

Abstract
Alternative sigma factors allow bacteria to reprogram global transcription rapidly and to adapt to changes in the environment. Here we report on growth- and cell division-dependent σ32 regulon activity in Escherichia coli in batch culture. By analyzing σ32 expression in growing cells, an increase in σ32 protein levels is observed during the first round of cell division after exit from stationary phase. Increased σ32 protein levels result from transcriptional activation of the rpoH gene. After the first round of bulk cell division, rpoH transcript levels and σ32 protein levels decrease again. The late-logarithmic phase and the transition to stationary phase are accompanied by a second increase in σ32 levels and enhanced stability of σ32 protein but not by enhanced transcription of rpoH. Throughout growth, σ32 target genes show expression patterns consistent with oscillating σ32 protein levels. However, during the transition to early-stationary phase, despite high σ32 protein levels, the transcription of σ32 target genes is downregulated, suggesting functional inactivation of σ32. It is deduced from these data that there may be a link between σ32 regulon activity and cell division events. Further support for this hypothesis is provided by the observation that in cells in which FtsZ is depleted, σ32 regulon activation is suppressed.
In Escherichia coli the regulation of the σ32 stress regulon, induced by heat shock or other stress conditions, has been extensively studied (15, 17, 31, 41). It is commonly accepted that environmental cues, such as elevated temperature or other conditions disturbing protein homeostasis, cause the accumulation of misfolded proteins and lead to transcriptional activation of stress genes encoding heat shock proteins (HSPs). HSPs are mainly chaperones and proteases that work in concert to maintain the structural and functional integrity of all living cells. To activate this response pathway, E. coli uses the alternative sigma factor σ32, which binds to core RNA polymerase to recognize σ32-specific promoters and initiate the transcription of about 90 genes, e.g., dnaK, groESL, and hslU (27). Regulation of the heat shock response is accomplished at different levels, affecting σ32 transcription, translation, protein stability, and activity (10, 11, 20, 33, 34). Two major chaperones, DnaK and GroEL, are involved not only in protein refolding/degradation but also in the regulation of σ32 (15, 34). In the absence of cellular stress and when cells adapt to stress conditions, DnaK and GroEL bind to σ32 and mediate its rapid degradation by the protease FtsH, a member of the AAA+ protease family (17, 33). Given the relatively slow degradation of σ32 by FtsH in vitro, the involvement of a still unknown factor in chaperone-mediated σ32 degradation in vivo has been postulated (39).
Many HSPs, such as GroEL (Hsp60) and DnaK (Hsp70), are essential for cellular survival under all growth conditions, and it has been shown that σ32 is essential for growth and cell division above 20°C (42). Moreover, a temperature-sensitive variant of σ32 (35) displays cell division defects at the nonpermissive temperature. Deletion of dnaK results in filamentous growth (5, 19); overexpression of DnaK leads to a cell division and growth defect (4). It has recently been shown that GroE-depleted E. coli cells grow in filaments as a consequence of impaired folding of the cell division protein FtsE (12). It has also been proposed that GroEL localizes to a midcell position in an FtsZ-dependent manner and has a role in cell division (28). All these observations suggest a link between the σ32 regulon and cell division.
However, activation of the heat shock regulon by cell division has never been demonstrated in E. coli, in contrast to the alphaproteobacterium Caulobacter crescentus, where expression of the groESL operon was found to be cell cycle controlled (1). During the past few years, major advances in the knowledge of prokaryotic cell division have been achieved (reviewed in references 6, 30, and 37). A picture emerges that shows that the process of prokaryotic cytokinesis is highly regulated and requires the coordinated activity of cytoskeletal proteins that guide the synthesis and rearrangements of the cell envelope, including localized peptidoglycan synthesis (6, 30, 37). To shed some light on a possible growth phase- and cell-cycle-dependent activation of the σ32 regulon in E. coli, we monitored the levels of σ32 protein, the transcription of several σ32-dependent genes, and the activity of the σ32-controlled groESL promoter. Our results show that the σ32 regulon of E. coli is highly responsive to growth phases and oscillates during exponential growth. Our results further indicate that the first round of cell division after exit from stationary phase triggers a transient activation of the σ32 regulon by enhanced transcription of rpoH. This activation occurs during cytokinesis, because we found that σ32 activation is suppressed by depletion of the essential tubulin-like cell division protein FtsZ. In late-exponential phase, σ32 protein is stabilized compared to its status during early-logarithmic growth, leading to a second peak of heat shock gene expression. When cells transit to stationary phase, σ32-dependent genes are rapidly inactivated despite the presence of high levels of full-length σ32 protein. At this stage, a stable degradation product of σ32 becomes apparent. Given the fact that σ32 is essential for the growth of E. coli at temperatures that are physiologically relevant (e.g., 37°C), we propose that the activation of σ32 after exit from stationary phase reactivates bacterial cells for normal progression through the cell cycle. On the other hand, the transition to stationary phase involves σ32 inactivation by a mechanism distinct from σ32 degradation.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The sequenced E. coli K-12 laboratory strain MG1655 (F− λ− ilvG rfb-50 rph-1) (3) was used for quantitative mRNA measurements and protein level determinations. For β-galactosidase assays, the MC4100 derivative SR6618 [MC4100 φ (groESL::lacZ)], equivalent to SR4499 (20), generously provided by S. Raina, was used. Stationary-phase cultures (optical density at 600 nm [OD600], 2.5 to 3.5) of these strains were obtained after growth for 20 to 24 h at 37°C in M9 minimal medium (26) supplemented with 3 g/liter Casamino Acids, 5 mM MgSO4, and 0.2% glucose. For all experiments, 10 ml of stationary-phase cultures in 100-ml Erlenmeyer flasks was prepared. For all experiments, unless otherwise indicated, 200 ml fresh, prewarmed M9 medium in a 1-liter Erlenmeyer flask was inoculated 1:30 with stationary-phase cells and incubated at 37°C in a shaking water bath (113 rpm). For β-galactosidase assays, 50-ml cultures were prepared. If appropriate, 10 μg/ml chloramphenicol or 5 μg/ml cefotaxime was added. For β-galactosidase assays and the corresponding growth curves, 50 ml of M9 medium in a 300-ml Erlenmeyer flask was inoculated 1:30 with stationary-phase cells.
