

Abstract
During development, human β-globin locus regulation undergoes two critical switches, the embryonic-to-fetal and fetal-to-adult hemoglobin switches. To define the role of the fetal Aγ-globin promoter in switching, human β-globin-YAC transgenic mice were produced with the Aγ-globin promoter replaced by the erythroid porphobilinogen deaminase (PBGD) promoter (PBGDAγ-YAC). Activation of the stage-independent PBGDAγ-globin strikingly stimulated native Gγ-globin expression at the fetal and adult stages, identifying a fetal gene pair or bigenic cooperative mechanism. This impaired fetal silencing severely suppressed both δ- and β-globin expression in PBGDAγ-YAC mice from fetal to neonatal stages and altered kinetics and delayed switching of adult β-globin. This regulation evokes the two human globin switching patterns in the mouse. Both patterns of DNA demethylation and chromatin immunoprecipitation analysis correlated with gene activation and open chromatin. Locus control region (LCR) interactions detected by chromosome conformation capture revealed distinct spatial fetal and adult LCR bigenic subdomains. Since both intact fetal promoters are critical regulators of fetal silencing at the adult stage, we concluded that fetal genes are controlled as a bigenic subdomain rather than a gene-autonomous mechanism. Our study also provides evidence for LCR complex interaction with spatial fetal or adult bigenic functional subdomains as a niche for transcriptional activation and hemoglobin switching.
Developmental and tissue-specific expression of multigene loci of higher eukaryotes involves concerted regulation from distal and proximal elements for gene transcriptional activation or silencing. To understand the dynamic control of specific expression patterns and to characterize chromosomal organization, the human β-globin locus is an excellent model system, based on several regulation and mutation studies. The human β-globin locus consists of a locus control region (LCR) and five β-like globin genes expressed sequentially in development, based on their order within the locus (5′-ɛ-Gγ-Aγ-δ-β-3′). The first gene, ɛ, is expressed in the yolk sac during primitive erythropoiesis, and then a switch to the duplicated fetal globin genes, Gγ and Aγ, initiates liver definitive erythropoiesis. At birth, a second globin switch occurs when the fetal genes are silenced and the adult genes, β and δ, in the bone marrow become activated. Expression of the β-globin cluster is enhanced by the major hypersensitive DNase I sites or LCR, situated upstream of the ɛ-globin gene.
The molecular bases of the human β-like globin gene-LCR interaction in developmental stage-specific expression are complex. Early studies of individual human γ- and β-globin genes in transgenic mice have delineated, despite low expression and integration position effects, that proximal regulatory sequences of these genes are sufficient for an appropriate developmental expression pattern in the mouse. Expression of the human Gγ- and Aγ-globin genes was confined to primitive erythroid cells, whereas that of the adult β-globin gene was present in definitive erythroid cells. Experiments using miniconstructs/miniloci with an intact or dissected LCR linked to β-like globin genes conferred high-level expression but also provided additional insights into the regulation of the locus. Regulation of the ɛ-globin gene linked to the LCR indicated an autonomous mechanism for both activation in primitive erythropoiesis and silencing in definitive erythropoiesis (47, 53). Fetal Gγ- and Aγ-globin genes with sufficient promoter lengths are also reported to undergo substantial gene-autonomous silencing in definitive erythroid cells (14), but an additional interplay between the γ-globin genes and the adult β-globin gene appears necessary for proper hemoglobin switching (36, 50, 54). Indeed, the adult β-globin gene linked to the LCR requires a γ-globin gene or additional regulatory elements for restricted adult activation (25), indicating regulation by reciprocal competition. When the entire β-globin locus with the LCR contained in a yeast artificial chromosome (YAC) is used as a transgene, the temporal and tissue gene regulation recapitulates the hemoglobin switching mechanism in the mouse, making this experimental system most relevant for studies on the mechanisms of the fetal-to-adult globin switch.
A model of competition between genes has been supported by a number of studies, including evidence for physical interactions between the LCR and one globin gene at a time by in situ hybridization analysis (63). Long-range physical interaction of the LCR with globin genes was proposed to form, through spatial looping, an active chromatin hub regulatory mechanism (15, 17, 43). Although temporal expression is in part regulated by interactions between the LCR and the globin genes, it is relatively well recognized that distance, polarity, or gene order relative to the LCR plays a critical role in the regulation of all β-like globin genes. Indeed, globin gene displacement or the use of marked globin genes at different positions in the locus or at various distances from the LCR influences the expression pattern (2, 10, 26). A tracking model is also supported by the production of distinct intergenic transcripts within the locus that would be implicated in remodeling/opening chromatin subdomains at different temporal stages to activate particular β-like globin genes (24, 46).
