with TRP-2 peptide, suggest that in mice, tolerance to TRP-2 can be broken and TRP-2-specifc CTLs mediate anti-tumor immunity (20-22). TRP-2(180-188) is an epitope recognized by both human and murine CTLs and is presented by the MHC class I haplotypes HLA-A*0201 and H-2 Kb, respectively (19, 21). We have shown that vaccination with GM-CSF-secreting irradiated (B16) melanoma cells, in combination with anti-CTLA-4 antibody, generated anti-B16 melanoma specific CTLs which showed unique specificity to this immunodominant epitope of TRP-2 (23). Interestingly, immunity to TRP-2 in these experimental murine models was associated with autoimmune depigmentation reminiscent of the vitiligo observed in melanoma patients undergoing immunotherapy (24, 25). Thus, TRP-2 is a strong candidate as a TAA relevant to anti-tumor immune responses.
Given the considerable evidence for a role for TRP-2 in immunity to melanoma, particularly the murine B16 melanoma, in this study, we sought to examine how priming of naïve TRP-2-specific T cells influences tumor immunity. To accomplish this, we examined the effector functions of TRP-2-specific CTLs using T cells from mice that bear a transgene encoding a TRP-2(180-188) peptide-specific TCR (TCR Tg). T cells from these mice matured into the peripheral repertoire at high frequency, expressed functional Vα17 and Vβ11 TCR chains, and displayed a naïve T cell phenotype. Following adoptive transfer into wild-type (W/T) C57BL/6 mice, the Tg T cells remained ignorant and failed to react to their cognate antigen expressed by either melanocytes or B16 melanoma cells. However after vaccination, the transferred Tg T cells migrated into the subcutaneous B16 tumor. Although they maintained effector function as tumor-infiltrating cells, the Tg T cells had no effect on B16 tumor growth. In contrast, in vitro-activated Tg T cells were capable of controlling B16 tumor growth. Our findings suggest that the context in which CTL are primed can be critical for determining the efficacy of TAA-reactive CTL for the immunotherapy of tumors.
Materials and Methods
Mice
C57BL/6 female mice were used at the age of 6-12 week for experiments. Mice were purchased from the National Cancer Institute Animal Production Area Facility (Frederick, MD), and maintained in a specific pathogen-free facility. Animal study protocols were approved by the Institutional Animal Care and Use Committees, NCI-Frederick, according to the “Guide for the Care and Use of Laboratory Animals” set forth by the Office of Laboratory Animal Welfare, NIH.
Generation of TRP-2 TCR Tg mice
TRP-2 TCR Tg mice were generated using TCR chains from a TRP-2(180-188) peptide-specific CD8+ T cell clone (clone 37) generated in our lab. Briefly, to generate this clone, mice were sensitized to the TRP-2180-188 epitope in IFA as described previously. Lymph node cells from these mice were prepared and stimulated every 14 days using the TRP-2180-188 peptide, IL-2 (20 IU/ml), and irradiated syngeneic splenocytes. T cell clones were prepared by limit dilution and confirmed to be TRP-2-reactive using the in vitro assays described below. Clone 37 was selected due to its strong reactivity in proliferation assays, potent killing of TRP-2-pulsed target cells, and clonal use of a single TcR-Vβ chain as revealed by spectratype analysis. Vα and Vβ TCR chain usage by clone 37 was determined by spectratype analysis (26). Clone 37 expressed two Vα chains (Vα3 and Vα17) and one Vβ11. Vα3, Vα17 and Vβ11 TCR chains were amplified by RT-PCR, cloned into pBluescript (Stratagene, La Jolla, CA) and sequenced. Functional TCR chain usage was confirmed by TRP-2-specific proliferation and cytokine secretion analyzed on splenocytes with retrovirus encoding Vα3 or Vα17 in combination with Vβ11 TCR chains. Functional Vα17 and Vβ11 TCR chains were cloned into the hCD2 mini-gene expression vector (generous gift D. Kioussis, National Institute of Medical Research, London, England and Dr. Joan Goverman, Department of Immunology, University of Washington, Seattle, WA). Embryos were injected with the linearized cDNA construct and founders were screened by Southern blot analysis as well as PCR for co-integration of Vα17 and Vβ11 mini-gene constructs. Mouse peripheral blood lymphocytes were screened for H2Kb/TRP-2(180-188) tetramer binding. TRP-2 tetramers were made by tetramer core facility, National Institute of Allergy and Infectious Diseases, National Institutes of Health. We studied one founder line referred as 37B7. 6–12 week-old TCR Tg mice were used in all experiments.
Cell lines and peptide
The B16-BL6 murine melanoma cell line was cultured in DMEM supplemented with heat-inactivated fetal bovine serum (10%), L-glutamine (2 mM), penicillin (100 U/ml), streptomycin (100 μg/ml), 1x non-essential amino acids, 1x essential amino acids, sodium pyruvate (1 mM), 10mM HEPES and 2-ME (50 μM) at 37°C in a humidified incubator with 5% CO2. TRP-2(180-188) (SVYDFFVWL) peptide was purchased from New England Peptide, Inc. (Gardner, MA) and was >90% pure as determined by HPLC.
CFSE labeling and adoptive transfer
Lymph node (LN) cells were dispersed into a single cell suspension and labeled with 5 μM 5, 6-carboxyfluorescein-diacetate succinimidyl ester (CFSE, Molecular Probes) in PBS for 10 minutes at room temperature. Cells were washed in DMEM supplemented with 2% fetal bovine serum. Finally, cells were resuspended in HBSS and 5×106 cells transferred into recipient mice by tail vein injection.
Proliferation assay and generation of CD8+ effectors
Lymph node cells or splenocytes were cultured with increasing doses of TRP-2(180-188) peptide in a 96 well round bottom plate. After 48 h, the cells were pulsed with 1 μCi/well of 3H-thymidine overnight. Cells were harvested on a filtermat and 3H-thymidine incorporation measured using a liquid scintillation counter.