Cell number determinations.
Cells were fixed directly in growth medium with 2.8% formaldehyde and 0.04% glutaraldehyde and were stored at 4°C. Fixed cells were counted using a CASY cell counter, model TTC (Innovatis AG, Reutlingen, Germany) with a 45-μm capillary.
Microscopy and cell length analyses.
Aliquots of the cultures were harvested at the given time points by centrifugation. Cells were washed with PBS buffer (200 mM sodium phosphate buffer [pH 7.2], 150 mM NaCl) and fixed with 2.8% formaldehyde and 0.04% glutaraldehyde in PBS buffer for 15 min at room temperature (8). Cells were washed three times with PBS buffer, resuspended in an appropriate volume of PBS buffer, spotted onto glass slides coated with poly-l-lysine (Sigma-Aldrich, St. Louis, MO), and incubated for 10 min before the supernatant was removed. For phase-contrast microscopy, a Zeiss Axioscope equipped with a 100× oil immersion objective was used. Cell lengths were determined by analyzing phase-contrast images using the Metamorph (version 5.1) software package (Molecular Devices, Downingtown, PA).
Western blot analysis.
Cells were harvested by centrifugation for 5 min at 4°C and 4,200 × g. Whole-cell lysates were separated on 12.5% sodium dodecyl sulfate (SDS)-polyacrylamide gels, and proteins were electrotransferred to an Immobilon P nitrocellulose membrane (Millipore, Bedford, MA). Primary antibodies against RNA polymerase subunits σ32, σ70, and σS were purchased from Neoclone (Madison, WI) and used at a 1:2,000 dilution in 1× TST (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.1% Tween 20) containing 1% dry milk powder. The FtsZ antibody was a kind gift from Tanneke Den Blaauwen. As a secondary antibody, a peroxidase-conjugated antibody against mouse immunoglobulin G (GE Healthcare, Little Chalfont, England) or against rabbit immunoglobulin G (Sigma-Aldrich, St. Louis, MO) was used at a 1:15,000 dilution, and the Amersham ECL system (GE Healthcare, Little Chalfont, England) was used for detection. Chemiluminescence signals were detected with AGFA Curix Ultra UV-G X-ray film (Agfa-Gevaert, Mortsel, Belgium) and were quantitated using a Molecular Dynamics densitometer and ImageQuant (version 5.1) software (Molecular Dynamics, Sunnyvale, CA).
In vivo stability of σ32.
The degradation of σ32 in E. coli cultures was monitored by Western blot analysis essentially as described previously (36). E. coli MG1655 was grown at 37°C as described above for growth experiments, and at the indicated time points (90, 180, and 210 min after inoculation), 200 μg/ml chloramphenicol was added to aliquots (50 ml) of the culture to stop protein synthesis. Cells were further incubated at 37°C, and 1.35-ml samples were taken at 10- or 30-s intervals and immediately precipitated with 10% trichloroacetic acid on ice for 30 min. After centrifugation for 15 min at 4°C and 14,000 × g, the pellets were washed with acetone and resuspended in 1× FSB (0.12 M dithiothreitol, 2% [wt/vol] SDS, 0.06 M Tris-HCl [pH 6.8], 8.7% [vol/vol] glycerol, 0.004% [wt/vol] bromophenol blue), and aliquots corresponding to 0.3 OD unit were subjected to Western blot analysis with anti-σ32 as described above.
RNA isolation.
At the given time points after inoculation, cells were harvested by centrifugation at 4°C for 4 min and 4,200 × g, quickly frozen in liquid nitrogen, and subsequently stored at −70°C. Total RNA was isolated with the RNeasy minikit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. RNA concentrations were determined photometrically.
Northern blot analyses.
Northern blot analysis was performed as described previously (40). In brief, 5 μg total RNA was denatured at 65°C in loading buffer containing formaldehyde and ethidium bromide, separated on a 1.2% agarose gel, and transferred to an Amersham Hybond-N membrane (GE Healthcare, Little Chalfont, England). Hybridization was performed with alkali-labile digoxigenin-11-dUTP-labeled probes diluted in DIG Easy Hyb (Roche Applied Science, Penzberg, Germany) according to the manufacturer's protocol. The oligonucleotides for PCR synthesis of the probes are listed in Table 1. After hybridization at 50°C, detection was performed using an alkaline phosphatase-conjugated anti-digoxigenin Fab fragment and ready-to-use CSPD (Roche Applied Science, Penzberg, Germany) as a substrate for the alkaline phosphatase. Chemiluminescence signals were detected with AGFA Curix Ultra UV-G X-ray film (Agfa-Gevaert, Mortsel, Belgium) and quantitated using a Molecular Dynamics densitometer and ImageQuant (version 5.1) software (Molecular Dynamics, Sunnyvale, CA).
TABLE 1.
Oligonucleotides
| Oligonucleotide | Sequence |
|---|---|
| q-RT-PCR | |
| groESL_fw | CGATGGCTACGGTGTGAAAT |
| groESL_rev | AGTGTCGTGCGCGGATTA |
| rpoA_fw | CCGAGGTTGAGATTGATGGT |
| rpoA_rev | CCTTTCAGGTTGAGCAGGAT |
| HEX_groESL | 5′-HEX-TCCGAAAGCGACATTCTGGCAATT-3′-BHQ-1 |
| FAM_rpoA | 5′-FAM-ACAGCACCAAAGAAGGCGTTCAGG-3′-BHQ-1 |
| Northern blotting | |
| groEL_probe_fw | CCATCTCCGCTAACTCCGAC |
| groEL_probe_rev | GCAACCACGCCTTCTTCTAC |
| dnaK_probe_fw | GACGAACCCGCAAAACAC |
| dnaK_probe_rev | TGACCACCAACGAGGATAAC |
| hslU_probe_fw | AACAGTGTGTAAACGACGAG |
| hslU_probe_rev | GGAAGAGATGACCAGCCAG |
| rpoH_probe_fw | CCCAGTTATCATCTTCAATGCC |
| rpoH_probe_rev | GCAGCTAAAACGCTGATCC |
| Cloning | |
| dicF_ecor1_fw | TAGAATTCGCCTGTCGTTAAATTTTCGTC |
| dicF_hind3_rev | TAAAGCTTACACCAATTTCAAAACAACCTTC |
q-RT-PCR.
