

Abstract
Whole-brain irradiation (WBI) can lead to cognitive impairment several months to years after irradiation. Studies on rodents have shown a rapid and sustained increase in activated microglia (brain macrophages) following brain irradiation, contributing to a chronic inflammatory response and a corresponding decrease in hippocampal neurogenesis. Thus, alleviating microglial activation following radiation represents a key strategy to minimize WBI-induced morbidity. We hypothesized that pre-treatment with peroxisomal proliferator-activated receptor (PPAR)α agonists would ameliorate the pro-inflammatory responses seen in the microglia following in vitro radiation. Irradiating BV-2 cells (a murine microglial cell line) with single doses (2-10 Gy) of 137Cs γ-rays led to increases in 1] the gene expression of IL-1β and TNFα, 2] Cox-2 protein levels and 3] intracellular ROS generation. In addition, an increase in the DNA-binding activity of redox-regulated pro-inflammatory transcription factors AP-1 and NF-κB was observed. Pre-treating BV-2 cells with the PPARα agonists, GW7647 and Fenofibrate significantly inhibited the radiation-induced microglial pro-inflammatory response, in part, via decreasing i] the nuclear translocation of the NF-κB p65 subunit and ii] phosphorylation of the c-jun subunit of AP-1 in the nucleus. Taken together, these data support the hypothesis that activation of PPARα can modulate the radiation-induced microglial pro-inflammatory response.
Keywords: ionizing radiation, microglia, PPARα, inflammation, radiation-induced brain injury, NF-κB, AP-1
INTRODUCTION
Brain metastases represent a significant cause of morbidity and mortality, and are the most common intracranial tumors in adults, occurring in 10% to 30% of adult cancer patients [1,2]. The annual incidence appears to be rising as a result of an aging population, improved treatment of systemic disease and the use of advanced imaging techniques such as magnetic resonance imaging to detect smaller metastases in asymptomatic patients [1]. Radiation therapy, administered in the form of large-field partial or whole brain irradiation (WBI), is the primary mode of treatment for brain metastases; over 170,000 patients will receive WBI/year in the US [3,4]. However, late delayed effects of brain irradiation characterized by a progressive cognitive impairment occur in up to 50% of brain tumor patients who are long-term survivors [5] (>6 months post-irradiation). Currently there are neither long- term treatments nor any preventive strategies to alleviate this radiation-induced morbidity [1].
Although the exact pathogenic mechanisms of radiation-induced brain injury are not known, a growing body of data suggests that oxidative stress/pro-inflammatory responses might play a role [6]. An acute molecular response characterized by increased expression of proinflammatory cytokines such as tumor necrosis factor alpha (TNFα), interleukin 1 beta (IL-1β), intracellular adhesion molecule-1 (ICAM-1), cyclooxygenase-2 (Cox-2) and activation of transcription factors such as nuclear factor kappa B (NF-κB) is observed within hours of irradiating the rodent brain [7-9]. In addition, a chronic elevation of TNFα has been observed in the mouse brain up to 6 months post-irradiation [10].
Microglia, the immune cells of the brain, are one of the key mediators of neuroinflammation. They represent about 10% of the total glial population in the central nervous system [11]. In the ramified state, microglia actively survey the microenvironment and ensure normal central nervous system activity by secreting neurotrophic factors such as neuronal growth factor (NGF) [12]. However, they can become activated by a variety of stimuli and release a host of pro-inflammatory cytokines, chemokines and reactive oxygen/nitrogen oxide species (ROS/RNOS) [13]. Although microglial activation plays an important role in phagocytosis of dead cells in the central nervous system, prolonged activation leads to a sustained inflammatory status in the central nervous system [14]. Microglial activation has been implicated in several neurodegenerative diseases such as multiple sclerosis, Alzheimer’s disease and Parkinson’s disease [14].
In vitro studies suggest that irradiating microglia leads to a marked increase in expression of proinflammatory genes including TNFα, IL-1β, IL-6 and Cox-2 [15-17]. Radiation-induced expression of microglial TNFα and IL-1β has been shown to enhance ICAM-1 expression in non-irradiated astrocytes [16]. These studies are supported by in vivo experiments in rodents which indicate that brain irradiation leads to a marked increase in microglial activation associated with both a concomitant decrease in neurogenesis in the sub-granular zone (SGZ) of the hippocampus and spatial memory retention deficits [18,19]. Further, administration of the antiinflammatory drug indomethacin decreased radiation-induced microglial activation and was associated with an improvement in hippocampal neurogenesis [20]. These data suggest that the efficacy of anti-inflammatory therapies to mitigate radiation-induced brain injury may involve inhibition of radiation-induced microglial activation.
Peroxisomal proliferator-activated receptor alpha (PPARα) is one of the three nuclear receptor subtypes belonging to the PPAR family [21]. Following activation, PPARs regulate gene transcription by binding to specific consensus sequences termed PPAR response elements (PPREs) in the promoter regions of genes as a heterodimer with the retinoid X receptor (RXR) [21]. PPARα is activated by both natural ligands such as certain long-chain fatty acids and eicosanoids and synthetic ligands such as hypolipidemic fibrates [22]. PPARα is predominantly expressed in tissues that catabolize high amounts of fatty acids such as the liver, kidney and heart [23], and regulates many metabolic pathways, including activation of fatty acid β-oxidation and apolipoprotein expression [22,24,25]. More recently, PPARα has been shown to play a major role in regulating inflammatory processes. Administration of fibrates to patients with a moderate hyperlipidemia decreased plasma concentrations of pro-inflammatory mediators such as IL-6, TNF-α, interferon-γ (IFNγ), fibrinogen, and C-reactive protein [25]. PPARα ligands can negatively impact atherogenesis and vascular thrombus formation, in part, by repressing Tissue factor and TNF-α expression in T lymphocytes and macrophages [26,27]. In addition, PPARα has been shown to mediate its anti-inflammatory activities, in part, via downregulation of activator protein-1 (AP-1) and NF-κB signaling pathways [28].
In the brain, PPARα is expressed in multiple cell types including the microglia [27]. PPARα agonists have been shown to inhibit the production of nitric oxide and secretion of proinflammatory cytokines including TNFα, IL-1β and IL-6 in both cytokine and LPS-stimulated microglia [29-31]. The role of PPARα in radiation-induced brain injury is unknown. We hypothesized that activation of PPARα could modulate the inflammatory and/or oxidative stress responses of the microglia following radiation. In the current study, we report that pre-treatment of microglial cells with PPARα agonists prevented the radiation-induced increases in TNFα and IL1β gene expression and Cox-2 protein levels, in part, by modulating the activity of AP-1 and NF-κB transcription factors.
MATERIALS AND METHODS
Cell culture and reagents
The immortalized BV-2 murine microglial cell line was cultured in high glucose DMEM (Invitrogen, Carlsbad, CA) containing 5% fetal bovine serum (Sigma-Aldrich, St. Louis, MO), 2 mM L-glutamine, 100 IU/mL penicillin and 100 mg/mL streptomycin. These cells display phenotypic and functional properties of reactive microglial cells and resemble non-activated primary microglial cells [32]. Cells were maintained at 37°C with 10% CO2/90% air mixture and the culture medium was replaced with serum-free media 24 h prior to irradiation. The PPARα agonists GW7647 and Fenofibrate were purchased from Sigma-Aldrich. The JNK inhibitor, SP 600125, the NF-κB inhibitors Bay-117082 and 6-Amino-4-(4-phenoxyphenylethylamino) quinazoline were purchased from Calbiochem (EMD Biosciences, La Jolla, CA). All drugs were dissolved in Me2SO4 (DMSO); for some experiments, Fenofibrate was dissolved in N, N-Dimethyl formamide (DMF). Goat anti-Cox-2 was purchased from Santa Cruz Biotechnologies (Santa Cruz, CA). Rabbit anti-p65 was purchased from Cell Signaling (Danvers, MA).
Irradiation
Cells were irradiated using a 137Cs irradiator (J.L. Shepherd and Associates, San Fernando, CA) at a dose rate of ~ 4.0 Gy/min. All irradiations were performed at room temperature; control cells received sham-irradiation. After irradiation, the culture dishes were returned to the incubator and maintained at 37°C in a 10% CO2/90% air mixture.
Measurement of intracellular ROS generation
2′7′-dichlorofluorescein diacetate (DCFH-DA) was used as an indicator of intracellular formation of ROS as described previously [33]. DCFH-DA is a cell-permeant probe that enters the cell followed by cleavage of the diacetate molecules by cellular esterases. The probe becomes fluorescent when it is oxidized in cells by ROS. In brief, BV-2 cells were plated onto 24-well plates at a density of 45,000 cells/well. Twenty four hours after plating, cells were washed twice with 1X PBS+ (1X PBS with 0.14 g/L CaCl2 and 0.1 g/L of MgCl2) and subsequently incubated in PBS+ containing 10 μM DCFH-DA (Invitrogen, CA/Molecular Probes, Eugene, OR) for 45 min. The cells were rinsed twice with 1X PBS+ to wash off the probe and then treated with a single dose of 2, 4, 6, 8 or 10 Gy of 137Cs γ rays ; controls were sham-irradiated. As a positive control, cells were treated with either 100 μM or 1 mM H2O2 for 1 h prior to fluorescence measurement. ROS generation was measured 1 h post-irradiation as relative fluorescent intensity using a FLUOstar OPTIMA multimodal plate reader (BMG LabTech, Germany) at excitation wavelength 485 nm and emission wavelength 530nm. Ten micromolar carboxy-DCFH-DA (Invitrogen/Molecular Probes) was used as a negative control for ROS assays. C-369 is a non-oxidizable fluorescent probe that does not change its fluorescence in the presence of ROS. For experiments with GW7647, cells were incubated with 10 μM GW7647 or vehicle (DMSO) for 24 h prior to being incubated with DCFH-DA. For experiments with Fenofibrate, cells were incubated with DCFH-DA for 45 min and subsequently treated with 100 μM Fenofibrate or vehicle (DMF) for 1 h prior to irradiation. Irradiation and fluorescence measurements were carried out as outlined above.