All of these mechanistic models postulate that sequences in proximity to or within the genes are required for developmental activation and repression of the β-like globin genes (58, 59). Studies of fetal gene regulatory elements have identified the binding of key transcription factors within the γ-globin proximal promoter (35, 41, 50) that frequently overlap with point mutations found in human hereditary persistence of fetal hemoglobin (HPFH) (reviewed in reference 19). In addition, hemoglobin switching studies on the Aγ-globin promoter/gene in a γ-β minilocus did not delineate whether only the γ-globin promoter or any active promoter could prevent activation of β-globin at the embryonic stage (1, 25, 51). While these experiments used promoters that can be active at all stages of development, it is not clear whether a heterologous (Tk) promoter of low transcriptional activity, similar to the γ-globin promoter, or a strong erythroid promoter (β-spectrin) independent of the LCR can fully explain the above-mentioned discrepancy. Furthermore, it cannot be excluded that the gene distance from the LCR, a truncated LCR, and/or the absence of some β-like globin genes may influence the mechanism of the fetal-to-adult hemoglobin switch.
To evaluate the role of the Aγ promoter and to clarify the interaction of the LCR with the β-like globin genes in hemoglobin switching, replacement of the Aγ promoter by the erythroid-specific and stage-independent porphobilinogen deaminase (PBGD) gene promoter was undertaken in a human β-globin YAC for production of transgenic mice. This promoter was selected because it has similar transcriptional potential to that of the γ promoter, it is regulated by a cognate group of trans-acting factors, like the globin promoters, and particularly, it can be activated by the LCR (16, 27, 38, 39). RNA/protein expression analysis of all human β-like globin genes in transgenic PBGDAγ-YAC mice showed major activation of the native Gγ gene at the fetal/adult stage and delayed activation of the adult β-globin gene that evoked the two hemoglobin switching patterns in humans. Our data show that both intact Gγ and Aγ promoters are required for appropriate adult silencing, supporting a fetal gene pair or bigenic control mechanism. Interactions of the LCR with β-like globin genes, determined by chromosome conformation capture (3C), suggest a model of hemoglobin switching regulation that consists of fetal and adult bigenic conformational subdomains creating a transcriptionally active niche or hub with the LCR complex.
MATERIALS AND METHODS
Oligonucleotide primers.
A list of all primers used in this study is provided in the supplemental material.
Production of PBGDAγ-YAC.
The yeast integrating plasmid (YIp) pRS-PBGDAγ was produced by first ligating an upstream Aγ MscI-SapI blunted fragment (positions −3423 to −838) isolated from pVZ-ES into NotI-digested and blunted pRS406 vector. Secondly, the 887-bp human PBGD promoter was obtained by nested PCR amplification from human DNA, digested with AvrII and BamHI, blunted, and ligated into the XbaI-BamHI blunted YIp, creating a ClaI site. Thirdly, a downstream Aγ BsaHI-EcoRV blunted fragment (positions +49 to +6558) obtained from the FC12 cosmid was ligated into the blunted ClaI site. By this strategy, a sequence (GGCCGCTCTAG) of the polylinker was located at the SapI-AvrII junction, between the 5′Aγ-globin region and the PBGD promoter, and 3 bp (CGC) was located at the BamHI-BsaHI junction, prior to the Aγ-globin ATG, which did not alter the Kozak sequence. After being sequenced, pRS-PBGDAγ linearized at the BspEI site was transformed into a spheroplasted 150-kb wild-type β-YAC-containing S. cerevisiae strain (A201F4) by the pop-in/pop-out method (44). Briefly, transformed yeast cells were selected on Trp− Lys− Ura− dropout plates, and pop-in clones were isolated and characterized by Southern blotting. Clones with proper integration of the YIp pRS-PBGDAγ were then plated on 5-fluoroorotic acid, and several pop-out PBGDAγ-YAC clones were isolated. The PBGDAγ-YAC obtained was characterized by Southern blotting, using five probes covering different genes of the locus, including HS2 (HpaI-HindIII; 1.6 kb), ɛ (ClaI-PvuII; 0.9 kb), γ (StuI-SphI; 2.6 kb), 5′Aγ (BanII-SapI; 1.3 kb), and β (BamHI-SphI; 1.1 kb). While the 5′Aγ probe was used on EcoO65I digests, all other globin probes were used on EcoRI, PstI, BamHI, and HindIII digests. The right and left arms of the YAC were analyzed using two specific probes on undigested or digested DNA (KpnI or SfiI/SalI fragments) separated by pulsed-field gel electrophoresis (PFGE).
Production and characterization of transgenic PBGDAγ-YAC mice.
The PFGE-purified PBGDAγ-YAC was microinjected into (C57BL/6J × CBA/J) F2 oocytes (60). The transgene was characterized by Southern blotting with EcoRI digestion, using HS2, ɛ, γ, and β probes as described above. The presence of the two arms of the YAC was assessed by PCR (9). Transgenic mice were produced on a mixed C57BL/6J and CBA/J background and selected as homozygous for the murine β-globin haplotype “diffuse” (60). A mouse line containing the parental 150-kb wild-type human β-globin locus (β-YAC) (9) was used as a control. All experiments conformed to the standards of the Canadian Council on Animal Care.