Quantitative reverse transcription real-time PCR (q-RT-PCR) analysis of total RNA was performed in a RotorGene 6000 machine (Corbett Life Science, Sydney, Australia) using the SensiMix one-step kit (Quantace, London, United Kingdom) in combination with the dually labeled oligonucleotide probes (with 6-carboxyfluorescein [FAM] or 6-carboxy-1,4-dichloro-2′,4′,5′,7′-tetra-chlorofluorescein (HEX) at the 5′ end and black hole quencher 1 (BHQ-1) at the 3′ end) listed in Table 1. For primer and probe design, Primer3 software, which is available online (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) (29), was used. Two sets of primers/probes were used in one reaction, with one probe labeled with FAM and the other with HEX. Total RNA was isolated as described for Northern blot analyses. To remove traces of DNA, DNase I digestion was performed. One microgram of total RNA was incubated for 30 min at 37°C with 1 U DNase I (Fermentas, Burlington, Canada) in the appropriate buffer with MgCl2. The incubation was stopped by the addition of EDTA to a final concentration of 2.5 mM and further incubation at 65°C for 10 min. q-RT PCR was performed in a total volume of 10 μl containing 4 ng of DNase I-treated RNA, the 2× SensiMix one-step mix, 5 mM MgCl2, 200 nM each primer, 100 nM each probe, and 2 U of RNase inhibitor. Relative mRNA concentrations, representing the change in gene expression compared to expression at a selected time point, were determined using the 2−ΔCT method as described elsewhere (23). In short, the threshold cycle (CT) of stationary-phase samples was subtracted from the CT of each time point, resulting in −ΔCT. Relative concentrations were calculated as 2−ΔCT.
β-Galactosidase assays.
The β-galactosidase activity of strain SR6618 was determined by the method in reference 26, with the modifications described in reference 40. Kinetic measurements to determine the change in optical density at 420 nm multiplied by 1,000 per min per OD600 unit (ΔmOD420/min/OD600) during the linear increase in absorption were taken in 96-well microtiter plates using a Bio-Rad microplate reader, model 550 (Bio-Rad Laboratories, Hercules, CA).
Cloning of dicF.
The DNA sequence encoding the DicF antisense RNA was PCR amplified from the MG1655 genome (GenBank accession no. U00096; region 1647239 to 1647689) using oligonucleotides dicF_ecor1_fw and dicF_hind3_rev, listed in Table 1. The dicF sequence was ligated into plasmid pGZ119EH (22) at the EcoRI and HindIII restriction sites, resulting in plasmid pGZdicF. Transcription of DicF antisense RNA was induced from the tac promoter by the addition of 70 μM isopropyl-β-d-thiogalactopyranoside (IPTG) to the growth medium.
RESULTS
σ32 protein levels during the growth cycle of E. coli.
To gain insight into the role of the alternative sigma factor σ32 in cells grown in batch cultures under nonstress conditions, we asked if σ32 responds to changes in the physiological status of the cells and if its level varies during the growth cycle. The growth characteristics and cell lengths of E. coli MG1655 grown in minimal medium at 37°C were determined (Fig. 1). During a relatively short period, the number of cells doubled from 1 × 108 per ml to 2 × 108 per ml (Fig. 1 A, 75 min and 105 min, respectively), suggesting that at 105 min, most cells had completed the first round of cell division after exit from stationary phase. This conclusion is corroborated by the fact that cell lengths increased only until 90 min and then decreased sharply by 105 min (Fig. 1B). A logarithmic increase in the cell mass, with a doubling time of 50 min, was observed until 165 min. The transition to stationary phase began after 165 min and was accompanied by a decline in the growth rate, the appearance of σS (Fig. 2), longer generation times, and continuous shortening of cells (Fig. 1; see also Fig. S1 in the supplemental material).
FIG. 1.

Growth characteristics of a batch culture of E. coli MG1655. Late-stationary-phase cells grown in M9 minimal medium at 37°C for at least 20 h were taken and inoculated into fresh, prewarmed M9 medium. (A) Growth curves showing increases in cell mass (OD600) and in cell numbers (cells/ml) as determined by counting cells in a cell counter. Data shown are averages for at least three independent experiments. (B) Box-and-whisker plot representing cell length distributions. Cell lengths were determined by morphometric analyses of phase-contrast microscopic images. At least 150 cells were measured per time point. Boxes represent the cell length distribution of 50% of all measured cells; horizontal lines within boxes represent median cell lengths. After a short lag phase of approximately 15 min, cells start to grow and elongate. Cell lengths reach a maximum at 90 min, reflecting the time point before the majority of the cells divide for the first time (between 90 and 105 min). The 165-min time point marks the end of exponential growth and the beginning of the transition to stationary phase. S, stationary phase.
FIG. 2.

σ32, σ70, and σS protein levels during a growth cycle of E. coli MG1655 in M9 medium. (A) Cells were harvested at the indicated time points, and whole-cell lysates corresponding to 0.2 OD600 unit (∼3 × 108 cells) were subjected to SDS-polyacrylamide gel electrophoresis and Western blotting with antibodies specific for σ32, σ70, or σS. Increased amounts of σ32 are detectable at 60 to 90 min and again at 165 min and later. σ70 protein levels show a distinctly different pattern, remaining similar throughout the growth cycle. The appearance of σS correlates with the transition to stationary phase and reaches a maximum at 195 min. (B) For quantification, Western blot signals were normalized to the amount of protein loaded. Bars represent the relative abundances of σ32 and σ70. S, stationary phase.