RNA isolation and Real-time qPCR
RNA was isolated from cells using Trizol reagent (Invitrogen) according to manufacturer’s specifications. DNA contamination from RNA was removed by subjecting it to Acid-Phenol Chloroform extraction (pH 4.6, 125:24:1, Ambion Inc., Austin, TX) followed by RQ1 DNase treatment (Promega, Madison, WI). The PCR amplifications were done in 25 μL reaction volume containing 2 μL cDNA, 12.5 μL of Platinum® Quantitative PCR SuperMix-UDG w/ROX (Invitrogen), unlabeled antisense primer, FAM-labeled sense primer and nuclease-free water. The PCR reaction was carried out in a ABI Prism® 7000 at 50° C for 2 min, 95 ° C for 2 min and 45 cycles of 95°C for 15 min, 55 ° C for 30 sec and 72 ° C for 30 sec. The fold changes in gene expression of TNFα and IL-1β were calculated using the comparative Ct (cross threshold) method. Briefly, the Ct of the housekeeping gene β-actin was subtracted from the Ct of TNFα or IL-1β to get Δ Ct. The Δ Ct value of sham-irradiated sample was then subtracted from the Δ Ct of the rest of the treatments to get the ΔΔ Ct value. Fold differences compared to sham-irradiated sample are obtained by calculating 2-ΔΔCt for each treatment group. Data represent the Mean ± S.E.M of three independent experiments.
Electromobility shift assay (EMSA)
Cells were lysed with cold Buffer A (10 mM HEPES pH 7.9, 1.5 mM MgCl2,10 mM KCl, 0.5 mM DTT) followed by homogenization using a Dounce homogenizer (B type pestle). Lysed cells were centrifuged at 12,000 rpm for 2 min to isolate nuclei. Nuclear protein was isolated by treating the nuclear pellet with Buffer C (5 mM HEPES pH 7.9, 1.5 mM MgCl2, 25% v/v Glycerol, 400 mM NaCl, 1 mM EDTA 0.5 mM DTT, 0.5 mM PMSF, 2 μg/mL aprotinin, 2 μg/mL leupeptin and 1 mM Na3VO4) for 30 min in ice followed by centrifugation at 12000 rpm for 2 min. Protein concentrations were calculated using the Bradford Assay (Bio-Rad, Hercules, CA) by measuring the absorbance at 595 nm. The EMSA procedure was carried out using the Promega Gel-Shift Core Assay System according to the manufacturer’s instructions. Briefly, 10 μg of nuclear protein in sterile water were incubated with 2 μL of Binding Buffer (Promega) for 10 min. Consensus NF-κB binding sequence: 5’-AGTTGAGGGGACTTTCCCAGG C-3’and 3’-TCA ACTCCCCTGAAAGGGTCCG-5’and AP-1 binding sequence: 5’-CGCTTGATGAGTCAGCCGGAA-3’ and 3’-GCGAACTACTCAGTCGGCCTT-5’ were labeled with 20 μCi γ-P32 (GE Healthcare, Piscataway, NJ) and T4 polynucleotide kinase (Promega) and subsequently incubated with the nuclear protein samples for 20 min and electrophoresed on a 4% non-denaturing polyacrylamide gel. The gel was subsequently stained in 7% acetic acid, washed twice with water, and vacuum dried (Thermo Scientific, Waltham, MA) for 20 min. An X-ray film was placed on top of the dried gel and the image allowed to develop at -80° C. The X-ray film was processed using a Kodak Processing System. Films were scanned and densitometry was performed to quantify the intensity of the signal (Scion Image, Frederick, MD). For supershift experiments, nuclear proteins were incubated with 2 μg of either anti-p65 or anti-c-Jun antibodies for 20 min after addition of radiolabeled probes. For competition assays, 1 μL of either NF-κB or AP-1 unlabelled oligos was added to the nuclear extracts prior to addition of radiolabeled probes.
Luciferase assay
8 × 104 BV-2 cells were plated on 24-well plates. Twenty four hours later, cells were co-transfected with 0.2 μg of either PPRE-ACOX (consensus PPRE for the rat acyl-CoA oxidase gene, a kind gift from Dr. Thomas McIntyre, Univ. of Utah) or control vector (pGL3, Promega) and a renilla plasmid (0.02 μg, pRL-SV40, a kind gift from Dr. Lee Yong Woo, Univ. of Virginia) using Effectene Reagent (Qiagen, Valencia, CA) according to the manufacturer’s protocol. Transfected cells were treated after 24 h with either vehicle or the PPARα agonists. Luciferase activity was then measured 24 h post-drug treatment using the Dual Luciferase assay kit (Promega) according to the manufacturer’s instructions. The change in luciferase activity is depicted as fold change in luminescence calculated as RLU of firefly/RLU of renilla luciferase.
Immunoblotting
Total cellular protein extraction was carried out using M-PER reagent (Thermo Scientific). Briefly, cells were washed and subsequently scraped in 1 mL of 1X PBS. Cell pellets were lysed and collected using M-PER mammalian lysis buffer (Pierce Biotechnology, Inc. Rockford, IL) containing 1 mg/mL aprotinin, 1 mg/mL leupeptin (Sigma-Aldrich), and 10 mg/mL phenylmethylsulfonyl fluoride (PMSF). Protein was quantified using the Bio-Rad DC protein assay kit (Bio-Rad). Thirty to 50 μg of protein were separated by SDS-PAGE. Proteins were transferred to a polyvinylidene fluoride (PVDF) membrane for 12–16 h at 30 V, and blocked in 5% skim milk in TBST (0.02 M Tris, 0.15 M NaCl, 0.05% Tween 20, pH 7.5). Following this, membranes were incubated with the respective primary antibodies diluted in 2% BSA in TBST overnight. Proteins were visualized using the ECL detection system (GE Healthcare, NJ) after incubation with the respective HRP-conjugated secondary antibodies. Films were scanned and densitometry was performed to quantify the intensity of the signals (Scion Image). Signal intensities were normalized using those of housekeeping gene products (β-actin for total cell lysates and total c-jun for nuclear protein extracts).
Statistical Analysis
All analyses were performed using SAS software (Cary, NC). Though the sample size within each treatment group is not large, we believe that the distribution for the outcome measure (e.g., ratio) is normally distributed in the population. Using our data, we also examined the distribution of the measures and the need for any transformations in order to minimize heterogeneity of variance. As deemed appropriate, either one-sample t test or the analysis of variance (ANOVA) was used for determining statistical significance between the experimental groups. Bonferroni and Tukey’s studentized range tests were used for the pair wise comparisons. The constant variance assumption was tested using Levene’s test for homogeneity of variance. If and when the assumption of constant variance was not valid, the Kruskal-Wallis test was performed.
RESULTS
BV-2 cells possess a functional PPARα
In order to confirm the suitability of the BV-2 cells for our studies, we co-transfected these cells with a PPRE-driven reporter plasmid construct along with a renilla vector and performed luciferase activity assays 24 h after treatment with the PPARα agonists GW7647 and Fenofibrate. Incubating BV-2 cells with GW7647 (1 μM and 10 μM) and Fenofibrate (100 μM) increased the luciferase activity 2-fold suggesting that PPARα is functional in these cells (data not shown). Since GW7647 and Fenofibrate were able to significantly activate PPARα at 1 μM and 100 μM, respectively, we chose these concentrations for the remainder of our studies.
Radiation leads to increases in intracellular ROS generation and induces a pro-inflammatory response in the microglia
Although several studies suggest that microglial cells show enhanced ROS generation following various inflammatory stimuli such as H2O2 [34] and lipopolysacharride [35], a radiation-induced increase in ROS generation has not been reported. When BV-2 cells were incubated with DCFH-DA for 45 min and then irradiated, we observed a dose-dependent increase in the intracellular ROS generation 1 h after irradiation (*p<0.05 vs. 0 Gy, Fig. 1A). The non-oxidizable control probe, carboxy-DCF (C369) did not show any radiation-induced difference in fluorescence (Fig. 1A)
Figure 1. Irradiating BV-2 cells leads to increases in intracellular ROS generation, TNFα and IL-1β gene expression as well as an increase in Cox-2 protein levels.
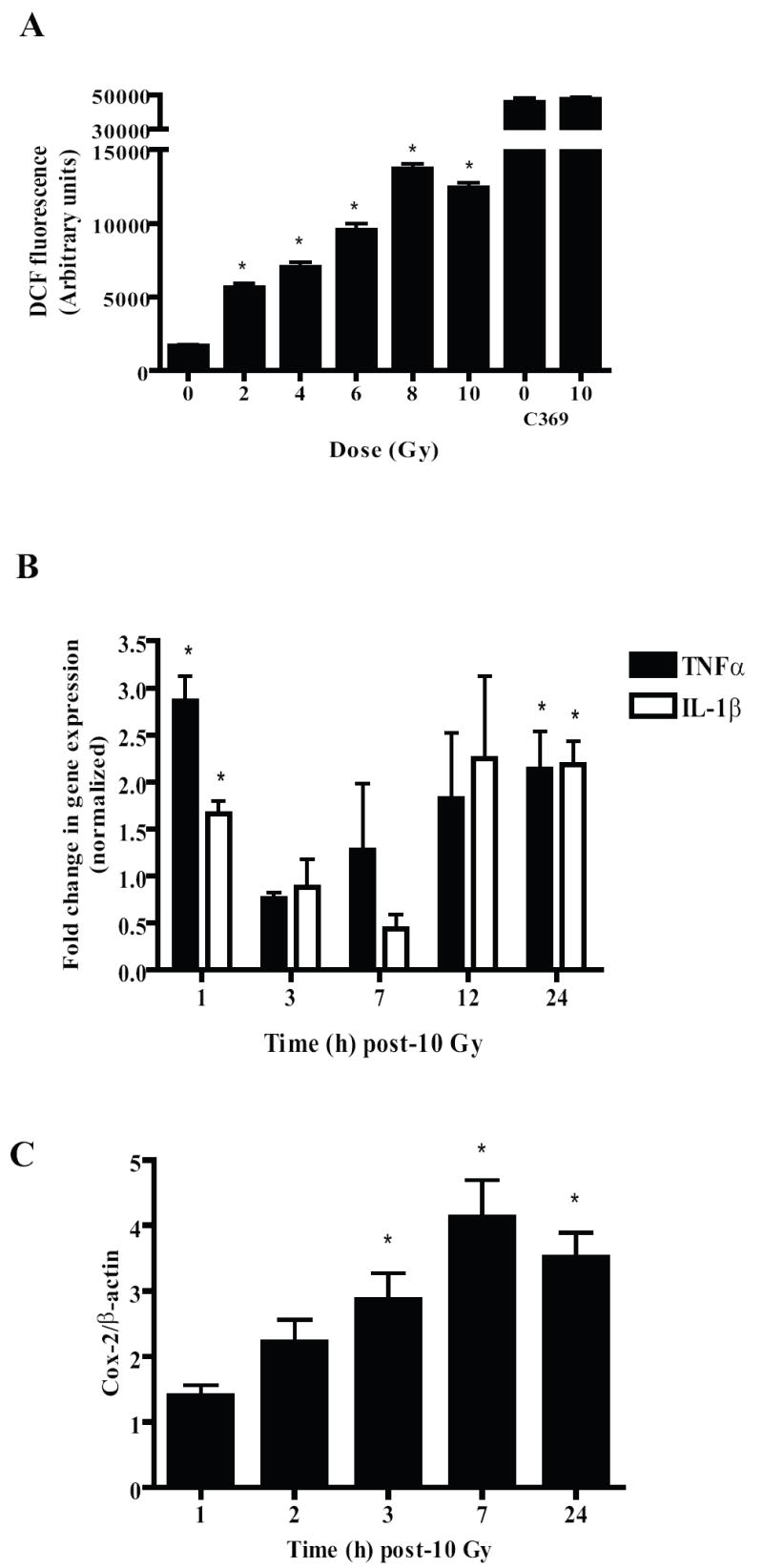
A, BV-2 cells were plated on 24-well plates. Twenty-four h later, cells were incubated with 10 μM DCFH-DA for 45 min. The probe was then washed off the cells using 1X PBS+ and the cells irradiated with a single dose of 2-10 Gy of 137Cs γ rays; control cells received sham-irradiation. Intracellular ROS were measured 1 h post-irradiation as described in Materials and methods. Results are presented as arbitrary fluorescence units for irradiated and sham-irradiated cells. Mean ± S.E.M; *, p< 0.05 vs. sham-irradiated cells; n = 3. C-369 served as a negative control for DCFH experiments. BV-2 cells were incubated with 10 μM carboxy-DCFDA (C-369), a non-oxidizable analog of DCFH and then were either treated with a single dose of 10 Gy or sham-irradiated. Mean ± S.E.M; n = 3. B and C, BV-2 cells were irradiated with a single dose of 10 Gy and RNA (B) or total cell lysates (C) were collected at the indicated times. Quantitative real-time PCR was used to detect changes in TNFα and IL-1β (B) gene expression. Results were calculated using the 2-ΔΔCt method (see Materials and Methods) and presented as fold changes compared to sham-irradiated cells after normalization with β-actin expression levels. Cox-2 levels (C) were measured using immunoblotting and normalized with β-actin protein levels. Mean ± S.E.M; *, p< 0.05 vs. sham-irradiated cells; n = 3.