For generation of TRP-2-specific CD8+ T effectors, pooled lymph node cells and splenocytes from TCR Tg mice were cultured in complete DMEM with 1uM of TRP-2 (180-188) peptide and human recombinant IL-2 (20 IU/ml, Chiron). Three days later, cells were expanded with media containing 20 IU/ml IL-2. On day 5, cells were centrifuged over ficoll to remove dead cells and used as effectors to treat mice with 3-day B16 pulmonary nodules. OVA-specific CD8+ T effectors were generated from OT-I/rag-/- mice (courtesy, Dr. Dennis M. Klinman, NCI-Frederick). Pooled OT-I lymph node cells and splenocytes were cultured with equal numbers of irradiated B6 splenocytes as antigen presenting cells in complete DMEM medium with 0.1μM of Ova (257-264) peptide and IL-2 (20IU/ml).
Generation of bone marrow-derived dendritic cells
On day 0, red blood cell-depleted bone marrow cells isolated from femurs and tibia were plated in 6-well tissue culture plates in complete RPMI 1640 medium supplemented with 10% supernatant from a GM-CSF-secreting EL-4 cell line (22). On day 2, non-adherent cells were washed from wells and fresh media containing GM-CSF was added. On day 4, cultures were refed with fresh medium supplemented with GM-CSF. Non-adherent cells were harvested from culture plates on day 7 and pulsed with TRP-2(180-188) peptide (15 uM) overnight. The following day, the non-adherent cells were harvested, washed 2x with HBSS and resuspended in HBSS. Mice were vaccinated subcutaneously with control (unpulsed) or peptide-pulsed dendritic cells (2.5×105/50μl of HBSS) on each of the left and right dorsal flanks.
Isolation of tumor infiltrating lymphocytes
Subcutaneous B16 tumors were excised from euthanized mice and incubated in dissociation solution (RPMI supplemented with 5% fetal bovine serum, collagenase type-I (200 U/ml), and DNAse I (200 μg/ml)) for 2 hours at 37°C. While incubating, tumors were mechanically sheared by repeated pipetting. In the last 30 min of incubation, EDTA was added to a final concentration of 10mM. After incubation, suspensions were passed through a 70 μM cell strainer and washed extensively with PBS. We initially attempted to re-isolate the TCR Tg T cells using anti-Thy1.1-coupled magnetic beads. However, we observed that this induced antigen-independent activation of the transferred T cells. Thus, cell suspensions were layered over lymphocyte separation medium (Biowhittaker) and centrifuged. The cells from interface were collected, washed and used as tumor infiltrating lymphocytes.
Flow cytometric analysis of TCR Tg T cells
Lymph nodes, spleen and thymus were harvested from both B6 mice and TCR Tg mice. Single cell suspensions were prepared and incubated with antibodies: CD8 (53-6.7), CD4 (GK1.5), Vb11 (RR3-15), CD25 (PC61.5), CD44 (IM7), CD62L (MEL-14), CD69 (H1.2F3) and Ly6c (AL-21) after blocking Fc receptors with 2.4G2 antibody. For tetramer staining, cells were first stained with anti-CD8 and additional antibodies, washed with FACS buffer (PBS+0.5% BSA+0.1% Sodium Azide), and then incubated with tetramer for 2hr at 4°C. After staining, cells were fixed with 1% paraformaldehyde/FACS buffer.
For analysis of adoptively transferred cells, mice were euthanized at the indicated time after cell transfer. Tumor-draining LNs (Axillary, Brachial and Inguinal, DLN), non-draining LNs (Mesenteric, NDLN), and spleens were collected and dispersed into single-cell suspensions. Alternatively, s.c. tumors were prepared as described above. Lymph node cells, splenocytes and tumor infiltrating lymphocytes were then stained with antibodies directed against Thy1.1 (OX-7), CD8 (5H10), CD44 (IM7), CD25 (PC61.5) and CD69 (H1.2F3) after blocking with 2.4G2 antibody. Stained suspensions were analyzed by flow cytometry (BD LSR II).
Intracellular IFN-γ, Granzyme B and CD107a degranulation
To test for IFN-γ and Granzyme B expression, lymph node cells or splenocytes (5×106/well of 24- well plate) were cultured with “Golgi-plug” (BD pharmingen) in the presence of peptide (1uM) overnight in complete DMEM. After overnight activation, cells were harvested, incubated with 2.4G2 antibody to block Fc-receptor cells, and incubated with anti-CD8 and anti-Thy1.1 antibodies. Cells were washed with FACS buffer, fixed in 2% paraformaldehyde at 4°C for 30 min and washed twice in PBS. After washing, cells were permeabilized using FACS buffer containing saponin (0.5% for 30 min at 4°C). Cells were then washed with buffer containing saponin and stained with anti-IFN-γ (XMG1.2) antibody, anti-Granzyme B antibody (16G6), or an isotype control antibody for 45 min at 4°C, washed, and fixed with paraformaldehyde.
To estimate CD107a mobilization, splenocytes (5×106/well of 24-well plate) were cultured with Golgi-stop (BD Pharmingen) in the presence of peptide (1uM) and either anti-CD107a (1D4B) antibody or control isotype antibody and incubated overnight at 37°C in complete medium. After overnight activation, cells were incubated with 2.4G2 antibody to block Fc-receptors and then incubated with anti-CD8 and anti-Thy1.1 antibodies. Cells were washed with FACS buffer and fixed in paraformaldehyde.
For analysing IFN-γ secretion or Granzyme B degranulation, tumor infiltrating lymphocytes (TILs), (5×105/well) were activated as described above for splenocytes and LN cells, with the exception that cultures were supplemented with splenocytes (4.5×106/well) as antigen presenting cells.
In vivo cytotoxicity assay
On day 0, lymph node cells from TCR Tg mice were adoptively transferred into C57BL/6 mice. On day 1, mice were vaccinated with control (unpulsed) or TRP-2 peptide-pulsed bone marrow-derived dendritic cells. Eleven days after vaccination, an equal mixture of splenocytes that had been labelled with 5 μM CFSE (peptide-pulsed, CFSEhi) or 0.5 μM CFSE (unpulsed, CFSElow), was injected i.v. The next day, vaccine-draining lymph nodes were analysed by flow cytometry for the presence of peptide-pulsed CFSEhi and control CFSElow B6 splenocytes, as an indication of cell lysis. The percent specific lysis was calculated using the following published formula (27):
Treatment of B16 subcutaneous and lung nodules
For adoptive transfer experiments, 5×106 LN cells from TCR Tg mice were transferred intravenously into W/T mice. The next day, mice were injected s.c. with 1×105 B16/BL6 tumor cells and were vaccinated s.c. 9 days later with TRP-2 peptide-pulsed bone marrow-derived dendritic cells. Tumor size was estimated by multiplying perpendicular diameters measured using a caliper. Mice were euthanized when tumor area exceeded 250 mm2 and was recorded as 250 mm2 thereafter.