Quantification of globin mRNA.
Total RNAs from embryonic day 10.5 (e10.5) yolk sacs, e12.5, e14.5, and e16.5 fetal livers, newborn spleens, and adult bone marrows of PBGDAγ-YAC lines (PY367, PY418, and PY465) and one β-YAC line were extracted using TRIzol reagent. Total RNA was DNase I treated and reverse transcribed with Moloney murine leukemia virus reverse transcriptase. Quantitative real-time PCR was performed with Quantitech SYBR green and 0.3 μM of each primer. Both S16 and glycophorin A were used as normalizers of input cDNA. Triplicates were done for each PCR amplification, carried out on an Mx4000 quantitative PCR machine, except for δ/β analysis, which was performed on an Mx3005P machine. Relative values were calculated using Mx4000 v4.20 or MxPro v3.00 software. Expression studies of globin mRNA were corrected for copy number (see Fig. 1C).
FIG. 1.

Production of PBGDAγ-YAC mice and characterization of transgenic lines. (A) The human β-globin locus in a YAC was modified by replacement of the 887-bp proximal Aγ promoter (pr. Aγ), from positions −838 to +49 (SapI to BsaHI fragment), with the equivalent region (887 bp) of the erythroid-specific PBGD promoter (pr. PBGD) that spans positions −815 to +72 (BamHI fragment). (B) Analysis of the PBGDAγ-YAC construct by use of different restriction enzymes (EcoRI, HindIII, BamHI, PstI, and BstEII) on the modified human β-globin YAC obtained by homologous recombination in yeast. Southern analysis was carried out on DNA isolated from yeast containing the PBGDAγ-YAC mutant (mut) or the wild-type human β-globin YAC (wt). The following five probes were used: (i) HS2 probe (EcoRI; 10.4 kb), (ii) ɛ probe (EcoRI; 3.7 kb) containing pSP73 vector sequences hybridizing to the YAC arm (Ya), (iii) 3′ Gγ probe (BstEII; 3.7 kb for wild type and 3.1 kb for PBGDAγ), (iv) γ probe hybridized to Gγ (EcoRI; 1.6 and 7.0 kb) and to Aγ (EcoRI; 2.6 kb for wild type and 2.1 kb for PBGDAγ), and (v) β probe (SphI fragment) for the β-globin (EcoRI; 5.5 kb and 3.8 kb) and δ-globin (EcoRI; 2.3 kb) genes. Analysis by PFGE was also carried out on KpnI or SfiI/SalI fragments, using probes for the YAC left arm (a), LCR (b), γ (c), and the YAC right arm (d), or on the full-length sequence (∼150 kb), using the γ probes (c). No rearrangements in the β-globin locus were detected from the Aγ promoter substitution homologous recombination steps. (C) Representative transgenic line (PY) analysis of transgene integrity by Southern blotting of EcoRI-digested genomic DNA compared to DNA of the wild-type β-YAC transgenic line (Y), monitored with probes i, ii, and iv (see above) and with a β probe (SphI-BamHI fragment) hybridized to the β-globin gene (5.5 kb) and to the δ-globin gene (2.3 kb). The presence of both the left and right arms of the YAC was detected by a multiplex PCR (204 and 183 bp for the left and right arms, respectively), as shown for both sides of the locus. Seven transgenic lines displayed all expected bands.
Quantification of globin chains.
Globin chains were separated by urea-Triton polyacrylamide gel electrophoresis (6) from peripheral blood lysates at e10.5, e12.5, e14.5, and e16.5 (β-YAC and three PBGDAγ-YAC lines [PY367, PY418, and PY465]); at postnatal day 0 (P0) to P21 (lines PY367 and PY465); and from adult mice (β-YAC and all seven PBGDAγ-YAC lines). The globin chains were then quantified by densitometry (ImageQuant v5.0 software). The following controls served to identify human globin chains: fetal cord blood (β, Gγ, and Aγ), Aγ-globin from μLCR-0.2 Aγ transgenic mouse blood (5), and ɛ-globin from human ɛ transgenic mouse blood (49).
FACS analysis.
Fluorescence-activated cell sorting (FACS) analysis was performed on blood from (C57BL/6J × CBA/J)F1, β-YAC, and seven PBGDAγ-YAC mouse lines according to the methods of Wallac/Isolab. Briefly, formaldehyde-glutaraldehyde-fixed cell pellets were suspended in 0.1% Triton X-100 solution (phosphate-buffered saline-0.1% bovine serum albumin [BSA]) and 1.5 μg of biotin-conjugated anti-HbA antibody (Perkin-Elmer) per 100-μl aliquot for 30 min. Pellets were washed in 0.01% Triton X-100 solution (phosphate-buffered saline-0.1% BSA) and suspended in 0.01% Triton X-100 solution and 2.5 μg of fluorescein isothiocyanate-conjugated anti-HbF antibody and 0.1 μg of allophycocyanin-conjugated streptavidin (BD Bioscience) per 100 μl of cells for 30 min. Cells were then washed in 0.01% Triton X-100 solution and analyzed on a FACSCalibur flow cytometer, using CellQuest Pro v4.0.2 and WinMDI v2.8 software.