To determine σ32 protein levels, cells were harvested at different time points during the growth cycle and whole-cell lysates were subjected to Western blot analysis (Fig. 2A). In the stationary and early-logarithmic phases, no σ32 protein, or very little, could be detected. At 90 min, σ32 abundance had increased at least fourfold over that at 30 min. Note that at 90 min, average cell lengths reached a maximum, suggesting that most cells were in a stage just prior to cell separation. This observation suggested that cell division after exit from stationary phase has an impact on the σ32 stress response pathway. After the first σ32 peak, the σ32 protein level decreased during continuing logarithmic growth (and following cell divisions). A second peak in σ32 abundance was reached at 165 min, corresponding to the end of logarithmic phase. During the transition to stationary phase, cell growth slowed down, but cell numbers still increased (Fig. 1A). σ32 protein levels gradually increased during further progression into stationary phase. Interestingly, a degradation intermediate of σ32 appeared. We termed this stable degradation intermediate RpoH* (Fig. 2A). In addition to the examination of σ32 levels, we monitored σ70, the housekeeping sigma factor in E. coli, to rule out any general effect of growth phases on protein levels. In contrast to those of σ32, levels of σ70 did not oscillate during logarithmic growth (Fig. 2A). Quantification of the signals obtained by Western blotting demonstrated that σ70 protein levels increased by a factor of 2 to 3 after the first round of cell division and then diminished when the cells entered stationary phase (Fig. 2B). As expected, and as shown by others (21, 38), the stationary-phase-specific sigma factor σS appeared only during the transition to stationary phase or in stationary phase (Fig. 2A). To show that the observed activation-and-inactivation pattern of σ32 was dependent on growth phases, but not on the growth rate, a time course experiment similar to that described above was performed in M9 minimal medium at 30°C (see Fig. S2 in the supplemental material). Despite a much longer doubling time (∼100 min), σ32 was barely detectable in freshly inoculated cells that were growing but not yet dividing. σ32 abundance increased at the time when the first round of cell division was proceeding. Again, the transition to stationary phase was accompanied by increased σ32 levels and the appearance of a stable degradation intermediate of σ32. From the results described above, we conclude that σ32 not only responds to stress stimuli, such as heat, but is subjected to growth phase- and cell division-dependent regulation.
The transcription of rpoH is upregulated during the first round of cell division.
To elucidate if the increased steady-state level of σ32 during the first round of cell division after exit from stationary phase is a result of increased transcription of rpoH or enhanced stability of σ32, we measured the abundance of rpoH transcripts and the stability of the σ32 protein (Fig. 3). Northern blot analysis with rpoH-specific probes revealed a three- to fivefold increase in the rpoH mRNA level during cell division (90 min) over that in predivisional cells (30 min) (Fig. 3A). Since during heat shock, transcriptional activation of the rpoH promoters plays a minor role but σ32 activation is achieved mainly through increased σ32 stability (31), we also investigated whether σ32 protein stability was enhanced. This was done by measuring σ32 protein half-lives after inhibition of protein synthesis (Fig. 3B and C). Surprisingly, calculated half-lives were extremely short (15 s for 90-min cultures). Thus, our results indicate that the increase in σ32 levels observed during the first round of cell division after the exit from stationary phase results not from protein stabilization but from transcriptional activation of rpoH. Elevated σ32 levels during the transition to stationary phase most likely are due to increased σ32 protein stability. The half-life of σ32 at 180 min (30 s) was found to be double that at 90 min (Fig. 3B and C).
FIG. 3.

(A) rpoH mRNA levels. Total RNA was isolated from E. coli MG1655 at the indicated time points (in minutes) during a growth cycle, and Northern blot analysis was performed. Analysis of 23S rRNA, used as a loading control, is shown in the lower panel. For quantification, rpoH signals were normalized to the amount of RNA loaded. Numbers at the bottom represent the rpoH mRNA level at each time point relative to the level at 120 min. S, stationary phase. (B and C) σ32 protein stability. (B) σ32 degradation after inhibition of protein synthesis. Aliquots from 90- and 180-min cultures were taken at the indicated time points (seconds) after inhibition of protein synthesis. σ32 was detected by Western blotting. A star (*) marks the stable degradation product of σ32. (C) Amounts of σ32 plotted versus time. Values were used to determine the half-lives (t1/2) of σ32 in cells from 90-min and 180-min cultures.
Transcription of σ32-dependent stress genes during the growth cycle of E. coli.
Increased levels of σ32 protein in E. coli cells should lead to increased transcription from σ32-dependent promoters. To investigate the activity of σ32 during the growth cycle, we monitored the transcription of representative members of the σ32 regulon. Total RNA was isolated from E. coli MG1655 cells, grown as described above, at different time points, and Northern blot analyses with specific probes were performed to detect σ32-dependent mRNAs. As can be seen in Fig. 4, the levels of groESL, dnaK, and hslU mRNAs mirror the levels of σ32 during growth except when the cells transit to stationary phase (see below). During late-stationary phase and up to 30 min after inoculation, the tested mRNAs were not detectable except for a faint signal corresponding to dnaK mRNA in stationary phase. The first signals corresponding to groESL, dnaK, and hslU mRNAs could be seen from 45 to 60 min, followed by pronounced increases at 75 min, corresponding to the onset of cell division after exit from stationary phase (Fig. 4A). Quantification of Northern blots revealed a three- to fourfold increase in the level of groESL mRNA in cells that had started to divide (75 min and 90 min) over that in cells prior to cell division (Fig. 4B). This burst in σ32-dependent mRNA levels occurred simultaneously with the three- to fourfold increase in the level of σ32 protein (Fig. 2), indicating that the increased transcription of the σ32 regulon is a direct consequence of increased σ32 levels. At 120 min, the time when most cells had completed the first round of cell division, mRNA dropped to levels comparable to those observed in predivisional cells. Again, this decrease corresponds well to the observed low level of σ32 at 120 min. A second maximum was reached at 150 min and continued until 180 min, when cells transited to stationary phase. All mRNA levels measured decreased at 195 min and became undetectable at 360 min. Until 180 min, a good correlation exists between σ32 protein levels and σ32-dependent mRNA levels. However, beginning with 195 min, mRNA levels of σ32-dependent genes and σ32 protein levels diverged (compare Fig. 2 and 4). Whereas the transcription of σ32-dependent genes dropped markedly, σ32 protein levels remained high. These findings suggest that during early-stationary phase, σ32 protein is present as a more stable yet functionally inactive form. There are several possibilities for how σ32 may become inactivated in early-stationary phase (see Discussion).