For treatment of pulmonary tumor nodules, mice received an i.v. injection of 2×105 B16/BL6 tumor cells. 3, 7 and 11 days after tumor challenge, mice received an i.v. injection of 1×107 in vitro-generated TRP-2-specific effector cells. Day 20 after tumor challenge, mice were euthanized and lung weight was measured and surface tumor nodules were counted using a binocular dissecting microscope.
Results
TRP-2-specific CD8+ T cells from TCR Tg mice display a naïve phenotype
We identified a TRP-2-specific T cell clone from mice that had been sensitized to the peptide epitope TRP-2180-188 (22). This T cell clone demonstrated cytotoxicity against TRP-2-bearing target cells and secreted IFN-γ in response to TRP-2-pulsed APCs. The TCR was cloned by R/T-PCR and the Vα17- and Vβ11-containing segments were identified as conferring specificity to TRP-2180-188. The cDNAs of these genes were subcloned into a human CD2 promoter-regulated expression vector (28) and used to generate transgenic mice on the C57BL/6 background. The mice were viable and showed no outward signs of autoimmunity or immune deficiency.
Given the endogenous expression of the cognate TRP-2 epitope by melanocytes, we initially studied Tg T cell development by examining the frequency of Tg T cells in central and peripheral lymphoid compartments. The number and frequency of thymocyte subpopulations in TCR Tg mice was compared to non-transgenic (W/T) mice. We observed that compared to W/T mice, TCR Tg mice have a decreased frequency of CD4+CD8- thymocytes and a skewing towards the CD4-CD8+ thymocyte population (Figure 1A), although the total number of thymocytes in TCR Tg mice was not different from W/T mice. Furthermore, tetramer staining revealed that approximately 45% of the CD4-CD8+ thymocytes were TRP-2-specific (Figure 1A). Consistent with these findings, we observed that approximately 60% of lymph node cells were CD8+ and all of these were tetramer positive and Vβ11+ (Figure 1B). Phenotypic analysis revealed that TRP-2-specific cells in lymph node were CD69neg and CD62Lhi (data not shown) and the majority of the cells were CD25neg, CD44low, and ly6Clow (Figure 1C). Taken together, these data demonstrate that TRP-2-specific TCR Tg T cells mature from the thymus to the periphery and persist as phenotypically naïve cells, despite expression of their cognate antigen in the skin.
Figure 1. TRP-2-specific T cells migrate to the periphery in TCR Tg mice and have a naïve phenotype.
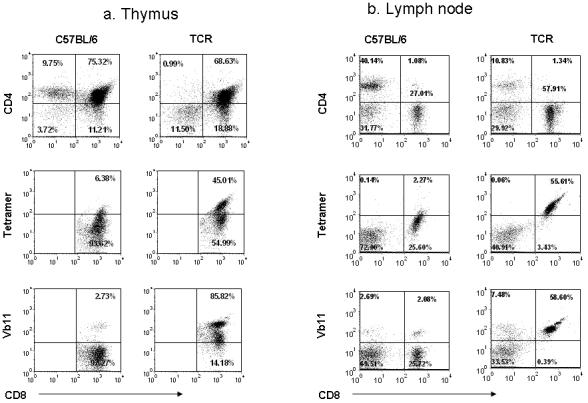

A. Thymocytes from TCR Tg mice and W/T C57Bl/6 mice were analyzed for CD4, CD8′ and Vβ11 transgene expression and tetramer binding. CD4-CD8+ thymocytes were gated and analyzed for TRP-2 tetramer binding (middle panel) and TCR Vβ11 expression (lower panel).
B. Lymph node cells from TCR Tg and W/T C57Bl/6 mice were analyzed for CD4, CD8′ and Vβ11 transgene expression and tetramer binding. C. TRP-2 tetramer positive CD8 cells from TCR Tg mice thick black line) and CD8+ cells from control W/T mice (think black line) were analyzed for activation markers; CD25 expression (upper panel, statistics presented as % positive), CD44 cells (middle panel, statistics presented as % CD44HI) and Ly6C (lower panel, statistics presented as % Ly6CHI) in M1 gate analyzed. Shaded histogram: control stain. Data are individual animals representative of at least five mice per condition.
TRP-2 specific CD8+ T cells can be activated in vitro and confer immunity to B16 pulmonary nodules
To determine whether endogenous TRP-2 expression tolerized TCR Tg T cells, lymph node cells from TCR Tg mice were stimulated in vitro with TRP-2(180-188) peptide and proliferative responses were tested by thymidine incorporation. As shown in Figure 2A, we observed that the Tg T cells proliferated extensively in vitro in the presence of their cognate antigen. In addition, in vitro stimulation of the Tg T cells with antigen and IL-2 resulted in potent, antigen-specific cytotoxicity (Figure 2A).
Figure 2. TRP-2-specific CD8+ T cells can be activated in vitro and develop effector function that controls B16 pulmonary tumors.
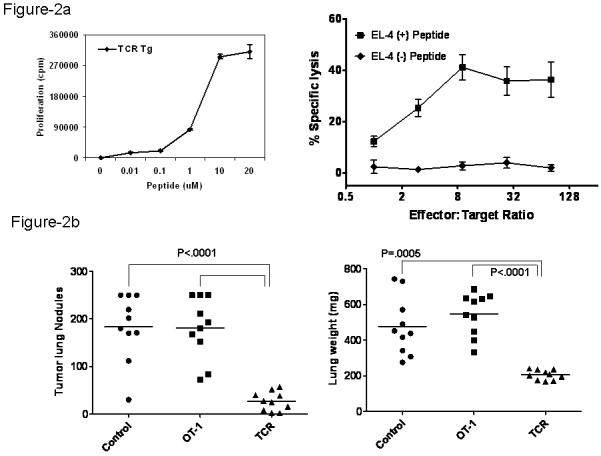

A. (right) In vitro proliferation of TRP-2 specific cells from TCR Tg mice. Lymph node cells from TCR Tg mice were cultured with irradiated splenocytes as antigen presenting cells and the indicated doses of TRP-2(180-188) peptide. Proliferation was determined by 3H-thymidine incorporation. (left) TCR Tg T cells were stimulated with TRP-2 peptide with IL-2 (20IU/ml). Six days later, effector cells were used in a 51Cr-release cytotoxicity assay using TRP-2 peptide-pulsed EL4 thymoma cells as targets. Data are representative of four experiments with similar results.