ChIP assay.
Chromatin immunoprecipitation (ChIP) analysis was carried out on e12.5 total fetal liver cells (n = 5 to 8/line) and on FACS-sorted adult erythroid marrow cells (n = 3/line) of β-YAC, PY367, and PY465 transgenic mice. Erythroid precursors were sorted using MoFlo cell sorter with phycoerythrin (PE)-conjugated anti-Ter119 antibody (3). ChIP was performed according to the manufacturer's protocols, using antibodies raised against histone H3, acetylated histones H3 (K9 and K14) and H4 (K5, K8, K12, and K16), and dimethylated histone H3 (K4) (Upstate Biotech). Chromatin fragments were PCR amplified using Mx4000 multiplex quantitative PCR and Quantitech SYBR green and normalized on the Zfp37 gene (7) (see the supplemental material).
Methylation analysis.
Genomic DNAs from e12.5 fetal livers of PBGDAγ-YAC (n = 4) and β-YAC (n = 3) mice were purified by phenol-chloroform extraction. DNA digestions were carried out in consecutive reactions, first with enzyme EcoRI and second with a selected methylation-sensitive enzyme according to the region analyzed, i.e., HhaI, BsaAI, or HpaII, and then compared to total digestion with MspI. Digested DNAs were analyzed by Southern blotting using a β probe (BamHI; positions −1461 to +476) that cross-hybridizes with δ and/or a Gγ probe (BglII/EcoRI; positions −1659 to +1429) that recognizes Aγ. Quantification was done by densitometry (ImageQuant v5.0), and the ratio of cut to uncut DNA was normalized for probe length.
3C assay.
3C assay was performed as described previously (13, 57), with modifications in cell preparation. Transgenic e12.5 fetal livers (n = 3 or 4) were pooled and suspended in Dulbecco's modified Eagle's medium-10% fetal calf serum, and total bone marrow erythroid cells were purified by positive selection on an autoMACS separator with PE-conjugated anti-Ter119 antibody and anti-PE microbeads, producing a >90% homogeneous population. As a control template, equimolar amounts of the PBGDAγ-YAC and the murine Pkd1-BAC (56) were digested with EcoRI and ligated. Ligation products (300 ng; n = 3 pools/line for e12.5 embryos or n = 3 mice/line for adults) from 3C experiments and a control template (20 ng) were PCR amplified. Samples were analyzed in 2.1% agarose gels, visualized by ethidium bromide, and quantified by densitometry (ImageQuant v5.0 software).
Statistical methods.
Values are expressed as means ± standard deviations (SD) or as medians ± interquartile ranges when the distribution did not pass the D'Agostino and Pearson omnibus normality test. Unpaired two-sample Student's t test or the Mann-Whitney test was used for statistical analysis when two groups were compared, repeated-measure analysis of variance (ANOVA) and Tukey's multiple comparison test or the Kruskal-Wallis test and Dunn's posttest were used when three groups were compared, and the F test of slope was used to assess globin gene expression kinetics. In all statistical tests, P values of <0.05 were considered significant.
RESULTS
Generation of human PBGDAγ-YAC transgenic mice.
To define the mechanistic implication of the Aγ promoter in globin switching, the human β-globin locus contained in a 150-kb YAC was modified by replacing the Aγ-globin 5′ untranslated region (5′-UTR)/promoter with the equivalently sized and stage-independent erythroid PBGD promoter, using homologous recombination. The PBGD promoter was chosen based on similarities in structural organization with the globin promoter and in regulatory factors involved in globin regulation (27, 38). This modification was generated with the yeast integrating plasmid (YIp) pRS-PBGDAγ (Fig. 1A). Once YIp integrity was confirmed by sequencing, it served to produce the modified YAC. Modified YAC integrants with proper integration of the PBGD promoter subsequently underwent homologous recombination for excision of the 5′-UTR/promoter of the Aγ-globin gene (Fig. 1A). Extensive genomic structure analysis of recombinants was carried out by Southern blotting. PBGDAγ-YAC recombinants (∼25%) showed the intended modification without alteration of other genes in the locus (Fig. 1B).
Transgenic PBGDAγ-YAC lines were produced and characterized by Southern blotting and PCR (Fig. 1C). Seven lines integrated 1 to 10 intact copies of the complete transgene. Transgenic mouse progenies homozygous at the endogenous β-globin haplotype “diffuse” (βMajor and βMinor) were maintained for RNA and protein analysis.