Irradiating BV-2 cells with a single dose of 10 Gy led to significant, biphasic increases in gene expression of the pro-inflammatory cytokines TNF-α and IL-1β at 1 and 24 h post-irradiation (Fig. 1B). A radiation-induced increase in protein levels of COX-2 was also observed, with peak levels being seen at 7 h post-irradiation (Fig. 1C). Since ionizing radiation has been shown to increase the activity of proinflammatory transcription factors such as NF-κB [17] and AP-1 [36], we determined their radiation response in the BV-2 cells. As shown in Figs. 2A and D, we observed marked increases in the DNA binding of NF-κB and AP-1 as early as 30 min post-irradiation; maximal increases were seen 1 h post-irradiation.
Figure 2. Irradiating BV-2 cells leads to increases in NF-κB and AP-1 activation.

BV-2 cells were irradiated at indicated times using a single dose of 10 Gy and nuclear proteins were collected. Ten micrograms of nuclear extracts were incubated with either γ-32P-labelled NF-κB (A) or AP-1 (D) oligonucleotides (see Materials and Methods). Electro-mobility shift assay was performed as described in Materials and methods. For supershift assays (B and E), nuclear extracts from irradiated samples was incubated with 2 μg of anti-p65 or anti-c-jun antibodies for 20 min after addition of the radiolabeled probe. For competition assays (C and F), nuclear extracts were incubated with either unlabelled NF-κB or AP-1 oligonucleotides prior to addition of radiolabeled probe; CC= Cold competitor, NSC= Non-specific competitor. Blots are representative of three independent experiments (n=3).
When nuclear extracts from irradiated BV-2 cells were incubated with α-p65 or α-c-jun antibodies, we observed a marked decrease in the DNA binding of the nuclear proteins to the NF-κB (Fig. 2B) and AP-1 (Fig. 2E) consensus oligos indicating that the antibodies interfered with the binding of p65 and c-jun to the radiolabeled consensus DNA probe. These findings suggest that the p65 subunit of NF-κB and c-jun subunit of AP-1 are involved in the radiation response. Consistent with these results, we observed a significant increase in p65 nuclear translocation and nuclear c-jun phosphorylation at site Ser63 following irradiation (data not shown).
Regulation of radiation-induced proinflammatory responses in the microglia by AP-1 and NF-κB
Although the expression of TNFα, Cox-2 and IL-1β in the microglia is known to be regulated by AP-1 and NF-κB [37-41], a direct link between the pro-inflammatory transcription factors and the pro-inflammatory mediators has not been established in the context of ionizing radiation. To test this, we treated the BV-2 cells with either a specific c-jun kinase inhibitor, SP600125 (SP; 5 μM)) or two NF-κB activation inhibitors, 6-Amino-4-(4-phenoxyphenylethylamino) quinazoline (Q; 100 nM)) and Bay-117082 (Bay; 5 μM) and determined the radiation-induced pro-inflammatory response in the microglia. As anticipated, pre-treating BV-2 cells with the Q or SP inhibitor prevented the radiation-induced increase in NF-κB and AP-1 DNA binding, respectively (Figs. 3A and B) Similar inhibition of the radiation-induced activation of NF-κB was observed with the Bay inhibitor (data not shown). Further, treatment of BV-2 cells with SP inhibited the radiation-induced increase in TNFα, IL-1β and Cox-2 expression (Fig. 3C). In contrast, treating BV-2 cells with the Q inhibitor led to inhibition of the radiation-induced increase in IL-1β expression but failed to inhibit the radiation-induced increases in either Cox-2 or TNFα (Fig. 3C). Similar results were obtained using the Bay, NF-κB inhibitor (data not shown). To rule out the possibility that the JNK inhibitor had a non-specific effect on the NF-κB pathway, BV-2 cells were treated with SP for 1 h, irradiated and 1 h later, nuclear proteins were collected and subjected to EMSA. The SP compound did not alter the radiation-induced increase in NF-κB DNA binding (Fig. 3A) suggesting that the effect of SP on TNFα, IL-1β and Cox-2 is independent of the NF-κB pathway. Together, these data suggest that in the BV-2 cells, the radiation-induced increases in TNFα and Cox-2 are regulated primarily by the AP-1 pathway, while IL-1β expression is controlled by both AP-1 and NF-κB pathways.
Figure 3. The radiation-induced pro-inflammatory response is regulated, in part, by NF-κB and AP-1.

A and B, BV-2 cells were treated with either SP 600125 (SP) or 6-Amino-4-(4-phenoxyphenylethylamino) quinazoline (Q) for 1 h, and then irradiated with a single dose of 10 Gy. Nuclear extracts were collected 1 h post-irradiation and subjected to EMSA. Data are presented as fold changes in DNA-binding of NF-κB (A) and AP-1 (B) compared to sham-irradiated cells. Mean ± S.E.M; *, p< 0.05 vs. sham-irradiated cells; #, p< 0.05 vs. 10 Gy; n.s, non-significant vs. 10 Gy; n = 3. C, BV-2 cells were treated with the inhibitors as above. TNFα (black bars) and IL-1β (dotted bars) gene expression was measured using real-time qPCR 1 h and 24 h post-irradiation, respectively. Cox-2 (checked bars) protein expression was measured 7 h post-irradiation. Data are presented as fold changes compared to sham-irradiated cells. Mean ± S.E.M; *, p< 0.05 vs. sham-irradiated cells; #, p< 0.05 vs. 10 Gy; n.s, non-significant vs. 10 Gy, n = 3.
Activation of microglial PPARα prevents the radiation-induced pro-inflammatory responses
Next we tested whether activation of PPARα could prevent the radiation-induced microglial pro-inflammatory responses. BV-2 cells were pre-treated with either 100 μM Fenofibrate or 1 μM GW7647 and the pro-inflammatory responses of the cells were measured following irradiation. Both PPARα agonists significantly inhibited the radiation-induced increase in TNFα and IL-1β gene expression (Fig. 4A). Similarly, the radiation-induced increase in Cox-2 protein was significantly inhibited when the cells were pre-treated with Fenofibrate (# p< 0.05 vs. 10 Gy, Fig. 4B) and GW7647 (Supplemental Fig. S1A). These data indicate that PPARα activation modulates the radiation-induced microglial pro-inflammatory response, in part, by inhibiting the radiation-induced increases in TNFα, IL1β and Cox-2.
Figure 4. PPARα agonists ameliorate radiation-induced microglial pro-inflammatory responses.