B. W/T mice were challenged i.v. with 2×105 B16 cells. 3,7, and 11 days later, mice were transferred with 1 × 107 TCR Tg effector cells or OVA-specific OT-I effector cells. 20 days later, mice were euthanized; lungs were weighed and the number of pulmonary nodules enumerated. Statistical analyses were performed using the Tukey’s Multiple Comparison model (tumor nodules) and Welch’s Modified 2-Sample t-Test (lung weight). Data are representative of two experiments with similar results.
C. Mice were treated as in (B), with the exception that B16 cells were inoculated s.c. Tumors were measured using a caliper and the tumor area estimated by calculating the product of bisecting diameter measurements. Data are representative of two experiments with similar results.
Given this strong TRP-2 reactivity, we next tested whether in vitro-generated effector T cells could control B16 tumors. TCR Tg T cells were stimulated for five days with antigen and IL-2 in vitro and used to treat pulmonary lung nodules. In this study, B6 mice were challenged with B16 tumor cells intravenously and on day 3, 7 and 11 days after tumor challenge, in vitro generated TRP-2 specific CD8+ effector T cells were transferred into mice. As a control, we transferred OVA-specific CD8+ effector T cells (OT-I cells). As shown in Figure 2B, the in vitro-generated TRP-2-specific effector cells were able to significantly reduce pulmonary B16 tumor burden, in contrast to mice transferred with OT-I effector cells. This was evident in both the number of pulmonary nodules detected as well as the lung weight. In contrast, the in vitro-generated effector cells had no impact on growth of subcutaneous B16 tumors (Figure 2C). Taken together, these findings validated the TRP-2 antigen as a B16 antigen and led us to investigate whether naïve Tg T cells could be primed in vivo and become potent anti-tumor effectors.
TRP-2 specific CD8+ T cells can be primed in vivo and develop effector function
To test their ability to be activated and develop effector functions in vivo, CFSE labeled lymph node cells from TCR Tg mice were adoptively transferred into W/T mice. The following day, mice were vaccinated with either bone marrow-derived dendritic cells pulsed with TRP-2 peptide or control, unpulsed dendritic cells. 5 days after vaccination, mice were euthanized and the TCR Tg cells were analyzed using the allelic marker Thy1.1 to distinguish the TCR transgenic cells from the endogenous CD8+ T cells. In the absence of an antigen-bearing vaccine, the transferred T cells showed no signs of activation, despite the presumed presence of melanocyte-derived TRP-2. This lack of activation is demonstrated by a lack of dilution of CFSE, no change in activation phenotype (CD44) and persistence in the secondary lymphoid tissues (Figure 3A, DC-CTRL). In contrast, following vaccination with the peptide-pulsed DC vaccine, most of the adoptively transferred T cells had diluted CFSE and displayed elevated levels of CD44 expression (Figure 3A). In addition, the majority of the CFSE-diluted cells expressed IFN-γ as determined by intracellular staining following overnight ex vivo stimulation with the TRP-2 peptide (Figure 3B).
Figure 3. TRP-2-specific CD8+ T cells can be activated in vivo and generate effector function.

A,B. On day 0, CFSE-labelled Thy1.1+ LN cells from TCR Tg mice were adoptively transferred into C57BL/6 mice. On day 1, mice were vaccinated subcutaneously with unpulsed (DC-CTRL) or peptide-pulsed (DC-TRP-2) bone marrow-derived dendritic cells. Five days after vaccination, vaccine draining LN cells were analyzed by gating on adoptively-transferred Thy1.1+ CD8+ T cells and assessing dilution of CFSE and CD44 (A) or IFN-γ expression (B). Data are presented for a single animal where identical results were obtained for 3 animals per group; the experiment was performed twice.
C. Lysis of TRP-2-pulsed target cells was assessed by injecting mice with CFSE-labelled splenocytes cells after vaccination with dendritic cell vaccines. The frequency of CFSELOW (unpulsed) and CFSEHI (TRP-2-pulsed) cells were analyzed in vaccine-draining LNs of mice transferred with TCR Tg cells (W/T-ACT) and untransferred mice (W/T). The percent specific lysis is presented in each panel. Data are presented for a single animal where identical results were obtained for 5 animals per group; the experiment was performed four times.
To determine the in vivo cytotoxic capacity of these activated T cells, after DC vaccination, mice were transferred with TRP-2-pulsed splenocytes labeled with 5uM of CFSE and control, unpulsed splenocytes labeled with 0.5uM of CFSE; specific lysis is indicated by a decreased frequency in CFSE-bright (antigen-pulsed) cells. Irrespective of Tg T cell transfer, control-vaccinated mice demonstrated little or no CTL acitvity against the antigen-pulsed target cells. W/T mice showed a weak response to the TRP-2-pulsed DC vaccine, as untransferred mice exhibited a low but detectable CTL response against the TRP-2 pulsed target cells. These findings are consistent with our previous findings demonstrating a weak, but detectable TRP-2 specific response in unmanipulated mice (22). In contrast, the TRP-2-pulsed target cells were efficiently lysed in mice transferred with the TCR Tg cells and vaccinated with the peptide-pulsed DC vaccine (Fig 3C), as evidenced by the high level (>95%) of specific lysis of the TRP-2-pulsed splenocytes. Taken together, these findings demonstrate that in vivo priming of the Tg T cells induces strong effector functions, including proliferation, IFN-γ production, and antigen-specific CTL activity.