Altered onset of expression and kinetics of hemoglobin switching in PBGDAγ-YAC mice.
To evaluate expression levels of the different genes of the locus, we performed quantitative real-time PCR on three PBGDAγ-YAC lines (PY367, PY418, and PY465) and on control wild-type β-YAC mice. Analyses were carried out at different stages of development, from e10.5 to adult, for embryonic, fetal, and adult genes (Fig. 2A and B; Table 1). The three PBGDAγ-YAC lines showed no significant difference throughout development in the ratio of the individual β-like globin genes as a function of total human β-like globin genes (P > 0.4).
FIG. 2.

Expression of human β-globin genes in PBGDAγ-YAC and β-YAC mice. (A) Patterns of human globin gene expression in β-YAC and PBGDAγ-YAC mice, from embryonic to newborn (NB) to adult (Ad) stages. Each human globin gene's expression was quantified and reported as a percentage relative to total human β-globin gene expression (± SD). Expression levels were determined for β-YAC mice at embryonic/fetal (n = 3), newborn (n = 3), and adult (n = 2) stages and for PBGDAγ-YAC mouse lines (PY367, PY418, and PY465) (mean of 2 or 3 samples/line) at embryonic/fetal (n = 9), newborn (n = 7), and adult (n = 8) stages. (B) The ratios of δ-globin/β-globin gene expression at embryonic to adult stages were quantified with specific primers, using the samples obtained for panel A from β-YAC mice and PBGDAγ-YAC lines. The P values were determined by Student's t test. *, P < 0.002; **, P < 0.0005; ***, P < 0.0001.
TABLE 1.
Globin gene expression in transgenic micea
| Transgenic line | ɛ-Globin gene expression
|
γ-Globin (Gγ:Aγ) gene expression
|
β-Globin gene expression
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| e10.5 | e12.5 | e14.5 | e10.5 | e12.5 | e14.5 | e16.5 | Newborns | Adults | e12.5 | e14.5 | e16.5 | Newborns | Adults | |
| YAC | 1.0 ± 0.2 | 1.0 ± 0.4 | 1.0 ± 0.6 | 1.0 ± 0.2 | 1.0 ± 0.3 | 1.0 ± 0.4 | 1.0 ± 0.2 | 1.0 ± 0.8 | 1.0 ± 0.9 | 1.00 ± 0.21 | 1.00 ± 0.39 | 1.00 ± 0.17 | 1.00 ± 0.12 | 1.00 ± 0.51 |
| PY367 | 0.8 ± 0.3 | 0.3 ± 0.1 | 1.5 ± 0.6 | 0.8 (0.6:0.2) ± 0.4 | 3.8 (2.7:1.1) ± 0.8 | 12.4 (8.8:3.6) ± 2.6 | 26.0 (15.1:10.9) ± 16.3 | 1.2 (0.6:0.6) ± 0.5 × 102 | 3.7 (2.3:1.4) ± 0.9 × 103 | 0.04 ± 0.01 | 0.04 ± 0.01 | 0.07 ± 0.04 | 0.11 ± 0.03 | 0.07 ± 0.01 |
| PY418 | 1.1 ± 0.4 | 1.0 ± 0.3 | 1.2 ± 0.5 | 1.1 (0.7:0.4) ± 0.5 | 4.2 (3.2:1.0) ± 1.2 | 34.3 (18.9:15.4) ± 9.9 | 31.4 (19.9:11.5) ± 13.9 | 2.8 (1.1:1.8) ± 0.5 × 102 | 11.8 (4.1:7.7) ± 11.5 × 103 | 0.03 ± 0.01 | 0.11 ± 0.05 | 0.46 ± 0.10 | 0.12 ± 0.03 | 0.63 ± 0.46 |
| PY465 | 3.7 ± 2.2 | 3.0 ± 2.1 | 3.3 ± 2.6 | 3.3 (2.4:0.9) ± 1.6 | 20.0 (14.3:5.7) ± 8.1 | 31.4 (22.7:8.7) ± 16.6 | 90.1 (61.4:28.7) ± 58.2 | 2.0 (0.9:1.0) × 102 | 37.9 (22.5:14.8) ± 38.3 × 103 | 0.17 ± 0.10 | 0.13 ± 0.05 | 0.16 ± 0.06 | 0.06 | 0.79 ± 0.58 |
Expression is expressed relative to that in β-YAC wild-type mice.
Embryonic ɛ-globin expression levels were measured as a function of total human β-like globin expression in erythroid cells from e10.5 yolk sacs; e12.5, e14.5, and e16.5 fetal livers; newborn spleens; and adult bone marrows. As shown in Fig. 2A, ɛ-globin expression in PBGDAγ-YAC lines was essentially comparable to that in wild-type β-YAC mice at all stages of development, indicating that ɛ-globin gene regulation was independent of PBGDAγ modification (Fig. 2A; Table 1).