FIG. 4.

Growth cycle-dependent oscillation of mRNA levels of σ32-controlled genes in E. coli MG1655. (A) Total RNA was isolated at the given time points and analyzed by Northern blotting. groESL, dnaK, and hslU mRNAs show similar fluctuation patterns during the course of the growth experiment. Transcript levels are low or below the detection limit in stationary phase, and an increase in the abundance of each transcript can be observed at 45 min, with a first peak at 75 to 90 min. After a decline with a minimum at 120 min, concomitant with the completion of the first round of cell division by most cells in the culture, a second peak appears at 150 min. The transition to stationary phase and the following reduction in cell division activity are accompanied by a marked reduction in transcript levels. 23S rRNA from the ethidium bromide-stained agarose gel is shown as a loading control. (B) Graphical representation of quantitated mRNA levels, showing the oscillation of the levels of σ32-dependent transcripts. The level of each mRNA at 120 min was set to 1. Experiments performed at least in triplicate produced similar results; one representative result is shown. S, stationary phase.
To confirm that the observed expression pattern of σ32-dependent genes was specific, we determined the expression of a σ70-dependent gene. For that purpose, we chose the σ70-controlled gene rpoA, which encodes the RNA polymerase α subunit. By using q-RT-PCR, the relative expression of rpoA mRNA was determined and compared to the σ32-dependent expression of groESL mRNA. As can be seen in Fig. 5, rpoA and groESL showed completely different expression patterns. rpoA mRNA was highly induced as early as 30 min after inoculation into fresh medium (about 40-fold compared to stationary phase). We interpreted this as a response to the nutritional upshift and the concomitant burst of metabolic activity. Most notably, at that time point, σ32-dependent groESL mRNA levels were still very low, close to the levels observed during stationary phase. At later time points, the rpoA mRNA level decreased gradually during growth, in sharp contrast to the oscillating pattern determined for groESL mRNA. During the transition to stationary phase (180 min), mRNA levels diminished further and finally reached stationary-phase levels (Fig. 5). This result shows that the observed growth phase-dependent expression pattern of σ32 regulon members is specific and—during exponential phase—completely different from the expression pattern of a typical σ70-dependent gene. Activation of the σ32 regulon occurs at the time when cells start to divide after recovery from stationary phase, followed by reduced σ32 activity after the first round of cell division and a second activation during late-exponential phase.
FIG. 5.
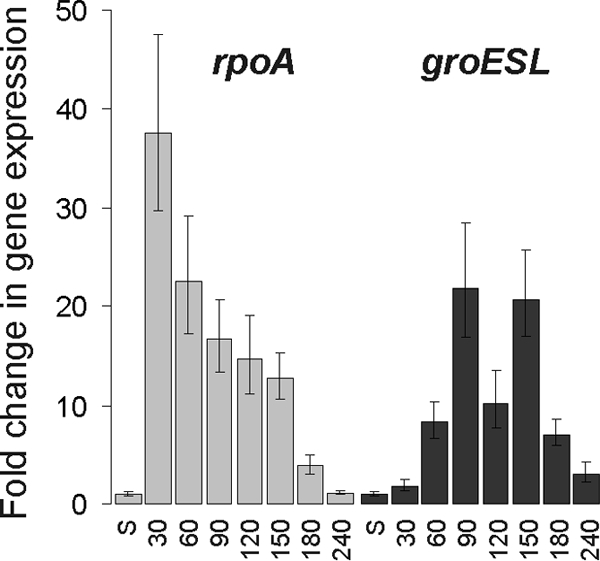
q-RT-PCR analysis of mRNA levels of σ70- and σ32-regulated genes in E. coli MG1655 shows that the observed oscillation pattern is specific for σ32-regulated genes. Total RNA was isolated at the time points shown. q-RT-PCR was performed, and the change in gene expression relative to that in stationary phase was calculated. The expression levels of rpoA (σ70 dependent) and groESL (σ32 dependent) mRNAs were distinctly different. A nutritional upshift caused an upregulation of rpoA expression (30 min, compared to stationary phase) whereas groESL expression was not affected by a nutritional upshift but oscillated in a pattern that correlated with cell division events. S, stationary phase.
Activation of the σ32-dependent groESL promoter.
To directly monitor the activation of a well-characterized σ32-dependent promoter, we used E. coli SR6618, a strain with a chromosomally integrated groESL::lacZ-fusion (20). groESL promoter activity was found to be very low from 15 to 60 min after cells were inoculated into fresh medium (Fig. 6). Ninety minutes after inoculation, groESL promoter activity increased two- to threefold, corresponding to the initiation of cell division in these cultures. This finding further confirmed our conclusions that the high mRNA levels of groESL (and of the other heat shock gene mRNAs investigated) that we observed during the first round of cell division after exit from stationary phase resulted from increased transcription from σ32-dependent promoters. β-Galactosidase activity increased further until the transition into stationary phase. Consistent with reduced σ32 levels and reduced mRNA levels of σ32-controlled genes, a reduction in β-galactosidase activity was observed during early stationary phase. In contrast to the dynamic changes in groESL, dnaK, and hslU mRNA levels, the comparatively long half-life of the reporter enzyme β-galactosidase did not allow oscillations in promoter activity to be detected. Nevertheless, a first burst of promoter activity during the first round of cell division after exit from stationary phase and a second burst at the end of exponential growth can be clearly seen.