TRP-2 specific CD8+ T cells are ignorant of endogenous and tumor-derived TRP-2
Our data presented in Figure 3 suggest that in the absence of priming, the TRP-2-specifc T cells do not respond to the endogenous TRP-2 antigen, as the T cells from the control-vaccinated mice did not exhibit any CFSE dilution or increase in CD44 expression. To study further the response of the TCR Tg T cells to both endogenous and tumor-derived TRP-2 antigen, CFSE-labeled Tg T cells were adoptively transferred into B6 mice. The following day, half of the mice were challenged with B16 tumor cells and 5, 15 and 30 days later, mice were vaccinated with bone marrow-derived DC pre-pulsed with TRP-2(180-188) peptide. Five days after the vaccine, adoptively-transferred cells were analyzed for proliferation by analyzing CFSE dilution and expression of the activation markers CD44, CD25 and CD69 on the Thy1.1+CD8+ cells .
As demonstrated in Figure 4A, in the absence of an antigen-specific priming, TCR Tg T cells neither diluted CFSE nor up-regulated CD44 expression. This was true for both naïve and tumor-bearing animals. This absence of activation was confirmed by the lack of expression of both CD25 and CD69 (data not shown). Even at a time when the mice had a large tumor burden (day 20 after cell transfer), Tg T cells neither diluted CFSE nor upregulated activation markers. These data demonstrate that the naïve Tg T cells are ‘ignorant’ of both endogenous and tumor-derived TRP-2 (29).
Figure 4. TRP-2-specific Tg T cells are ignorant of endogenous and B16-derived TRP-2.

CFSE-labelled Thy1.1+ LN cells from TCR Tg mice were adoptively transferred into W/T Thy1.2+ mice. The next day, half of the mice were injected s.c. with 1×105 B16/BL6 tumor cells. Mice were vaccinated with unpulsed (DC-CTRL, A) or peptide-pulsed (DC-TRP-2, B) bone marrow-derived dendritic cells 5, 15 or 30 days after transfer. Five days after each vaccination, adoptively-transferred Thy1.1+CD8+ T cells were analyzed for dilution of CFSE and CD44 expression; data is presented as the percent of CD8+Thy1.1+ cells that are CD44hiCFSElow. Results are shown as the mean +/- standard deviation at least three mice at each time point. Data are representative of three similar studies.
In contrast, sensitization of mice with TRP-2-pulsed dendritic cells induced strong proliferative responses by the transferred Tg T cells. Following vaccination, adoptively-transferred T cells in the spleen and lymph nodes of both naïve and tumor-bearing mice diluted CFSE and up-regulated expression of CD44 (Figure 4B). Interestingly, tumor growth did not influence the activation of adoptively transferred TRP-2-specific CD8+ T cell, as there was no difference in the frequency of cells that diluted CFSE or upregulated CD44 as a function of tumor growth. Even 35 days after transfer, TRP-2 specific Tg T cells in tumor-free were comparably activated by the vaccine. Similar results were observed in tumor-bearing mice, 20 days after transfer (tumor-bearing mice could not be followed to 35 days due to excessive tumor burden). These data suggest that the developing B16 tumor does not profoundly influence responsiveness of these in vivo-primed tumor antigen-specific T cells.
Vaccination induces infiltration of TRP-2 specific T cells but does not affect tumor growth
Given our observation that the TRP-2-pulsed DC vaccine could prime the adoptively transferred TCR Tg T cells, we next sought to determine whether this approach could reduce B16 tumor growth. Mice were treated with the Tg T cells and vaccinated 9 days after subcutaneous tumor challenge. As shown in Figure 5A, s.c. B16 tumor growth in adoptively-transferred mice sensitized to the TRP-2-pulsed vaccine was similar to growth in control-vaccinated or untransferred mice. Similar results were obtained in mice challenge intravenously (i.v.) with the B16 tumor cells, which results in pulmonary nodule formation (Figure 5B).
Figure 5. TRP-2-specific T cells infiltrate the tumor but do not affect tumor growth.

A. 5 × 106 LN cells from TCR Tg mice were adoptively transferred into W/T mice. The following day, transferred mice (ACT) or naïve mice (naïve) were challenged with 1×105 B16/BL6 tumor cells s.c. 9 days later, mice were vaccinated with unpulsed (DC-CTRL) or peptide-pulsed (DC-TRP-2) bone marrow-derived dendritic cells. Tumor size was estimated by calculating the product of bisecting perpendicular diameters. Results are presented for one of three similar studies using five animals per group. A similar study was performed in (B), with the exception that mice were vaccinated 3 days after i.v. challenge with B16 tumor cells. Tumor nodules were enumerated on day 24 after tumor challenge. P>0.05, using Tukey’s test. Data from one of two similar studies is presented, using five mice per group. One mouse in the transferred and vaccinated group (ACT, DC/TRP-2) was lost due to factors unrelated to the study.
C. Tumors from mice challenged as described in (A) were excised 5 days after vaccination and dispersed into single cell suspensions and enriched over a ficoll gradient. Tumor-infiltrating cells were analyzed for Thy1.1 and CD8 expression (left and center panels). The Thy1.1+CD8+ cells in the DC-TRP-2-vaccinated group were gated and further analyzed for CFSE dilution and CD44 expression (right panel). Data are representaive of three similar studies.
These findings suggested that in vivo priming of the TRP-2-specific TCR Tg T cells was not sufficient to induce tumor regression, despite TRP-2-specific CTL activity (Figure 3C). To determine whether in vivo priming of the Tg T cells resulted in tumor infiltration, we analyzed the dissociated tumors for the presence of the transferred Tg T cells using the allelic marker, Thy1.1, which permitted discrimination of the transferred Tg T cells from the endogenous, host-derived T cells. As demonstrated in Figure 5C, the unpulsed DC vaccine did not induce any tumor infiltration by the Tg T cells, consistent with their lack of activation and effector function (Figure 3). In contrast, the TRP-2-pulsed DC induced marked infiltration of the Tg T cells (Figure 5C). The Tg T cells were detected as early as 5 days after vaccination with the TRP-2 peptide-pulsed DC vaccine and they persisted throughout tumor growth. During that period, they maintained high levels of CD44 expression. Thus, despite effective in vivo priming, activation, and tumor infiltration, these tumor antigen-specific T cells were incapable of altering tumor progression.