Each fetal gene's expression was analyzed individually in PBGDAγ-YAC mice by determining the presence of the 5′-UTR of PBGD. As predicted for the PBGD promoter, Aγ expression in PBGDAγ-YAC mice was stable throughout development, from e10.5 to adulthood (Fig. 2A). This sustained expression differed from the repression of both indistinguishable γ-globin genes in β-YAC mice at e12.5 and their near absence from e14.5 onward (Fig. 2A). The expression pattern of the unmodified fetal Gγ gene in PBGDAγ-YAC mice was expected to follow that in β-YAC mice, but the gene was in fact significantly upregulated. While Gγ expression at e10.5 appeared normal, expression at e12.5 and e14.5 was not decreased, as observed in β-YAC mice, but instead was increased by 50% in all three of the PBGDAγ-YAC mouse lines relative to the level at e10.5 (Fig. 2A). This activation of the Gγ globin gene at e12.5 and e14.5 contrasts with the γ-globin (sum of Gγ and Aγ) major repression of 10- and 25-fold, respectively, at these stages in β-YAC mice. In PBGDAγ-YAC mice prior to birth, the Gγ expression pattern displayed a progressive decrease without ever being silenced completely, even at several months of age (Fig. 2A). This striking incomplete fetal globin switching was observed for all three transgenic lines, in contrast to the absence of expression by e14.5 in β-YAC mice (P ≤ 0.0005) (Fig. 2A; Table 1).
Expression analysis of adult genes showed altered switching and kinetic patterns. Analysis of δ-globin gene and β-globin gene expression with specific primers was carried out at all stages of development. At e10.5, expression of the δ-globin gene was virtually undetectable for both PBGDAγ-YAC and β-YAC mice. Interestingly, in wild-type β-YAC mice, the δ-globin gene relative to the adult β-globin gene (or total β-like globin genes) was expressed at low but stable levels from e12.5 until birth and increased ∼10-fold in adulthood (Fig. 2B). In the three PBGDAγ-YAC transgenic lines, δ-globin gene expression was detected from e12.5 to adulthood and was maintained at similar levels. However, the expression level was ∼10-fold lower than that measured in control β-YAC mice until birth (P < 0.0005). Since δ-globin expression was not activated in adult PBGDAγ-YAC bone marrow, the level was decreased even further, ∼100-fold, compared to that in adult controls (P < 0.002), showing that the PBGDAγ-globin gene interfered with both the δ-globin expression level and pattern. Quantitative adult β-globin gene expression at e10.5 as a function of total β-like globin was estimated at ∼5% for β-YAC mice (Fig. 2A), whereas very low levels (<0.1%) were detected in PBGDAγ-YAC mice (Fig. 2A), suggesting an impact of PBGDAγ-globin at this early stage. At e12.5, adult β-globin gene expression (80%) was predominant over that of all β-like globin genes in control β-YAC mice. By then, adult globin switching had essentially occurred. In contrast, adult β-globin gene expression was only becoming activated in PBGDAγ-YAC mice at the same stage. The delay in activation from e12.5 until birth corresponded to a fivefold decrease in kinetics of adult β-globin gene expression for the PBGDAγ-YAC transgenic mouse lines relative to control mice (P < 0.0001). At birth, the adult β-globin gene expression levels (∼65%) were below those measured at e12.5 for β-YAC mice, thereby undergoing a delay of >8 days. In fact, the maximum level (80%) of adult β-globin gene expression in the three PBGDAγ-YAC lines was reached only in adulthood (Fig. 2A; Table 1). Although adult β-globin gene expression in PBGDAγ-YAC mice was severely impaired (P < 0.0005), that of δ-globin was even more markedly repressed, suggesting that PBGD promoter-driven Aγ-globin had a profound impact on adult gene regulation.
In parallel to the globin RNA expression pattern, we also monitored globin chain protein expression from peripheral red blood cells (RBCs) at all development stages for β-YAC mice and three PBGDAγ-YAC lines (n = 2 to 5 per line), including P2 to P21 (alternating days) for two PBGDAγ-YAC lines (n = 3). The delay in RNA to protein expression for fetal-to-adult switching was ∼3 to 4 days for β-YAC mice (∼e11.5 to e15.5) and ∼11 to 14 days for PBGDAγ-YAC mice (∼e17.5 to P10) (Fig. 3A and B and data not shown). For both β-YAC and PBGDAγ-YAC fetuses, ɛ-globin protein levels were detectable until e14.5, indicating that some peripheral RBCs of embryonic origin from yolk sacs were still circulating, as observed previously (20).
FIG. 3.