FIG. 6.

Transcriptional activation of the σ32-controlled groESL promoter. Expression from the groESL promoter was determined using the E. coli groESL::lacZ reporter strain SR6618 and β-galactosidase assays. A significant increase in β-galactosidase levels (▴) (P < 0.05) was observed between 60 and 90 min. Cell division was monitored by determination of cell counts (*) (cells/ml). Promoter activity is given as ΔmOD420/min/OD600. Means and standard deviations for three independent experiments, with each sample measured in duplicate, are plotted.
Depletion of FtsZ completely abolishes the activation of the σ32 regulon.
Since the activation of the σ32 regulon coincided with the start of cell division, we reasoned that cell division itself is the “stress signal” for the σ32 stress response. We predicted that inhibition of cell division would reduce or even abolish this cytoplasmic stress response. To test this hypothesis, we inhibited cell division at an early stage. This was accomplished through depletion of FtsZ, a tubulin-like cytoskeletal protein that is essential for the formation of the divisome (14, 24). FtsZ was depleted using the antisense RNA DicF, which inhibits the translation of ftsZ mRNA (32). As expected, expression of dicF from a plasmid with an inducible promoter led to cell division inhibition and filamentous growth. The filaments contained multiple clearly separated nucleoids (see Fig. S3 in the supplemental material), showing that neither DNA replication nor chromosome segregation nor elongation of cells was blocked in cells depleted of FtsZ. As expected, Western blot analysis demonstrated a specific reduction in FtsZ protein levels in cells expressing DicF antisense RNA (Fig. 7A). Concomitant with reduced FtsZ levels and a block in cell division, we observed that σ32 protein was not detectable at 90 min or at 120 min (Fig. 7A). Due to low basal expression of DicF, which is in good agreement with the lower FtsZ levels in the uninduced control (Fig. 7A), the σ32 response was not completely suppressed but delayed. The unchanged σ70 levels show that the observed effect was specific for σ32. In accordance with the absence of σ32, no increase in promoter activity in the groESL::lacZ fusion strain SR6618 was detectable when dicF expression was induced (Fig. 7B). Basal expression led to a delay in groESL promoter activation, confirming the Western blot data. In contrast, transcription of a σ70-controlled gene (rpoA) was not affected by the expression of DicF antisense RNA (data not shown). Our results indicate that FtsZ accumulation, subsequent FtsZ ring formation, or FtsZ ring constriction triggers σ32 activation. To corroborate the effect of FtsZ depletion, we carried out an experiment in which we induced the expression of DicF during exponential growth. Figure 7C shows that σ32 levels after induction of DicF in asynchronously growing cells gradually decrease with time. Finally, after 120 min, σ32 is not detectable. Inhibition of cell division in these cultures was confirmed by light microscopy. Cells did not divide but formed filaments after the addition of the inducer (data not shown). This result shows that inhibition of cell division during exponential growth by FtsZ depletion also leads to a specific reduction in σ32 levels.
FIG. 7.

Inhibition of an early step in cell division abolishes the σ32-dependent stress response in E. coli. (A) Steady-state levels of σ32, FtsZ, and σ70 proteins in cells expressing DicF antisense RNA (+) and in control cells with the vector (c) or the uninduced plasmid (−) were compared by Western blotting. As a loading control, a section of the Coomassie-stained gel (Co) is shown. (B) The activities of the σ32-dependent groESL promoter in SR6618 expressing DicF from plasmid pGZdicF to deplete FtsZ, compared to those in controls, are shown. ▴, pGZdicF induced with 70 μM IPTG; ▵, pGZdicF without IPTG; □, pGZ119EH (vector control) with 70 μM IPTG. (C) Inhibition of σ32 by DicF expression in asynchronously growing cells. MG1665 harboring pGZdicF or pGZ119EH (control) was inoculated to an OD600 of 0.01 and grown to early-logarithmic phase (OD600, ∼0.1). IPTG was added to induce DicF expression, and samples taken before (0) and after the addition of IPTG were analyzed by Western blotting.
DISCUSSION
The σ32 response described here—-a transient activation of the σ32 regulon during cell division after exit from stationary phase in the bacterium E. coli—is reminiscent of cell cycle-dependent oscillations of regulators and stress proteins in Caulobacter crescentus (18, 25). Hence, HSP activation during cell division may be a common theme among prokaryotic and eukaryotic organisms. In gram-negative bacteria, heat shock regulon activation during cell division after exit from stationary phase may ensure that the cellular supply of chaperones and other HSPs is sufficient to sustain the necessary rearrangements, which occur mainly in the bacterial cell envelope. The reported cell division defects caused by σ32 and HSP mutations support the idea that HSPs are important factors for progression through certain steps of cell division in E. coli. A direct involvement of GroEL in the folding of the cell division protein FtsE has been reported recently (12). In addition, it was shown in early investigations on σ32 that an rpoH deletion strain develops filaments at normal growth temperatures (42), indicating a requirement for the heat shock sigma factor during cell division. GroEL, which is essential under all growth conditions, also confers a filamentous phenotype when depleted (12) or mutated (7). In addition, FtsZ-dependent localization of GroEL at the division site (28) may suggest that chaperones of the σ32 regulon are involved in the assembly of the cytoplasmic part of the divisome. We show here that FtsZ depletion suppresses the activation of the σ32 regulon. This finding again points to cell division as a key in triggering the σ32 regulon. FtsZ is an essential tubulin-like cell division protein that is found in almost all bacterial species. In rod-shaped cells, FtsZ polymerizes after chromosome replication and segregation at the cytoplasmic membrane at midcell and forms a ring structure, the Z-ring (2). The E. coli Z-ring acts as a scaffold for at least 10 additional proteins that assemble into a cytokinetic machinery, the divisome (13). Our observations suggest that the divisome at some stage generates a signal that is transmitted to the σ32 regulon. Since our analyses of the half-life of σ32 during the first round of cell division at 90 min did not reveal increased stability compared to that of heat-shocked cells or to that at later time points, we concluded that the observed increased transcription of rpoH is mainly responsible for increased σ32 levels during cell division after exit from stationary phase. Since transcription from the rpoH P1 promoter is positively affected by phosphorylated CpxR (40) and transcription from the P3 promoter is strictly σE dependent (9), transcriptional activation of both promoters elicited by cytokinetically induced envelope stress could account for the increased rpoH mRNA levels during cell division. This hypothesis, however, remains to be tested.