Tumor-infiltrating TCR Tg T cells maintain antigen reactivity despite progressive tumor growth
Given the lack of control of B16 tumor by the infiltrating TCR Tg T cells, we hypothesized that the developing tumor may tolerize the tumor-infiltrating cells (TILs). Thus, we sequentially analyzed their reactivity after vaccination. We isolated the mononuclear cells by ficoll density centrifugation and placed the infiltrating cells or splenocytes in culture overnight with TRP-2180-188 peptide. We then tested the cultures for IFN-γ and Granzyme B (GrB) expression as well as CD107a mobilization (a marker for CTL granule exocytosis) (30) by flow cytometric analysis, gating in the CD8+Thy1.1+CFSELow cells. As a positive control, splenocytes from tumor-free mice vaccinated with the same TRP-2-pulsed DC vaccine were tested for antigen-specific responses.
As indicated in Figure 6, TILs were highly responsive to antigen 5 days after vaccination. Expression of IFN-γ and GrB and mobilization of CD107a by tumor-infiltrating cells was similar to that of splenocytes of tumor-bearing mice, and was only slightly reduced compared to splenocytes from tumor-free mice. At later time points (10 and 15 days after vaccination), a small but statistically significant reduction in IFN-γ production was observed in the TILs when compared to Tg T cells from the spleen of tumor-free and tumor-bearing mice (due to large tumor burden, TILs could not be assessed at points later than 15 days after vaccination). Similarly, a small decrease in CD107a mobilization was noted in the TIL population (Figure 6B), but not in the splenocyte population. Finally, no change in GrB expression was detected at either 10 or 15 days after vaccination (Figure 6C). Taken together, these data demonstrate that while there was a modest decrease in some effector functions, the TCR Tg T cells maintained relatively strong reactivity for TRP-2 and were potent effector cells based on their expression of GrB and their ability to mobilize CD107a. Nevertheless, these data also suggest that the tumor micro-environment impedes the efficacy of tumor-reactive T cells.
Figure 6. TRP-2-specific CD8+ T cells in tumor retain effector functions.

CFSE-labelled Thy1.1+ LN cells from TCR Tg mice were adoptively transferred into W/T mice. The following day, mice were injected s.c. with 1×105 B16/BL6 tumor cells; another cohort of mice remained naïve to tumor challenge as a control group (naïve). 9 days later, mice were vaccinated with TRP-2-pulsed bone marrow-derived dendritic cells. At the indicated time after vaccination, mice were euthanized and splenocytes (SPL) or tumor-infiltrating cells (TIL) were analyzed for IFN-γ expression (A), CD107a mobilization (B) and Granzyme B expression (C) as described in the Materials and Methods section. *P<0.0001 in compared groups, using a Tukey multiple comparison analysis. In (A), data from three experiments was pooled. In B and C, data is presented from one of two identical experiments using three mice per group.
Discussion
Our data demonstrate that expression of a transgenic TCR that recognizes a peripheral antigen may not always result in tolerization of the transgenic T cell population. The presence of the Tg T cells at a high frequency in the secondary lymphoid tissues indicates that thymic deletion was not efficient. Positive selection of T cells in the thymus depends on antigen presentation by medullary epithelial cells, or thymic dendritic cells (31, 32). Thus, the TRP-2-specific cells may be positively selected because the level of antigen presented in thymus was not sufficient to delete the Tg T cells, and these cells persist in the secondary lymphoid tissues in a naïve, responsive state.
Our data further demonstrate that in the absence of exogenous priming, the TCR Tg T cells show no indication of priming by either the endogenous or the tumor-derived TRP-2 antigen in the periphery; i.e., they are ignorant of antigen from both sources. Recognition of cognate antigen in the periphery may be regulated by factors such as antigen dose (33) or the avidity of responding T cells (34). Since the TRP-2-specific Tg T cells did not respond to cognate antigen expressed by melanocytes or the B16 tumor, it is possible that the level of TRP-2 antigen presentation in skin-draining lymph node is too low (35) or that the responding T cells are of insufficient avidity. Further studies using tumors expressing higher levels of TRP-2 or higher avidity Tg T cells are being used to address such issues.
The efficacy of an anti-tumor immune response depends on the activation and infiltration of tumor antigen-specific CD8+ T cells. Ochsenbein et al. showed B16 tumors are capable of avoiding contact with naïve CD8+ T cells, resulting in ignorance to tumor-derived epitopes (36). It is relevant that we demonstrated that after vaccination with peptide-pulsed DC, the TRP-2-specific Tg T cells migrated to the tumor and persisted there as potent effector cells, despite progressive tumor growth, also suggestive of ignorance to tumor antigen. However, we cannot rule out the possibility that the infiltration by these tumor antigen-specific T cells is antigen-independent, as Palmer et al previously reported that melanoma-specific T cells can infiltrate into antigen-negative tumors (37). Other melanoma-specific TCR Tg model systems have been used to demonstrate that the use of altered T cell epitopes can elicit immunity to B16 tumors, indicating that ignorance is avoidable (38).
While cytotoxic effector function of tumor-infiltrating T cells is generally considered critical for tumor destruction, in this study, primary subcutaneous tumor growth was unaffected by TRP-2-specific CD8+ T cells primed in vitro or in vivo and demonstrated to be armed with potent effector functions. In contrast, in vitro-generated TRP-2-specific CD8+ effector T cells significantly reduced B16 pulmonary nodules but not subcutaneous tumors. This differential therapeutic effect on tumor growth at different sites may be due to several factors, including differential penetration of tumor by T cells and different tumor microenvironments (cellular infiltrates, tumor cell composition, stroma, etc.) which, in the case of the lung, may promote stronger inflammatory responses than at the primary, subcutaneous tumor site (22). Alternatively, adoptively-transferred effector cells may traffic more efficiently to the lung, as a simple function of blood flow.