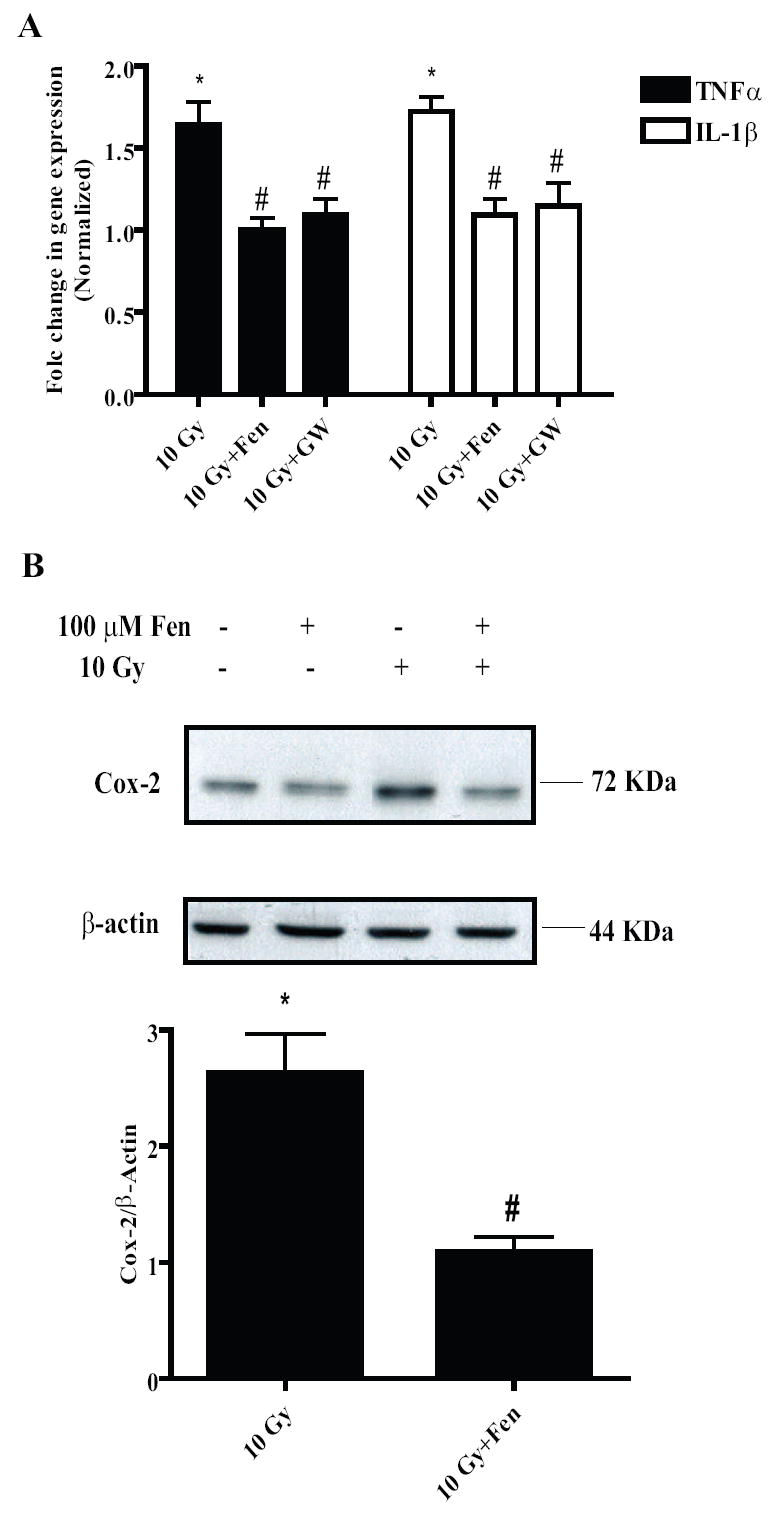
A, BV-2 cells were treated with either vehicle, GW7647 (1 μM) for 24 h or Fenofibrate (100 μM) for 1 h, irradiated and RNA was isolated for qPCR analysis of TNFα (black bars) and IL-1β (white bars) gene expression 24 h post-irradiation. Mean ± S.E.M; *, p< 0.05 vs. sham-irradiated cells; #, p< 0.05 vs. 10 Gy; n = 3. B, BV-2 cells were treated with Fenofibrate as above; total cell lysates were collected 7 h post-irradiation and subject to immunoblotting for Cox-2 and β-actin. B, upper panel shows representative blot of three independent experiments; lower panel shown densitometric analysis. Results are shown as fold changes compared to sham-irradiated cells. Mean ± S.E.M; *, p< 0.05; #, p< 0.05 vs. 10 Gy; n = 3.
PPARα activation does not alter the radiation-induced increase in microglial intracellular ROS generation
Studies have shown that the ROS generated by microglial cells following inflammatory insults such as thrombin [42] and LPS [43] play a role as second messengers in increasing the expression of several pro-inflammatory factors. Thus, we hypothesized that the modulation of the microglial pro-inflammatory responses by PPARα agonists could be due, in part, to inhibition of radiation-induced intracellular ROS generation. Using the DCFH-DA assay, we observed that incubating BV-2 cells with either Fenofibrate (100 μM) or GW7647 (1 and 10 μM) prior to irradiation did not affect ROS generation (Figs. 5A and 5B). These data suggest that in the BV-2 cells, PPARα ligands modulate the radiation-induced microglial pro-inflammatory response without altering the radiation-induced increase in intracellular ROS generation. This led us to hypothesize that inhibition of the radiation-induced increases in TNFα, IL-1β and Cox-2 by PPARα agonists might be through regulation of signaling events downstream of ROS generation.
Figure 5. PPARα agonists do not inhibit the radiation-induced increase in intracellular ROS generation.
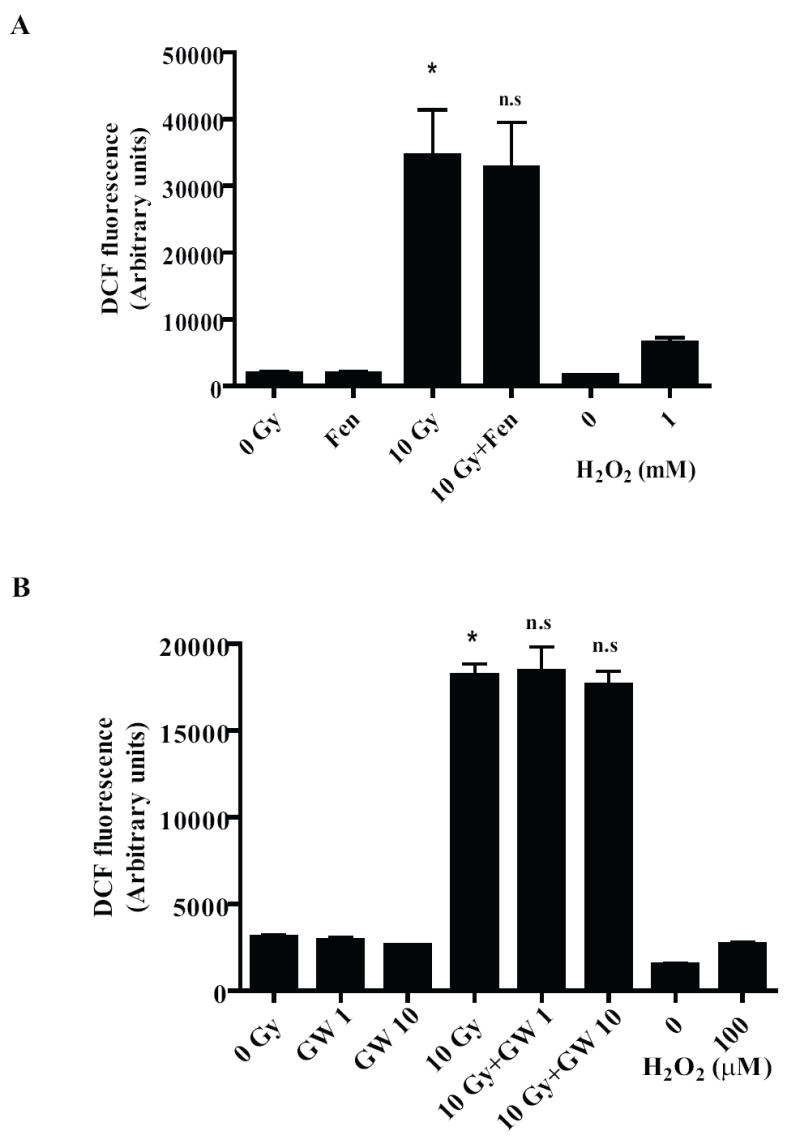
BV-2 cells were treated with either vehicle, Fenofibrate (A; 100 μM, 1 h) or GW7647 (B; 1 and 10 μM, 24 h) and intracellular ROS were measured as described above. As a positive control, cells were treated for 1 h with either 1mM (A) or 100 μM (B) H2O2 prior to ROS measurement. Results are presented as arbitrary fluorescence units for non-irradiated and irradiated samples. Mean ± S.E.M; *, p< 0.05 vs. sham-irradiated cells; n.s, non-significant vs. 10 Gy; n = 3.
Fenofibrate inhibits NF-κB and AP-1 transactivation properties, in part, by inhibiting p65 translocation and c-jun phosphorylation, respectively
Since we observed that the radiation-induced microglial pro-inflammatory response is regulated, in part, by NF-κB and AP-1, we wanted to determine whether the ability of PPARα agonists to prevent the radiation-induced increases in TNFα, IL-1β and Cox-2 was via inhibiting activation of these two transcription factors. Indeed, we observed a significant inhibition of NF-κB and AP-1 DNA binding activity when the BV-2 cells were pre-treated with Fenofibrate (# p<0.05 vs. 10 Gy, Figs. 6A and 7A) and GW7647 (Supplemental Fig. S1B).
Figure 6. PPARα activation in the microglia leads to negative regulation of the NF-κB pathway.
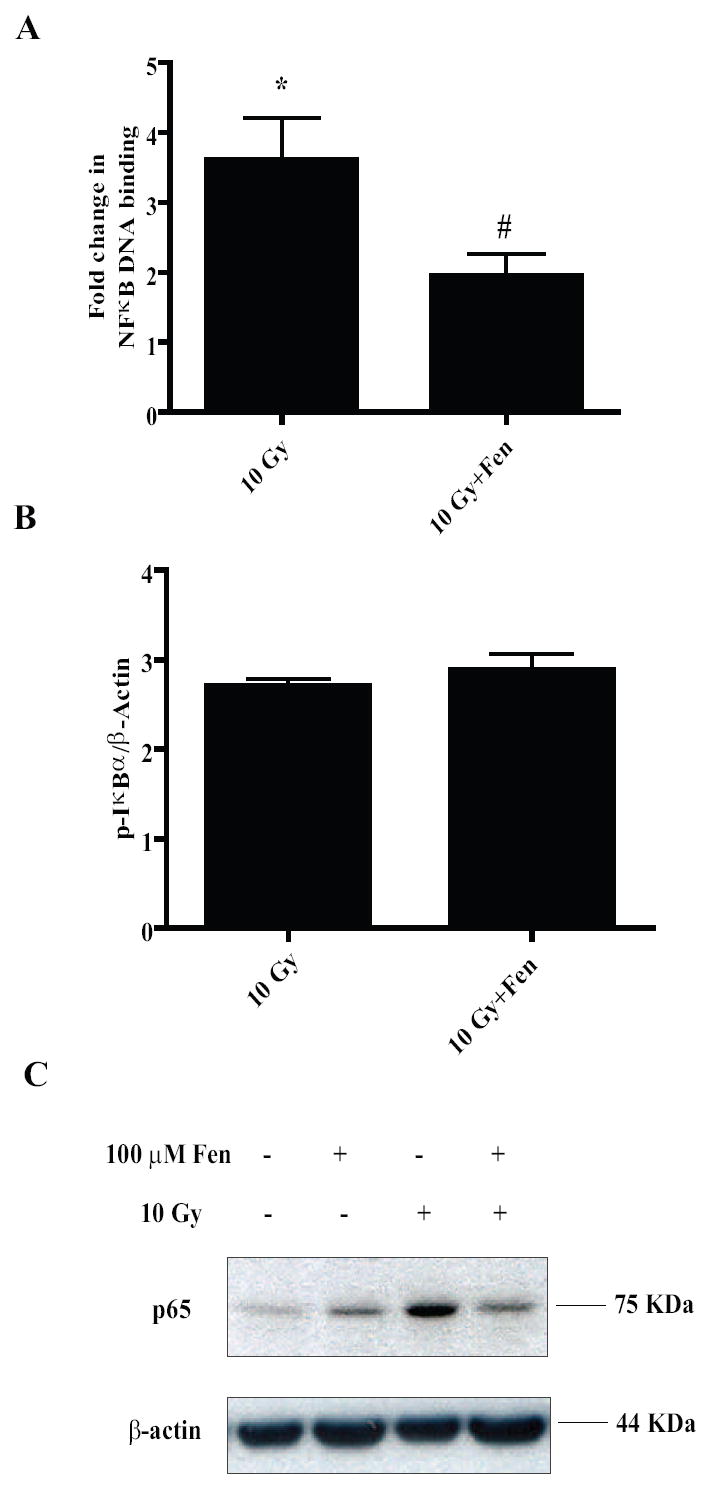
A, BV-2 cells were treated with vehicle or 100 μM Fenofibrate, irradiated and nuclear proteins were collected 1 h post-irradiation and used for EMSA using radiolabeled NF-κB consensus oligos. Results are presented as fold changes compared to sham-irradiated cells. Mean ± S.E.M; *, p< 0.05 vs. sham-irradiated cells; #, p< 0.05 vs. 10 Gy; n = 3. B, BV-2 cells were treated as above and total cell lysates were collected 1 h post-irradiation and subject to immunoblotting for p-IκBα. β-actin was used as the loading control. Results are presented as fold changes compared to sham-irradiated cells; and are representative of two independent experiments (n = 2). C, Nuclear extracts from BV-2 cells treated with Fenofibrate and/or radiation were subject to immunoblotting for p65 subunit of NF-κB. β-actin was used as the loading control. Results are representative of two independent experiments (n = 2).