A second important observation is the functional inactivation of σ32 when cells in the batch culture transit to stationary phase. Despite high σ32 levels, transcription of σ32-dependent genes rapidly diminishes to low levels. Functional inactivation of σ32 can be achieved by sequestration by the chaperonin DnaK, which binds with high affinity to σ32, or by phosphorylation of σ32 (20, 34). Alternatively, the observed stable truncated fragment of σ32, which lacks approximately 60 N-terminal amino acids (unpublished observations), may interfere with the function of full-length σ32. Functional inactivation of σ32 coincides with the appearance of σS, the sigma factor that governs the transcription of genes during stationary phase (16). Further investigations in our laboratory are aimed at understanding how σ32 is functionally inactivated during this transition phase from growing and dividing to nongrowing and nondividing cells.
Supplementary Material
Acknowledgments
We thank S. Raina for providing strain SR6618 and for helpful suggestions and T. den Blaauwen for the gift of the FtsZ antiserum. We thank J. Reidl for critical reading of the manuscript.
This work was funded by the Austrian Science Fund (FWF), project P17857-B12.
Footnotes
Published ahead of print on 29 December 2008.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Avedissian, M., and S. Lopes Gomes. 1996. Expression of the groESL operon is cell-cycle controlled in Caulobacter crescentus. Mol. Microbiol. 1979-89. [DOI] [PubMed] [Google Scholar]
- 2.Bi, E. F., and J. Lutkenhaus. 1991. FtsZ ring structure associated with division in Escherichia coli. Nature 354161-164. [DOI] [PubMed] [Google Scholar]
- 3.Blattner, F. R., G. Plunkett III, C. A. Bloch, N. T. Perna, V. Burland, M. Riley, J. Collado-Vides, J. D. Glasner, C. K. Rode, G. F. Mayhew, J. Gregor, N. W. Davis, H. A. Kirkpatrick, M. A. Goeden, D. J. Rose, B. Mau, and Y. Shao. 1997. The complete genome sequence of Escherichia coli K-12. Science 2771453-1474. [DOI] [PubMed] [Google Scholar]
- 4.Blum, P., J. Ory, J. Bauernfeind, and J. Krska. 1992. Physiological consequences of DnaK and DnaJ overproduction in Escherichia coli. J. Bacteriol. 1747436-7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bukau, B., and G. C. Walker. 1989. Cellular defects caused by deletion of the Escherichia coli dnaK gene indicate roles for heat shock protein in normal metabolism. J. Bacteriol. 1712337-2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cabeen, M. T., and C. Jacobs-Wagner. 2007. Skin and bones: the bacterial cytoskeleton, cell wall, and cell morphogenesis. J. Cell Biol. 179381-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chapman, E., G. W. Farr, R. Usaite, K. Furtak, W. A. Fenton, T. K. Chaudhuri, E. R. Hondorp, R. G. Matthews, S. G. Wolf, J. R. Yates, M. Pypaert, and A. L. Horwich. 2006. Global aggregation of newly translated proteins in an Escherichia coli strain deficient of the chaperonin GroEL. Proc. Natl. Acad. Sci. USA 10315800-15805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Den Blaauwen, T., N. Buddelmeijer, M. E. Aarsman, C. M. Hameete, and N. Nanninga. 1999. Timing of FtsZ assembly in Escherichia coli. J. Bacteriol. 1815167-5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Erickson, J. W., and C. A. Gross. 1989. Identification of the σE subunit of Escherichia coli RNA polymerase: a second alternate σ factor involved in high-temperature gene expression. Genes Dev. 31462-1471. [DOI] [PubMed] [Google Scholar]
- 10.Erickson, J. W., V. Vaughn, W. A. Walter, F. C. Neidhardt, and C. A. Gross. 1987. Regulation of the promoters and transcripts of rpoH, the Escherichia coli heat shock regulatory gene. Genes Dev. 1419-432. [DOI] [PubMed] [Google Scholar]
- 11.Fujita, N., and A. Ishihama. 1987. Heat-shock induction of RNA polymerase σ32 synthesis in Escherichia coli: transcriptional control and a multiple promoter system. Mol. Gen. Genet. 21010-15. [DOI] [PubMed] [Google Scholar]
- 12.Fujiwara, K., and H. Taguchi. 2007. Filamentous morphology in GroE-depleted Escherichia coli induced by impaired folding of FtsE. J. Bacteriol. 1895860-5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goehring, N. W., and J. Beckwith. 2005. Diverse paths to midcell: assembly of the bacterial cell division machinery. Curr. Biol. 15R514-R526. [DOI] [PubMed] [Google Scholar]
- 14.Goehring, N. W., F. Gueiros-Filho, and J. Beckwith. 2005. Premature targeting of a cell division protein to midcell allows dissection of divisome assembly in Escherichia coli. Genes Dev. 19127-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guisbert, E., C. Herman, C. Z. Lu, and C. A. Gross. 2004. A chaperone network controls the heat shock response in E. coli. Genes Dev. 182812-2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hengge-Aronis, R. 2002. Signal transduction and regulatory mechanisms involved in control of the σS (RpoS) subunit of RNA polymerase. Microbiol. Mol. Biol. Rev. 66373-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herman, C., D. Thevenet, R. D'Ari, and P. Bouloc. 1995. Degradation of σ32, the heat shock regulator in Escherichia coli, is governed by HflB. Proc. Natl. Acad. Sci. USA 923516-3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holtzendorff, J., D. Hung, P. Brende, A. Reisenauer, P. H. Viollier, H. H. McAdams, and L. Shapiro. 2004. Oscillating global regulators control the genetic circuit driving a bacterial cell cycle. Science 304983-987. [DOI] [PubMed] [Google Scholar]