The inability of in vivo-primed Tg T cells to confer immunity to the B16 tumor, despite demonstrated CTL function, may be further explained by two possible mechanisms. The first may be an ‘intrinsic’ mechanism that restricts the interactions between effector T cells and the tumor. This could include low levels of antigen/MHC complexes on the tumor cell surface, poor formation of immunological synapses due to diminished expression of adhesion molecules, or transient inhibitory signals that reduce T cell effector functions of the TILs, but are not detectable using ex vivo assays of T cell effector function. Alternatively, an ‘extrinsic’ mechanism may create an environment that reduces anti-tumor efficacy of the primed Tg T cells. This could include the presence of immunosuppressive cells like regulatory T cells, myeloid-derived suppressor cells, or immature DC, all of which have been associated with B16 tumors. Alternatively, the tumor itself may express or secrete factors (e.g., catabolic enzymes or immunosuppressive cytokines) that restrict the effector activity of the DC-primed Tg T cells. Similarly, the expression of inhibitory receptors on T cells, including PD-1 and CTLA-4, may restrict primed effector cells from clearing the B16 tumors. Although B16 tumors do not express CTLA-4 ligands, they have been demonstrated to express PD-L1 (39). The in vitro-generated effector cells may be endowed with an as-yet undefined component of effector function that is more resistant to these intrinsic and extrinsic mechanisms.
Taken together, our findings confirm that the simple provision of tumor antigen-specific T cells may not be sufficient to induce potent tumor immunity. While tumor antigen-specific T cells can traffic into the tumor bed and retain effector function, this may be insufficient to control tumor growth. These findings are consistent with a study of melanoma patients undergoing vaccination with gp100 peptides that demonstrated a high frequency of gp100-reactive T cells, despite tumor progression (40). By elucidating the mechanisms that this poorly immunogenic tumor uses to reduce its susceptibility to infiltrating effector T cells, future studies using this murine model system will help to identify novel immunotherapeutic approaches that may be applicable for managing cancer in humans.
Acknowledgements
The authors appreciate the critical review of this manuscript by Drs. Joost Oppenheim, Scott Durum, and Frank Ruscetti and the technical support provided by Terri Stull and Jami Willette-Brown.
This work was supported by the Intramural Research Program, NCI, NIH.
Footnotes
Financial Disclosure: All authors have declared there are no financial conflicts of interest in regards to this work.
References
- 1.Kawakami Y, Eliyahu S, Sakaguchi K, et al. Identification of the immunodominant peptides of the MART-1 human melanoma antigen recognized by the majority of HLA-A2-restricted tumor infiltrating lymphocytes. J Exp Med. 1994;180:347–352. doi: 10.1084/jem.180.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bakker AB, Schreurs MW, de Boer AJ, et al. Melanocyte lineage-specific antigen gp100 is recognized by melanoma-derived tumor-infiltrating lymphocytes. J Exp Med. 1994;179:1005–1009. doi: 10.1084/jem.179.3.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brichard V, Van Pel A, Wolfel T, et al. The tyrosinase gene codes for an antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J Exp Med. 1993;178:489–495. doi: 10.1084/jem.178.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang RF, Robbins PF, Kawakami Y, et al. Identification of a gene encoding a melanoma tumor antigen recognized by HLA-A31-restricted tumor-infiltrating lymphocytes. J Exp Med. 1995;181:799–804. doi: 10.1084/jem.181.2.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang RF, Appella E, Kawakami Y, et al. Identification of TRP-2 as a human tumor antigen recognized by cytotoxic T lymphocytes. J Exp Med. 1996;184:2207–2216. doi: 10.1084/jem.184.6.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Engelhard VH, Bullock TN, Colella TA, et al. Antigens derived from melanocyte differentiation proteins: self-tolerance, autoimmunity, and use for cancer immunotherapy. Immunol Rev. 2002;188:136–146. doi: 10.1034/j.1600-065x.2002.18812.x. [DOI] [PubMed] [Google Scholar]
- 7.Kawai K, Ohashi PS. Immunological function of a defined T-cell population tolerized to low-affinity self antigens. Nature. 1995;374:68–69. doi: 10.1038/374068a0. [DOI] [PubMed] [Google Scholar]
- 8.Theobald M, Biggs J, Hernandez J, et al. Tolerance to p53 by A2.1-restricted cytotoxic T lymphocytes. J Exp Med. 1997;185:833–841. doi: 10.1084/jem.185.5.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bouneaud C, Kourilsky P, Bousso P. Impact of negative selection on the T cell repertoire reactive to a self-peptide: a large fraction of T cell clones escapes clonal deletion. Immunity. 2000;13:829–840. doi: 10.1016/s1074-7613(00)00080-7. [DOI] [PubMed] [Google Scholar]
- 10.Zehn D, Bevan MJ. T cells with low avidity for a tissue-restricted antigen routinely evade central and peripheral tolerance and cause autoimmunity. Immunity. 2006;25:261–270. doi: 10.1016/j.immuni.2006.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morgan DJ, Kreuwel HT, Fleck S, et al. Activation of low avidity CTL specific for a self epitope results in tumor rejection but not autoimmunity. J Immunol. 1998;160:643–651. [PubMed] [Google Scholar]
- 12.Sandberg JK, Franksson L, Sundback J, et al. T cell tolerance based on avidity thresholds rather than complete deletion allows maintenance of maximal repertoire diversity. J Immunol. 2000;165:25–33. doi: 10.4049/jimmunol.165.1.25. [DOI] [PubMed] [Google Scholar]
- 13.Fields LE, Loh DY. Organ injury associated with extrathymic induction of immune tolerance in doubly transgenic mice. Proc Natl Acad Sci USA. 1992;89:5730–5734. doi: 10.1073/pnas.89.13.5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hernandez J, Aung S, Redmond WL, et al. Phenotypic and functional analysis of CD8(+) T cells undergoing peripheral deletion in response to cross-presentation of self-antigen. J Exp Med. 2001;194:707–717. doi: 10.1084/jem.194.6.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohlen C, Kalos M, Hong DJ, et al. Expression of a tolerizing tumor antigen in peripheral tissue does not preclude recovery of high-affinity CD8+ T cells or CTL immunotherapy of tumors expressing the antigen. J Immunol. 2001;166:2863–2870. doi: 10.4049/jimmunol.166.4.2863. [DOI] [PubMed] [Google Scholar]