Figure 7. PPARα activation in the microglia leads to negative regulation of the AP-1 pathway.
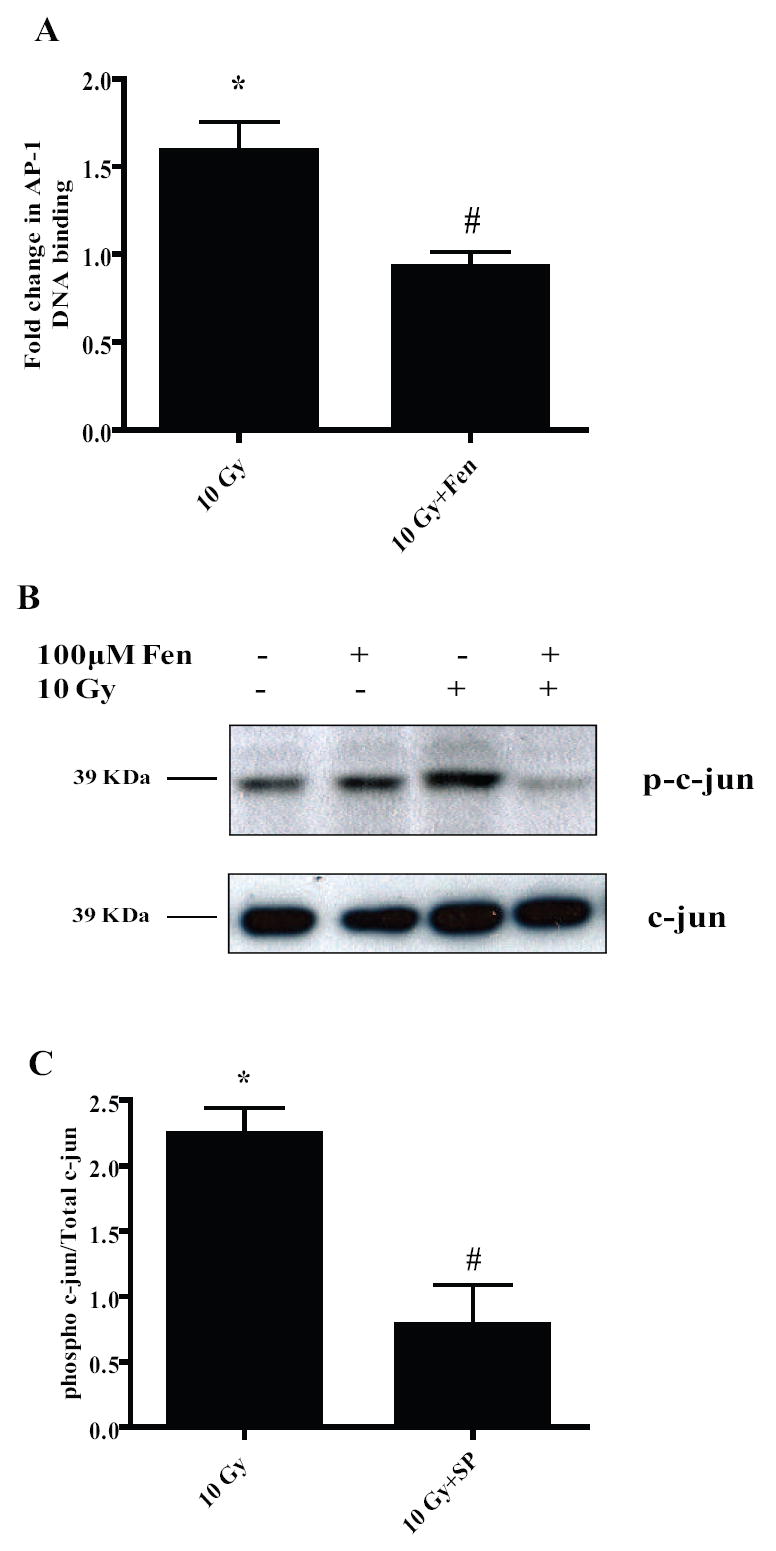
A, BV-2 cells were treated with vehicle or 100 μM Fenofibrate, irradiated and nuclear proteins were collected 1 h post-irradiation and used for EMSA using radiolabeled AP-1 consensus oligos. Results are presented as fold changes compared to sham-irradiated cells. Mean ± S.E.M; *, p< 0.05 vs. sham-irradiated cells; #, p< 0.05 vs. 10 Gy; n = 3. B, Nuclear extracts from A were subject to western blotting for phospho-c-jun. Total c-jun was used as the loading control. Results are representative of two independent experiments (n = 2). C, BV-2 cells were treated with vehicle or 5 μM SP600125 for 1 h, irradiated using a single dose of 10 Gy and nuclear proteins isolated and subjected to immunoblotting for phospho-c-jun. Total c-jun was used as the loading control; results are presented as fold changes compared to sham-irradiated cells. Mean ± S.E.M; *, p< 0.05 vs. sham-irradiated cells; #, p< 0.05 vs. 10 Gy; n = 3.
Exploring further into the inhibition of NF-κB, we tested whether phosphorylation of the inhibitor of NF-κB, IκBα was affected by Fenofibrate treatment. Irradiating the BV-2 cells led to an approximately 2.5-fold increase in phosphorylation of IκBα at Ser 32 which appeared to be unaffected by Fenofibrate treatment (Fig. 6B). Next we asked whether PPARα activation inhibits NF-κB DNA binding by decreasing the nuclear translocation of p65. Indeed, we observed that Fenofibrate treatment inhibited the radiation-induced increase in nuclear p65 levels (Fig. 6C). These data suggest that Fenofibrate modulates the radiation-induced expression of NF-κB dependent pro-inflammatory genes such as IL-1β, in part, by inhibiting p65 translocation into the nucleus.
To gain further insight into the signaling mechanism(s) involved in the inhibition of AP-1 DNA binding by PPARα agonists, we examined the phosphorylation status of c-jun in the nucleus following Fenofibrate treatment. We observed a marked increase in phosphorylation of nuclear c-jun at Ser63 residue which was inhibited when BV-2 cells were pre-treated with Fenofibrate (Fig. 7B). In order to determine the kinase responsible for the c-jun phosphorylation, we examined the effect of the JNK inhibitor, SP, on radiation-induced ser63c-jun phosphorylation. SP abrogated the phosphorylation of Ser63 of c-jun (# p< 0.05 vs. 10 Gy, Fig. 7C) suggesting that the radiation-induced activation of c-jun/AP-1 is mediated predominantly by JNK. Overall, these data suggest that Fenofibrate modulates the radiation-induced microglial pro-inflammatory response, in part, by preventing the activating phosphorylation of c-jun in the nucleus.
DISCUSSION
These studies indicate that irradiating BV-2 cells leads to an increase in intracellular ROS generation. Increases in ROS levels could amplify the pro-inflammatory responses of the microglia though effects on kinase signaling pathways and transcription factor activation [44]. Consistent with this, we also observed increased TNFα and IL-1β gene expression and Cox-2 protein levels following irradiation, extending previous reports [15-17]. Further, the current data not only confirm that irradiating microglial cells can increase the DNA binding activity of NF-κB [17] but demonstrate a similar radiation-induced increase in AP-1 activation. We hypothesized that pre-treatment of the microglial cells with PPARα agonists would prevent the radiation-induced pro-inflammatory responses. Indeed, PPARα activation effectively prevented the radiation-induced increases in TNFα, IL-1β and Cox-2, in part, by negatively regulating NF-κB and AP-1. To our knowledge, this is the first report examining the role of PPARα in modulating radiation-induced changes in microglial cell phenotype.
Previous studies have demonstrated that irradiating either primary murine microglial cells or immortalized murine BV-2 microglial cells led to increases in the gene expression of TNFα, IL-1β and Cox-2 [15-17]. However, these studies were limited, using either a single dose of 25 Gy [15,16], one or two time points following irradiation, or analysis of changes in gene expression using semi-quantitative RT-PCR [15-17]. The current studies confirm and extend considerably these previous findings. Thus, we observed increases in gene expression of TNFα and IL-1β using quantitative real-time PCR in addition to an increase in Cox-2 protein levels in the BV-2 cells following irradiation. Of interest, the increase in TNFα and IL-1β gene expression demonstrated a biphasic pattern following radiation. However, the biological importance of this biphasic response remains to be determined. Cox-2 mediated production of Prostaglandin E2, TNFα and IL-1β from the conditioned media of irradiated BV-2 cells has been shown to be important for alterations in astrocyte phenotype in vitro [17]. Radiation-induced increases in microglial TNFα and IL-1β have been proposed to be responsible for the increase in leukocyte adhesion in the brain via upregulation of ICAM-1 in astrocytes [16]. In vivo, brain irradiation leads to increases in gene expression of TNFα, IL-1β and Cox-2 acutely (4-24 h) [9, 15] and that of TNFα chronically (6 months) [8].