- 19.Kang, P. J., and E. A. Craig. 1990. Identification and characterization of a new Escherichia coli gene that is a dosage-dependent suppressor of a dnaK deletion mutation. J. Bacteriol. 1722055-2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klein, G., C. Dartigalongue, and S. Raina. 2003. Phosphorylation-mediated regulation of heat shock response in Escherichia coli. Mol. Microbiol. 48269-285. [DOI] [PubMed] [Google Scholar]
- 21.Lange, R., and R. Hengge-Aronis. 1991. Identification of a central regulator of stationary-phase gene expression in Escherichia coli. Mol. Microbiol. 549-59. [DOI] [PubMed] [Google Scholar]
- 22.Lessl, M., D. Balzer, R. Lurz, V. L. Waters, D. G. Guiney, and E. Lanka. 1992. Dissection of IncP conjugative plasmid transfer: definition of the transfer region Tra2 by mobilization of the Tra1 region in trans. J. Bacteriol. 1742493-2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCΤ method. Methods 25402-408. [DOI] [PubMed] [Google Scholar]
- 24.Lutkenhaus, J. 1993. FtsZ ring in bacterial cytokinesis. Mol. Microbiol. 9403-409. [DOI] [PubMed] [Google Scholar]
- 25.McAdams, H. H., and L. Shapiro. 2003. A bacterial cell-cycle regulatory network operating in time and space. Science 3011874-1877. [DOI] [PubMed] [Google Scholar]
- 26.Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 27.Nonaka, G., M. Blankschien, C. Herman, C. A. Gross, and V. A. Rhodius. 2006. Regulon and promoter analysis of the E. coli heat-shock factor, σ32, reveals a multifaceted cellular response to heat stress. Genes Dev. 201776-1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogino, H., M. Wachi, A. Ishii, N. Iwai, T. Nishida, S. Yamada, K. Nagai, and M. Sugai. 2004. FtsZ-dependent localization of GroEL protein at possible division sites. Genes Cells 9765-771. [DOI] [PubMed] [Google Scholar]
- 29.Rozen, S., and H. Skaletsky. 2000. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 132365-386. [DOI] [PubMed] [Google Scholar]
- 30.Shih, Y. L., and L. Rothfield. 2006. The bacterial cytoskeleton. Microbiol. Mol. Biol. Rev. 70729-754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Straus, D. B., W. A. Walter, and C. A. Gross. 1987. The heat shock response of E. coli is regulated by changes in the concentration of σ32. Nature 329348-351. [DOI] [PubMed] [Google Scholar]
- 32.Tétart, F., and J. P. Bouché. 1992. Regulation of the cell-cycle gene ftsZ by DicF antisense RNA. Division does not require a fixed number of FtsZ molecules. Mol. Microbiol. 6615-620. [DOI] [PubMed] [Google Scholar]
- 33.Tomoyasu, T., J. Gamer, B. Bukau, M. Kanemori, H. Mori, A. J. Rutman, A. B. Oppenheim, T. Yura, K. Yamanaka, H. Niki, et al. 1995. Escherichia coli FtsH is a membrane-bound, ATP-dependent protease which degrades the heat-shock transcription factor σ32. EMBO J. 142551-2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tomoyasu, T., T. Ogura, T. Tatsuta, and B. Bukau. 1998. Levels of DnaK and DnaJ provide tight control of heat shock gene expression and protein repair in Escherichia coli. Mol. Microbiol. 30567-581. [DOI] [PubMed] [Google Scholar]
- 35.Tsuchido, T., R. A. VanBogelen, and F. C. Neidhardt. 1986. Heat shock response in Escherichia coli influences cell division. Proc. Natl. Acad. Sci. USA 836959-6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Urech, C., S. Koby, A. B. Oppenheim, M. Munchbach, H. Hennecke, and F. Narberhaus. 2000. Differential degradation of Escherichia coli σ32 and Bradyrhizobium japonicum RpoH factors by the FtsH protease. Eur. J. Biochem. 2674831-4839. [DOI] [PubMed] [Google Scholar]
- 37.Vicente, M., A. I. Rico, R. Martinez-Arteaga, and J. Mingorance. 2006. Septum enlightenment: assembly of bacterial division proteins. J. Bacteriol. 18819-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weichart, D., R. Lange, N. Henneberg, and R. Hengge-Aronis. 1993. Identification and characterization of stationary phase-inducible genes in Escherichia coli. Mol. Microbiol. 10407-420. [PubMed] [Google Scholar]
- 39.Yura, T., E. Guisbert, M. Poritz, C. Z. Lu, E. Campbell, and C. A. Gross. 2007. Analysis of σ32 mutants defective in chaperone-mediated feedback control reveals unexpected complexity of the heat shock response. Proc. Natl. Acad. Sci. USA 10417638-17643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zahrl, D., M. Wagner, K. Bischof, and G. Koraimann. 2006. Expression and assembly of a functional type IV secretion system elicit extracytoplasmic and cytoplasmic stress responses in Escherichia coli. J. Bacteriol. 1886611-6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao, K., M. Liu, and R. R. Burgess. 2005. The global transcriptional response of Escherichia coli to induced σ32 protein involves σ32 regulon activation followed by inactivation and degradation of σ32 in vivo. J. Biol. Chem. 28017758-17768. [DOI] [PubMed] [Google Scholar]
- 42.Zhou, Y. N., N. Kusukawa, J. W. Erickson, C. A. Gross, and T. Yura. 1988. Isolation and characterization of Escherichia coli mutants that lack the heat shock sigma factor σ32. J. Bacteriol. 1703640-3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.