- 16.Ohlen C, Kalos M, Cheng LE, et al. CD8(+) T cell tolerance to a tumor-associated antigen is maintained at the level of expansion rather than effector function. J Exp Med. 2002;195:1407–1418. doi: 10.1084/jem.20011063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bendle GM, Holler A, Pang LK, et al. Induction of unresponsiveness limits tumor protection by adoptively transferred MDM2-specific cytotoxic T lymphocytes. Cancer Res. 2004;64:8052–8056. doi: 10.1158/0008-5472.CAN-04-0630. [DOI] [PubMed] [Google Scholar]
- 18.Bloom MB, Perry-Lalley D, Robbins PF, et al. Identification of tyrosinase-related protein 2 as a tumor rejection antigen for the B16 melanoma. J Exp Med. 1997;185:453–459. doi: 10.1084/jem.185.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parkhurst MR, Fitzgerald EB, Southwood S, et al. Identification of a shared HLA-A*0201-restricted T-cell epitope from the melanoma antigen tyrosinase-related protein 2 (TRP2) Cancer Res. 1998;58:4895–4901. [PubMed] [Google Scholar]
- 20.Bronte V, Apolloni E, Ronca R, et al. Genetic vaccination with “self” tyrosinase-related protein 2 causes melanoma eradication but not vitiligo. Cancer Res. 2000;60:253–258. [PMC free article] [PubMed] [Google Scholar]
- 21.Schreurs MW, Eggert AA, de Boer AJ, et al. Dendritic cells break tolerance and induce protective immunity against a melanocyte differentiation antigen in an autologous melanoma model. Cancer Res. 2000;60:6995–7001. [PubMed] [Google Scholar]
- 22.Ji Q, Gondek D, Hurwitz AA. Provision of granulocyte-macrophage colony-stimulating factor converts an autoimmune response to a self-antigen into an antitumor response. J Immunol. 2005;175:1456–1463. doi: 10.4049/jimmunol.175.3.1456. [DOI] [PubMed] [Google Scholar]
- 23.van Elsas A, Sutmuller RP, Hurwitz AA, et al. Elucidating the autoimmune and antitumor effector mechanisms of a treatment based on cytotoxic T lymphocyte antigen-4 blockade in combination with a B16 melanoma vaccine: comparison of prophylaxis and therapy. J Exp Med. 2001;194:481–489. doi: 10.1084/jem.194.4.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosenberg SA, White DE. Vitiligo in patients with melanoma: normal tissue antigens can be targets for cancer immunotherapy. J Immunother Emphasis Tumor Immunol. 1996;19:81–84. [PubMed] [Google Scholar]
- 25.Steitz J, Bruck J, Lenz J, et al. Peripheral CD8+ T cell tolerance against melanocytic self-antigens in the skin is regulated in two steps by CD4+ T cells and local inflammation: implications for the pathophysiology of vitiligo. J Invest Dermatol. 2005;124:144–150. doi: 10.1111/j.0022-202X.2004.23538.x. [DOI] [PubMed] [Google Scholar]
- 26.Currier JR, Robinson MA.Spectratype/immunoscope analysis of the expressed TCR repertoire Curr Protoc Immunol 2001. Chapter 10:Unit 1028. [DOI] [PubMed] [Google Scholar]
- 27.Wonderlich J, Shearer G, Livingstone A, et al. Induction and measurement of cytotoxic T lymphocyte activity Curr Protoc Immunol 2006. Chapter 3:Unit 311. [DOI] [PubMed] [Google Scholar]
- 28.Zhumabekov T, Corbella P, Tolaini M, et al. Improved version of a human CD2 minigene based vector for T cell-specific expression in transgenic mice. J Immunol Methods. 1995;185:133–140. doi: 10.1016/0022-1759(95)00124-s. [DOI] [PubMed] [Google Scholar]
- 29.Ochsenbein AF, Klenerman P, Karrer U, et al. Immune surveillance against a solid tumor fails because of immunological ignorance. Proc Natl Acad Sci USA. 1999;96:2233–2238. doi: 10.1073/pnas.96.5.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Betts MR, Brenchley JM, Price DA, et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 31.Gallegos AM, Bevan MJ. Central tolerance to tissue-specific antigens mediated by direct and indirect antigen presentation. J Exp Med. 2004;200:1039–1049. doi: 10.1084/jem.20041457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bonasio R, Scimone ML, Schaerli P, et al. Clonal deletion of thymocytes by circulating dendritic cells homing to the thymus. Nat Immunol. 2006;7:1092–1100. doi: 10.1038/ni1385. [DOI] [PubMed] [Google Scholar]
- 33.Kurts C, Sutherland RM, Davey G, et al. CD8 T cell ignorance or tolerance to islet antigens depends on antigen dose. Proc Natl Acad Sci USA. 1999;96:12703–12707. doi: 10.1073/pnas.96.22.12703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heath WR, Karamalis F, Donoghue J, et al. Autoimmunity caused by ignorant CD8+ T cells is transient and depends on avidity. J Immunol. 1995;155:2339–2349. [PubMed] [Google Scholar]
- 35.Spiotto MT, Yu P, Rowley DA, et al. Increasing tumor antigen expression overcomes “ignorance” to solid tumors via crosspresentation by bone marrow-derived stromal cells. Immunity. 2002;17:737–747. doi: 10.1016/s1074-7613(02)00480-6. [DOI] [PubMed] [Google Scholar]
- 36.Ochsenbein AF, Sierro S, Odermatt B, et al. Roles of tumour localization, second signals and cross priming in cytotoxic T-cell induction. Nature. 2001;411:1058–1064. doi: 10.1038/35082583. [DOI] [PubMed] [Google Scholar]
- 37.Palmer DC, Balasubramaniam S, Hanada K, et al. Vaccine-stimulated, adoptively transferred CD8+ T cells traffic indiscriminately and ubiquitously while mediating specific tumor destruction. J Immunol. 2004;173:7209–7216. doi: 10.4049/jimmunol.173.12.7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Overwijk WW, Theoret MR, Finkelstein SE, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blank C, Brown I, Peterson AC, et al. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004;64:1140–1145. doi: 10.1158/0008-5472.can-03-3259. [DOI] [PubMed] [Google Scholar]
- 40.Rosenberg SA, Sherry RM, Morton KE, et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Immunol. 2005;175:6169–6176. doi: 10.4049/jimmunol.175.9.6169. [DOI] [PubMed] [Google Scholar]