While the exact role of these pro-inflammatory mediators in the pathogenesis of radiation-induced brain injury is still under investigation, a hint to their function is suggested by studies with other brain injury models. Increased levels of pro-inflammatory cytokines have been associated with a number of neuro-inflammatory conditions such as Alzheimer’s disease [45], Parkinson’s disease [46] and multiple sclerosis [47]. Transgenic overexpression of TNFα, IL-1β and Cox-2 has been shown to induce behavioral and memory impairments in rodents [48-50]. TNFα and IL-1β have been shown to be potent inducers of apoptosis in oligodendrocytes and neural progenitor cells [51-53]. Thus, inhibiting the pro-inflammatory response following radiation appears a promising strategy to minimize radiation-induced brain injury.
PPARα, while classically known to be involved in fatty acid oxidation, has recently been demonstrated to be an important anti-inflammatory mediator [21]. PPARα ligands have been shown to inhibit both cytokine- and LPS-induced increases in pro-inflammatory mediators such as TNFα, IL-1β, Cox-2 and IL-6 in a variety of cell types including microglia [28-31]. Thus, we hypothesized that activation of PPARα in the microglia would inhibit the radiation-induced proinflammatory response. Indeed, we demonstrated that the radiation-induced increases in TNFα, IL-1β gene expression and Cox-2 protein were significantly inhibited by the PPARα agonists, GW7647 and Fenofibrate. Our findings emphasize the pleiotropic effects of PPARα agonists in response to inflammation as they target multiple pro-inflammatory microglial cytokines that might be involved in the development and progression of radiation-induced brain injury.
The promoter regions of TNFα, IL-1β and Cox-2 contain numerous transcription factor binding sites including AP-1 and NF-κB and several reports suggest that their expression in the microglia is regulated by these transcription factors [37-41]. Consistent with this, we observed marked increases in the DNA binding activity of AP-1 and NF-κB as early as 30 min post-irradiation in the microglial cells. Inhibiting JNK activation prevented the radiation-induced increase in microglial TNFα, IL-1β and Cox-2; inhibiting NF-κB prevented only the radiation-induced increase in IL-1β expression. These data indicate that, in the context of the radiation response of BV-2 cells, the expression of TNFα, and Cox-2 appears to be regulated by AP-1 and that IL-1β expression is regulated by both NF-κB and AP-1. Overall, these results highlight the importance of these two transcription factors in mediating the microglial pro-inflammatory response following radiation. This led us to hypothesize that the mechanism by which PPARα prevented the radiation-induced increases in TNFα, IL-1β and Cox-2 could involve, in part, inhibition of AP-1 and NF-κB activation.
Several lines of evidence suggest that PPARα mediates its anti-inflammatory effects via negative regulation of NF-κB. In human aortic smooth muscle cells (HASMCs), PPARα has been shown to inhibit NF-κB transactivation function via direct interaction with the Rel homology domain of the p65 subunit, which mediates its DNA binding and dimerization activity [28]. In addition, PPARα ligands upregulated the expression of the NF-κB inhibitor protein IκBα in both HASMCs and microglial cells leading to decreased DNA binding of NF-κB and p65-mediated gene expression [31, 54]. We observed that the PPARα ligands significantly diminished the radiation-induced increase in NF-κB DNA binding. This negative regulation of NF-κB appears to be downstream of IκBα degradation; Fenofibrate did not affect Ser32IκBα phosphorylation following irradiation, consistent with previous findings [54]. A number of post-translation events such as acetylation and phosphorylation activate p65 following its release from IκBα [55]. Thus, it is possible that Fenofibrate might negatively impact such modifications thereby leading to the retention of inactive p65 in the cytoplasm.
In addition to NF-κB, PPARα has been shown to downregulate the pro-inflammatory response by interfering with components of the AP-1 signaling pathway [28]. In the liver, Fenofibrate inhibited IL-6- induced acute phase gene expression by decreasing both total c-jun and Ser73phosphorylated c-jun (activating phosphorylation) in the nucleus [56]. In HASMCs, PPARα inhibited c-jun transactivation by directly binding to its c-jun kinase (JNK) phosphorylation domain [28]. The transactivation function of c-jun has been shown to depend on the phosphorylation of residues Ser73 and Ser63 [57]. Our studies indicate that in the microglial cells, the PPARα ligands significantly inhibited the radiation-induced increase in AP-1 DNA binding, in part, by inhibiting the phosphorylation of c-jun at Ser63 in the nucleus. Whether c-jun phosphorylation is affected due to direct binding of PPARα to c-jun or inhibition of upstream signaling events such as JNK activation remains to be determined in future research.
Previous studies indicate that the promoter regions of primary antioxidant enzymes including catalase and CuZnSOD possess PPREs [58, 59]. WY-14643, a PPARα ligand, has been shown to protect neurons from Aβ-induced toxicity, in part, by upregulating catalase protein levels [60]. However, we failed to detect any changes in the expression of antioxidant enzymes in BV-2 cells incubated with PPARα agonist (data not shown). Indeed, PPARα activation also failed to inhibit the increase in intracellular ROS determined in irradiated BV-2 cells. Thus, PPARα appeared to modulate the radiation-induced pro-inflammatory responses in microglial cells primarily by inhibiting AP-1 and/or NF-κB signaling.
To start to determine the specific ROS involved in the increased DCF oxidation following radiation, we incubated the BV-2 cells with L-NAME (a nitric oxide inhibitor, 1mM) for 1h prior to radiation. We observed that the radiation-induced increase in intracellular ROS was not inhibited by L-NAME (Supplemental Fig. S2). These results are consistent with previous observations that nitrite levels and inducible nitric oxide synthase gene expression were unchanged in the BV-2 cells following radiation [17]. Recently, we have shown that the radiation-induced ROS generation and subsequent pro-inflammatory response in rat brain microvascular endothelial cells is dependent, in part, on NADPH oxidase-dependent superoxide generation [61]. Whether NADPH oxidase is one of the key players involved in the radiation-induced microglial ROS generation is currently being studied in our laboratory.
Based on the current findings, we propose a working model for the role of PPARα in modulating the radiation-induced microglial inflammation as outlined in Figure 8. Radiation, through the generation of intracellular ROS, leads to activation of NF-κB and AP-1 in the microglia. These transcription factors increase the gene expression of TNFα, IL-1β and Cox-2 all of which contribute to the neuro-inflammatory phenotype of the microglia. Activation of PPARα by Fenofibrate and GW7647 appears to prevent the radiation-induced pro-inflammatory response, in part, by inhibiting the transactivation functions of NF-κB and AP-1. Mechanistically, inactivation of these transcription factors by PPARα agonists appears to be via decreased nuclear translocation of NF-κB/p65 subunit and reduced phosphorylation of nuclear c-jun/AP-1.
Figure 8. Proposed model outlining the role of PPARα in the modulation of radiation-induced microglial pro-inflammatory response.
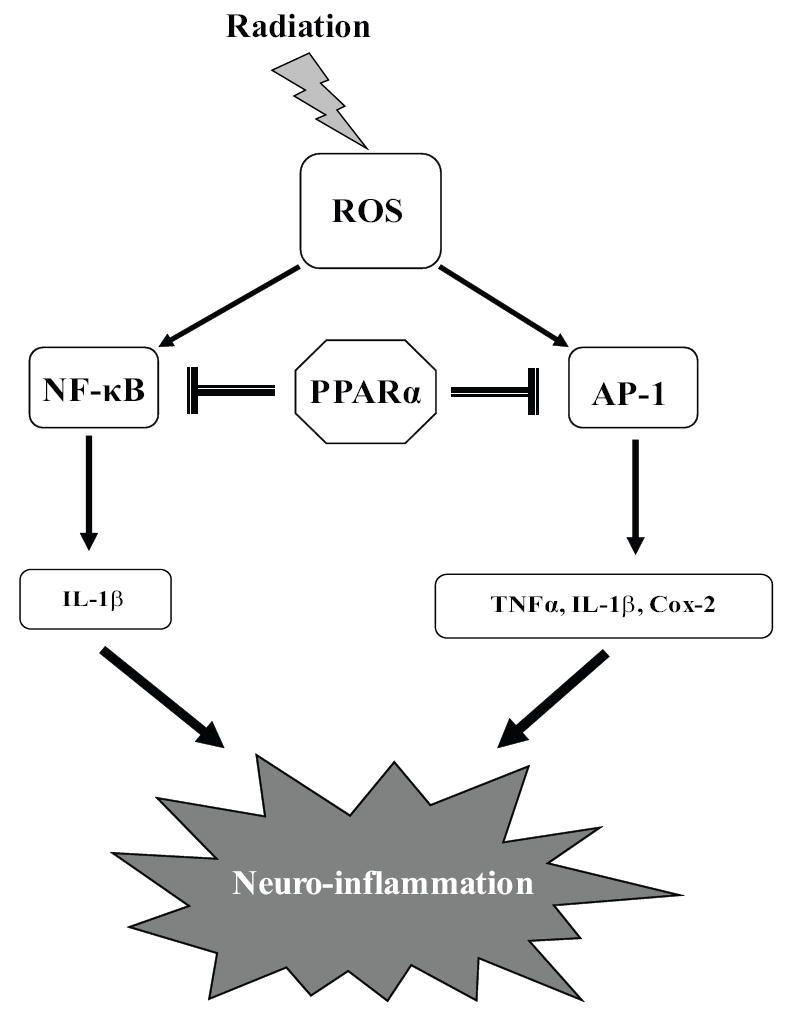
In the microglial cells, radiation leads to an increase in expression of pro-inflammatory mediators TNFα, IL-1β and Cox-2 all of which might contribute to augmented neuroinflammation. In addition, these mediators appear to be regulated by the increased activities of NF-κB and AP-1 following radiation. Pre-treating the microglial cells with PPARα ligands, Fenofibrate and GW 7647, inhibit the radiation-induced increase in TNFα, IL-1β and Cox-2, in part, by preventing the activation of NF-κB and AP-1. Thus, PPARα ligands represent a novel class of agents that could inhibit the radiation-induced microglial pro-inflammatory response and the resulting neuroinflammation.