Globin chain analysis of PBGDAγ-YAC and β-YAC RBCs. (A) Globin chains from total blood lysates of wild-type mice (wt), β-YAC mice (YAC), and PBGDAγ-YAC mice (PY) at different developmental stages were separated by urea-Triton polyacrylamide gel electrophoresis to distinguish the ɛ (arrow or upper band; the lower band is βH1), Aγ, Gγ, βhu, and αmu (endogenous murine α) globin chains. (B) Patterns of human globin chain expression at embryonic to adult stages for β-YAC mice or PBGDAγ-YAC mice. Quantification of each globin chain ratio is given as a percentage of the total human β-like globins (± SD). Levels of expression were determined for the β-YAC line (n = 2 to 5) and for three PBGDAγ-YAC lines, PY367, PY418, and PY465 (n = 9 to 11 [two to five mice per line]) at different stages.
Unlike the case for RNA analysis of β-YAC mice, Aγ and Gγ chains can be distinguished by protein expression analyses for both wild-type β-YAC mice and PBGDAγ-YAC transgenic mouse lines (Fig. 3). At e10.5, both wild-type and mutant mice displayed similar patterns of higher Gγ and Aγ chain levels. Interestingly, Gγ was the major protein chain at the fetal stage, as in humans, supporting the observation that PBGDAγ substitution had no marked impact on fetal globin expression in primitive erythropoiesis. From e12.5 on, however, the pattern of fetal chains in PBGDAγ-YAC mice differed from that in β-YAC mice. In β-YAC mice, Gγ protein expression levels were more elevated than those of Aγ chains, but at e14.5 both fetal chains decreased, reaching equivalent levels, and were gradually replaced by the adult β-globin chains. By e16.5, both γ chains had barely detectable levels in β-YAC mice. In contrast, the PBGDAγ-YAC mice showed significant activation of Gγ compared to Aγ from e12.5 on, and Gγ remained the major protein even after birth, until ∼P10 (data not shown). Noticeably, at several months of age, levels of both γ chains from peripheral blood represented 40 to 50% of human β-like chains.
Concomitant with sustained fetal chain expression throughout development, the onset of β protein expression was delayed in PBGDAγ-YAC mice compared to that in control β-YAC mice (Fig. 3). Furthermore, the kinetics of β-globin chain expression were altered. Maximum levels were obtained in adulthood. While expression of the δ-globin chain was readily detectable in adult β-YAC mice, representing 2.8% ± 0.7% of total β-globin, it was virtually undetectable in PBGDAγ-YAC mice at all stages, suggesting severely impeded δ-globin regulation.
Pancellular expression of fetal and adult β-globin in adults.
To assess whether the expression of γ- and β-globin genes observed in adult PBGDAγ-YAC mice was pancellular or in two distinct cell populations, RBCs of all seven transgenic lines were analyzed by flow cytometry. Two antibodies, anti-human HbA and anti-human HbF, were used to specifically recognize hybrid tetramers α2muβ2hu and α2muγ2hu, respectively, but not murine hemoglobin, as shown by analysis of wild-type nontransgenic mice (Fig. 4). Expression of human β-globin (HbA+) at the adult stage was detected in 70 to 80% of RBCs from all transgenic lines as well as from β-YAC mice, suggesting that expression distribution is not copy number dependent. Noticeably, the expression of human γ-globin (HbF+) was observed in 85 to 100% of RBCs from the seven PBGDAγ-YAC lines. Lines with two copies of the transgene and above displayed a slight increase in the proportion of γ-expressing cells, likely due to a shift to higher HbF signal intensities (Fig. 4B).
FIG. 4.

Cellular distribution of γ- and β-globin expression in adult peripheral blood. (A) Representative flow cytometry assays on peripheral blood of wild-type control (C57BL/6J × CBA/J)F1, β-YAC, and PBGDAγ-YAC mice. Specific antibodies that do not cross-hybridize with endogenous hemoglobins were used to recognize the human/mouse hybrid hemoglobins α2muγ2hu (α-HbF) and α2muβ2hu(α-HbA). (B) Distributions of cells expressing HbF and/or HbA at the adult stage. All seven PBGDAγ-YAC (PY) lines were compared to the β-YAC line and to the wild type (WT) as controls. The RBC percentages represent averages for two to four mice per line.
Chromatin modification of globin genes in PBGDAγ-YAC and β-YAC mice.