In summary, our data indicate that PPARα agonists can prevent the pro-inflammatory responses of the microglia following radiation. While this study addresses the response of the microglia to radiation in vitro, it does not adequately represent the in vivo scenario in which multiple cells types in the brain are irradiated and likely contribute to the pathogenesis of radiation-induced brain injury. Given the reports that PPARα ligands inhibit the proinflammatory responses of astrocytes [62] and are also neurotrophic [63], PPARα may mediate its anti-inflammatory effects in vivo in more than one cell type. Moreover, these in vitro studies utilize microglial cells grown under conditions of 21% oxygen concentration, a value much higher than would occur in vivo. The impact of “physiological” oxygen concentrations on the radiation-induced inflammatory response of microglial cells remains to be determined. However, animal studies have shown clearly that PPARα ligands can cross the blood-brain barrier and appear to be neuroprotective following ischemia-reperfusion [64], experimental autoimmune encephalomyelitis [65] (EAE) and stroke [66]. Overall, our in vitro data suggest that PPARα ligands offer promise as potent agents that could prove efficacious in the treatment and/or prevention of radiation-induced brain injury.
Supplementary Material
Acknowledgments
This work was supported by NIH grant CA112593 (MER). We thank Dr. Linda van Eldik, Northwestern University, USA, for generously providing the BV-2 cells, originally developed by Dr.V.Bocchini , Univeristy of Perugia, Italy. We thank Denise Gibo [Brain Tumor Center of Excellence Wake Forest University School of Medicine (WFUSM)] and Dr. Carol Milligan (Department of Neurobiology and Anatomy, WFUSM) for providing antibodies against total c-jun and phosphorylated c-jun, respectively. We also thank Sowmya Kootala for helpful comments and discussion.
LIST OF ABBREVIATIONS
- WBI
Whole-brain irradiation
- PPARα
peroxisomal proliferator activated receptor alpha
- TNFα
tumor necrosis factor alpha
- IL-1β
Interleukin 1 beta
- Cox-2
cyclooxygenase-2
- ROS
reactive oxygen species
- Gy
Gray
- NF-κB
nuclear factor kappa B
- AP-1
activator protein-1
- JNK
c-jun N-terminal kinase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Khuntia D, Brown P, Li J, Mehta MP. Whole-brain radiotherapy in the management of brain metastasis. J Clin Oncol. 2006;24:1295–1304. doi: 10.1200/JCO.2005.04.6185. [DOI] [PubMed] [Google Scholar]
- 2.Wen PY, Black PM, Loeffler JS. Metastatic Brain Cancer. In: DeVita V, Hellman S, Rosenberg SA, editors. Cancer: Principles and Practice of Oncology. 6. Philadelphia: Lippincott, Williams & Wilkins; 2001. pp. 2655–2670. [Google Scholar]
- 3.Mehta MP, Tremont-Lukats I. Radiosurgery for single and multiple brain metastasis. In: Sawaya R, editor. Intracranial metastases: Current management strategies. Malden: Blackwell publishing,Futura division; 2004. pp. 139–164. [Google Scholar]
- 4.Greenberg H, Chandler WF, Sandler HM. Brain Metastases. In: Greenberg H, Chandler WF, Sandler HM, editors. Brain tumors. New York: Oxford University Press; 1999. pp. 299–317. [Google Scholar]
- 5.Crossen JR, Garwood D, Glatstein E, Neuwelt EA. Neurobehavioral sequelae of cranial irradiation in adults: a review of radiation-induced encephalopathy. J Clin Oncol. 1994;12:627–642. doi: 10.1200/JCO.1994.12.3.627. [DOI] [PubMed] [Google Scholar]
- 6.Zhao W, Diz DI, Robbins ME. Oxidative damage pathways in relation to normal tissue injury. Br J Radiol. 2007;80(Spec No 1):S23–S31. doi: 10.1259/bjr/18237646. [DOI] [PubMed] [Google Scholar]
- 7.Raju U, Gumin GJ, Tofilon PJ. NF kappa B activity and target gene expression in the rat brain after one and two exposures to ionizing radiation. Radiat Oncol Investig. 1999;7:145–152. doi: 10.1002/(SICI)1520-6823(1999)7:3<145::AID-ROI2>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 8.Chiang CS, Hong JH, Stalder A, Sun JR, Withers HR, McBride WH. Delayed molecular responses to brain irradiation. Int J Radiat Biol. 1997;72:45–53. doi: 10.1080/095530097143527. [DOI] [PubMed] [Google Scholar]
- 9.Chiang CS, McBride WH. Radiation enhances tumor necrosis factor alpha production by murine brain cells. Brain Res. 1991;566:265–269. doi: 10.1016/0006-8993(91)91707-8. [DOI] [PubMed] [Google Scholar]
- 10.Hong JH, Chiang CS, Campbell IL, Sun JR, Withers HR, McBride WH. Induction of acute phase gene expression by brain irradiation. Int J Radiat Oncol Biol Phys. 1995;33:619–626. doi: 10.1016/0360-3016(95)00279-8. [DOI] [PubMed] [Google Scholar]
- 11.van Rossum D, Hanisch UK. Microglia. Metab Brain Dis. 2004;19:393–411. doi: 10.1023/b:mebr.0000043984.73063.d8. [DOI] [PubMed] [Google Scholar]
- 12.Elkabes S, DiCicco-Bloom EM, Black IB. Brain microglia/macrophages express neurotrophins that selectively regulate microglial proliferation and function. J Neurosci. 1996;16:2508–2521. doi: 10.1523/JNEUROSCI.16-08-02508.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- 14.Kim SU, de Vellis J. Microglia in health and disease. J Neurosci Res. 2005;81:302–313. doi: 10.1002/jnr.20562. [DOI] [PubMed] [Google Scholar]
- 15.Kyrkanides S, Moore AH, Olschowka JA, Daeschner JC, Williams JP, Hansen JT, Kerry OM. Cyclooxygenase-2 modulates brain inflammation-related gene expression in central nervous system radiation injury. Brain Res Mol Brain Res. 2002;104:159–169. doi: 10.1016/s0169-328x(02)00353-4. [DOI] [PubMed] [Google Scholar]
- 16.Kyrkanides S, Olschowka JA, Williams JP, Hansen JT, O’Banion MK. TNF alpha and IL-1beta mediate intercellular adhesion molecule-1 induction via microglia-astrocyte interaction in CNS radiation injury. J Neuroimmunol. 1999;95:95–106. doi: 10.1016/s0165-5728(98)00270-7. [DOI] [PubMed] [Google Scholar]
- 17.Hwang SY, Jung JS, Kim TH, Lim SJ, Oh ES, Kim JY, Ji KA, Joe EH, Cho KH, Han IO. Ionizing radiation induces astrocyte gliosis through microglia activation. Neurobiol Dis. 2006;21:457–467. doi: 10.1016/j.nbd.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 18.Rola R, Raber J, Rizk A, Otsuka S, VandenBerg SR, Morhardt DR, Fike JR. Radiation-induced impairment of hippocampal neurogenesis is associated with cognitive deficits in young mice. Exp Neurol. 2004;188:316–330. doi: 10.1016/j.expneurol.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 19.Monje ML, Mizumatsu S, Fike JR, Palmer TD. Irradiation induces neural precursor-cell dysfunction. Nat Med. 2002;8:955–962. doi: 10.1038/nm749. [DOI] [PubMed] [Google Scholar]
- 20.Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- 21.Willson TM, Brown PJ, Sternbach DD, Henke BR. The PPARs: from orphan receptors to drug discovery. J Med Chem. 2000;43:527–550. doi: 10.1021/jm990554g. [DOI] [PubMed] [Google Scholar]
- 22.Forman BM, Chen J, Evans RM. The peroxisome proliferator-activated receptors: ligands and activators. Ann N Y Acad Sci. 1996;804:266–275. doi: 10.1111/j.1749-6632.1996.tb18621.x. [DOI] [PubMed] [Google Scholar]
- 23.Kliewer SA, Forman BM, Blumberg B, Ong ES, Borgmeyer U, Mangelsdorf DJ, Umesono K, Evans RM. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc Natl Acad Sci U S A. 1994;91:7355–7359. doi: 10.1073/pnas.91.15.7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Auwerx J, Schoonjans K, Fruchart JC, Staels B. Regulation of triglyceride metabolism by PPARs: fibrates and thiazolidinediones have distinct effects. J Atheroscler Thromb. 1996;3:81–89. doi: 10.5551/jat1994.3.81. [DOI] [PubMed] [Google Scholar]
- 25.Madej A, Okopien B, Kowalski J, Zielinski M, Wysocki J, Szygula B, Kalina Z, Herman ZS. Effects of fenofibrate on plasma cytokine concentrations in patients with atherosclerosis and hyperlipoproteinemia IIb. Int J Clin Pharmacol Ther. 1998;36:345–349. [PubMed] [Google Scholar]
- 26.Neve BP, Corseaux D, Chinetti G, Zawadzki C, Fruchart JC, Duriez P, Staels B, Jude B. PPARalpha agonists inhibit tissue factor expression in human monocytes and macrophages. Circulation. 2001;103:207–212. doi: 10.1161/01.cir.103.2.207. [DOI] [PubMed] [Google Scholar]
- 27.Marx N, Kehrle B, Kohlhammer K, Grub M, Koenig W, Hombach V, Libby P, Plutzky J. PPAR activators as antiinflammatory mediators in human T lymphocytes: implications for atherosclerosis and transplantation-associated arteriosclerosis. Circ Res. 2002;90:703–710. doi: 10.1161/01.res.0000014225.20727.8f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Delerive P, De Bosscher K, Besnard S, Vanden Berghe W, Peters JM, Gonzalez FJ, Fruchart JC, Tedgui A, Haegeman G, Staels B. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J Biol Chem. 1999;274:32048–32054. doi: 10.1074/jbc.274.45.32048. [DOI] [PubMed] [Google Scholar]
- 29.Drew PD, Xu J, Storer PD, Chavis JA, Racke MK. Peroxisome proliferator-activated receptor agonist regulation of glial activation: relevance to CNS inflammatory disorders. Neurochem Int. 2006;49:183–189. doi: 10.1016/j.neuint.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 30.Xu J, Storer PD, Chavis JA, Racke MK, Drew PD. Agonists for the peroxisome proliferator-activated receptor-alpha and the retinoid X receptor inhibit inflammatory responses of microglia. J Neurosci Res. 2005;81:403–411. doi: 10.1002/jnr.20518. [DOI] [PubMed] [Google Scholar]
- 31.Jana M, Jana A, Liu X, Ghosh S, Pahan K. Involvement of phosphatidylinositol 3-kinase-mediated up-regulation of I kappa B alpha in anti-inflammatory effect of gemfibrozil in microglia. J Immunol. 2007;179:4142–4152. doi: 10.4049/jimmunol.179.6.4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol. 1990;27:229–237. doi: 10.1016/0165-5728(90)90073-v. [DOI] [PubMed] [Google Scholar]
- 33.Smith PS, Zhao W, Spitz DR, Robbins ME. Inhibiting catalase activity sensitizes 36B10 rat glioma cells to oxidative stress. Free Radic Biol Med. 2007;42:787–797. doi: 10.1016/j.freeradbiomed.2006.11.032. [DOI] [PubMed] [Google Scholar]
- 34.Hou RC, Wu CC, Huang JR, Chen YS, Jeng KC. Oxidative toxicity in BV-2 microglia cells: sesamolin neuroprotection of H2O2 injury involving activation of p38 mitogen-activated protein kinase. Ann N Y Acad Sci. 2005;1042:279–285. doi: 10.1196/annals.1338.050. [DOI] [PubMed] [Google Scholar]
- 35.Kim HS, Ye SK, Cho IH, Jung JE, Kim DH, Choi S, Kim YS, Park CG, Kim TY, Lee JW, Chung MH. 8-hydroxydeoxyguanosine suppresses NO production and COX-2 activity via Rac1/STATs signaling in LPS-induced brain microglia. Free Radic Biol Med. 2006;41:1392–1403. doi: 10.1016/j.freeradbiomed.2006.07.018. [DOI] [PubMed] [Google Scholar]
- 36.Chastel C, Jiricny J, Jaussi R. Activation of stress-responsive promoters by ionizing radiation for deployment in targeted gene therapy. DNA Repair (Amst) 2004;3:201–215. doi: 10.1016/j.dnarep.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 37.Waetzig V, Czeloth K, Hidding U, Mielke K, Kanzow M, Brecht S, Goetz M, Lucius R, Herdegen T, Hanisch UK. c-Jun N-terminal kinases (JNKs) mediate proinflammatory actions of microglia. Glia. 2005;50:235–246. doi: 10.1002/glia.20173. [DOI] [PubMed] [Google Scholar]
- 38.Liu L, Li Y, Van Eldik LJ, Griffin WS, Barger SW. S100B-induced microglial and neuronal IL-1 expression is mediated by cell type-specific transcription factors. J Neurochem. 2005;92:546–553. doi: 10.1111/j.1471-4159.2004.02909.x. [DOI] [PubMed] [Google Scholar]
- 39.Jeon YJ, Han SH, Lee YW, Lee M, Yang KH, Kim HM. Dexamethasone inhibits IL-1 beta gene expression in LPS-stimulated RAW 264.7 cells by blocking NF-kappa B/Rel and AP-1 activation. Immunopharmacology. 2000;48:173–183. doi: 10.1016/s0162-3109(00)00199-5. [DOI] [PubMed] [Google Scholar]
- 40.Deng WG, Montero AJ, Wu KK. Interferon-gamma suppresses cyclooxygenase-2 promoter activity by inhibiting C-Jun and C/EBPbeta binding. Arterioscler Thromb Vasc Biol. 2007;27:1752–1759. doi: 10.1161/ATVBAHA.107.144352. [DOI] [PubMed] [Google Scholar]
- 41.Swantek JL, Cobb MH, Geppert TD. Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) is required for lipopolysaccharide stimulation of tumor necrosis factor alpha (TNF-alpha) translation: glucocorticoids inhibit TNF-alpha translation by blocking JNK/SAPK. Mol Cell Biol. 1997;17:6274–6282. doi: 10.1128/mcb.17.11.6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choi SH, Lee DY, Kim SU, Jin BK. Thrombin-induced oxidative stress contributes to the death of hippocampal neurons in vivo: role of microglial NADPH oxidase. J Neurosci. 2005;25:4082–4090. doi: 10.1523/JNEUROSCI.4306-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang T, Qin L, Liu B, Liu Y, Wilson B, Eling TE, Langenbach R, Taniura S, Hong JS. Role of reactive oxygen species in LPS-induced production of prostaglandin E2 in microglia. J Neurochem. 2004;88:939–947. doi: 10.1046/j.1471-4159.2003.02242.x. [DOI] [PubMed] [Google Scholar]
- 44.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 45.Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW, Mrak RE. Glial-neuronal interactions in Alzheimer’s disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol. 1998;8:65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mogi M, Harada M, Narabayashi H, Inagaki H, Minami M, Nagatsu T. Interleukin (IL)-1 beta, IL-2, IL-4, IL-6 and transforming growth factor-alpha levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neurosci Lett. 1996;211:13–16. doi: 10.1016/0304-3940(96)12706-3. [DOI] [PubMed] [Google Scholar]
- 47.Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 48.Tobinick EL, Gross H. Rapid cognitive improvement in Alzheimer’s disease following perispinal etanercept administration. J Neuroinflammation. 2008;5:2. doi: 10.1186/1742-2094-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Song C, Horrobin D. Omega-3 fatty acid ethyl-eicosapentaenoate, but not soybean oil, attenuates memory impairment induced by central IL-1beta administration. J Lipid Res. 2004;45:1112–1121. doi: 10.1194/jlr.M300526-JLR200. [DOI] [PubMed] [Google Scholar]
- 50.Andreasson KI, Savonenko A, Vidensky S, Goellner JJ, Zhang Y, Shaffer A, Kaufmann WE, Worley PF, Isakson P, Markowska AL. Age-dependent cognitive deficits and neuronal apoptosis in cyclooxygenase-2 transgenic mice. J Neurosci. 2001;21:8198–8209. doi: 10.1523/JNEUROSCI.21-20-08198.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ye P, D’Ercole AJ. Insulin-like growth factor I protects oligodendrocytes from tumor necrosis factor-alpha-induced injury. Endocrinology. 1999;140:3063–3072. doi: 10.1210/endo.140.7.6754. [DOI] [PubMed] [Google Scholar]
- 52.Takahashi JL, Giuliani F, Power C, Imai Y, Yong VW. Interleukin-1beta promotes oligodendrocyte death through glutamate excitotoxicity. Ann Neurol. 2003;53:588–595. doi: 10.1002/ana.10519. [DOI] [PubMed] [Google Scholar]
- 53.Sheng WS, Hu S, Ni HT, Rowen TN, Lokensgard JR, Peterson PK. TNF-alpha-induced chemokine production and apoptosis in human neural precursor cells. J Leukoc Biol. 2005;78:1233–1241. doi: 10.1189/jlb.0405221. [DOI] [PubMed] [Google Scholar]
- 54.Delerive P, Gervois P, Fruchart JC, Staels B. Induction of IkappaBalpha expression as a mechanism contributing to the anti-inflammatory activities of peroxisome proliferator-activated receptor-alpha activators. J Biol Chem. 2000;275:36703–36707. doi: 10.1074/jbc.M004045200. [DOI] [PubMed] [Google Scholar]
- 55.Perkins ND. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene. 2006;25:6717–6730. doi: 10.1038/sj.onc.1209937. [DOI] [PubMed] [Google Scholar]
- 56.Gervois P, Kleemann R, Pilon A, Percevault F, Koenig W, Staels B, Kooistra T. Global suppression of IL-6-induced acute phase response gene expression after chronic in vivo treatment with the peroxisome proliferator-activated receptor-alpha activator fenofibrate. J Biol Chem. 2004;279:16154–16160. doi: 10.1074/jbc.M400346200. [DOI] [PubMed] [Google Scholar]
- 57.Herdegen T, Waetzig V. AP-1 proteins in the adult brain: facts and fiction about effectors of neuroprotection and neurodegeneration. Oncogene. 2001;20:2424–2437. doi: 10.1038/sj.onc.1204387. [DOI] [PubMed] [Google Scholar]
- 58.Kim YH, Park KH, Rho HM. Transcriptional activation of the Cu,Zn-superoxide dismutase gene through the AP2 site by ginsenoside Rb2 extracted from a medicinal plant, Panax ginseng. J Biol Chem. 1996;271:24539–24543. doi: 10.1074/jbc.271.40.24539. [DOI] [PubMed] [Google Scholar]
- 59.Girnun GD, Domann FE, Moore SA, Robbins ME. Identification of a functional peroxisome proliferator-activated receptor response element in the rat catalase promoter. Mol Endocrinol. 2002;16:2793–2801. doi: 10.1210/me.2002-0020. [DOI] [PubMed] [Google Scholar]
- 60.Santos MJ, Quintanilla RA, Toro A, Grandy R, Dinamarca MC, Godoy JA, Inestrosa NC. Peroxisomal proliferation protects from beta-amyloid neurodegeneration. J Biol Chem. 2005;280:41057–41068. doi: 10.1074/jbc.M505160200. [DOI] [PubMed] [Google Scholar]
- 61.Collins-Underwood JR, Zhao W, Sharpe JG, Robbins ME. NADPH oxidase mediates radiation-induced oxidative stress in rat brain microvascular endothelial cells. Free Radic Biol Med. 2008 doi: 10.1016/j.freeradbiomed.2008.06.024. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Storer PD, Xu J, Chavis J, Drew PD. Peroxisome proliferator-activated receptor-gamma agonists inhibit the activation of microglia and astrocytes: implications for multiple sclerosis. J Neuroimmunol. 2005;161:113–122. doi: 10.1016/j.jneuroim.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 63.Bento-Abreu A, Tabernero A, Medina JM. Peroxisome proliferator-activated receptor-alpha is required for the neurotrophic effect of oleic acid in neurons. J Neurochem. 2007;103:871–881. doi: 10.1111/j.1471-4159.2007.04807.x. [DOI] [PubMed] [Google Scholar]
- 64.Deplanque D, Gele P, Petrault O, Six I, Furman C, Bouly M, Nion S, Dupuis B, Leys D, Fruchart JC, Cecchelli R, Staels B, Duriez P, Bordet R. Peroxisome proliferator-activated receptor-alpha activation as a mechanism of preventive neuroprotection induced by chronic fenofibrate treatment. J Neurosci. 2003;23:6264–6271. doi: 10.1523/JNEUROSCI.23-15-06264.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lovett-Racke AE, Hussain RZ, Northrop S, Choy J, Rocchini A, Matthes L, Chavis JA, Diab A, Drew PD, Racke MK. Peroxisome proliferator-activated receptor alpha agonists as therapy for autoimmune disease. J Immunol. 2004;172:5790–5798. doi: 10.4049/jimmunol.172.9.5790. [DOI] [PubMed] [Google Scholar]
- 66.Inoue H, Jiang XF, Katayama T, Osada S, Umesono K, Namura S. Brain protection by resveratrol and fenofibrate against stroke requires peroxisome proliferator-activated receptor alpha in mice. Neurosci Lett. 2003;352:203–206. doi: 10.1016/j.neulet.2003.09.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.