Differences in globin gene regulation patterns, in particular that of the Gγ gene, in PBGDAγ-YAC and β-YAC mice prompted us to analyze chromatin status and its potential role. Since our results at e12.5 showed that PBGDAγ-YAC mice expressed essentially γ, whereas β-YAC mice expressed β (Fig. 2), we selected fetal liver cells at e12.5 during development and erythroid bone marrow precursors in adults for these experiments. ChIP analysis was performed to monitor the methylation and acetylation status of both fetal and adult promoters (Fig. 5A). Analysis was carried out with two PBGDAγ-YAC lines (PY367 and PY465) and β-YAC mice. Total occupancy by histone H3 for both the Gγ and PBGDAγ promoters was slightly but significantly reduced (37% and 87%) in comparison to that for β-YAC mice. However, relative enrichments of histone H3 modification in fetal Gγ and PBGDAγ promoters were detected for K9/K14 acetylation (∼2- and 4-fold, respectively) and for K4 dimethylation (∼2- and 3-fold, respectively) compared to the two γ promoters in the β-YAC mouse (Fig. 5A). For the adult β-globin gene promoter in PBGDAγ-YAC and β-YAC mice, similar levels of histone H3 occupancy and of histone H3 and H4 modification were detected. In addition, similar analysis was performed on FACS-sorted erythroid precursors (CD71+ Ter119+) from adult bone marrows of PBGDAγ-YAC transgenic mice (PY367 and PY465) and control β-YAC mice (n = 3 for each line). No significant difference was measured in chromatin status for the native Gγ or β promoter for PBGDAγ-YAC or β-YAC lines (data not shown). These data for PBGDAγ-YAC mice suggest that chromatin modifications may contribute to altered fetal gene regulation.
FIG. 5.

Epigenetic analysis at fetal and adult globin promoters in e12.5 fetal livers. (A) ChIP assays performed on e12.5 fetal livers from two PBGDAγ-YAC lines (PY367 and PY465) (white bars) (n = 9 to 13) and the control β-YAC line (black bars) (n = 7), using specific antibodies for histone H3 (H3), the acetylated forms of histone H3 (H3ac) and H4 (H4ac), and the K4-dimethylated form of histone H3 (H3me). Enrichment of histone acetylation/methylation (y axis), determined at the γ-globin promoters (for γ, positions −158 to +53; and for PBGDAγ, positions −167 to +47) and at the β-globin promoter (positions −146 to +36) for β-YAC and PBGDAγ-YAC lines, is depicted in the histogram as the median of ratios relative to the control gene Zfp37 ± the interquartile range, normalized on total H3 for both H3ac and H3me modifications. The P values were determined by Kruskal-Wallis and Mann-Whitney tests. *, P < 0.01; **, P < 0.002; ***, P < 0.0001. (B) γ- and β-globin promoter DNA methylation status was assessed in e12.5 fetal livers from two PBGDAγ-YAC lines (PY418 and PY367) (n = 4 [2/line]) and the control β-YAC line (n = 3). The histograms show CpG dinucleotide methylation at each locus coordinate, with localization relative to the +1 initiation site indicated in parentheses, as percentages ± SD of the undigested methylated HpaII (H), HhaI (Hh), and BsaAI (B) sites from EcoRI (E) globin promoter fragments. The P values were determined by Student's t test. *, P < 0.002.
To evaluate whether differences in transcriptional regulation patterns were associated with DNA modification, the methylation profile of CpG dinucleotides was monitored in fetal and adult globin gene regions. The percentage of methylation (5-methylcytosine [m5C]) was quantified from e12.5 DNA fetal livers at characterized methylation-sensitive restriction sites (HpaII, HhaI, or BsaAI) of Gγ, Aγ, δ, or β promoter regions. The Gγ distal −1410 site (HhaI) was heavily methylated to similar levels for both PBGDAγ-YAC and β-YAC mice (Fig. 5B). In contrast, the Gγ proximal −54 site (HpaII) showed a significant threefold decrease in m5C for the PBGDAγ-YAC compared to β-YAC fetal livers, consistent with the ongoing active transcription in PBGDAγ-YAC mice (Fig. 5B). Notably, the PBGD CpG-rich promoter also displayed very low m5C levels, comparable to those at the Gγ proximal site in PBGDAγ-YAC mice, whereas high m5C levels were detected at the barely active Aγ or Gγ proximal −54 site (HpaII) in β-YAC fetal livers (Fig. 5B and data not shown). At the δ coding sequence site (+469), methylation was above 90% in both PBGDAγ-YAC and β-YAC mice. The β-globin gene was monitored at three sites (−1791, −268, and +471), and the two distal sites did not show significant differences in m5C levels between PBGDAγ-YAC and β-YAC fetal livers. While the +471 site in the β-globin coding sequence remained highly methylated in PBGDAγ-YAC mice, correlating with virtual inactivity of β-globin, it was ∼50% demethylated in the β-YAC mice expressing essentially adult human β-globin. For both PBGDAγ-YAC and β-YAC mice, the β-like globin gene DNA demethylation profile corresponded to the gene activation pattern.
Sequestration of the LCR at the PBGD promoter leads to sustained activation of Gγ-globin.
To gain insight into the process underlying sustained activation of fetal genes at the expense of adult β-globin expression in PBGDAγ-YAC mice, we carried out 3C analysis. Interactions of individual globin genes of the locus with the LCR were analyzed in e12.5 fetal liver cells and adult bone marrow erythroid precursors of two PBGDAγ-YAC transgenic lines (PY367 and PY465) and compared to those for the control β-YAC transgenic line. The frequency of genomic interactions was quantified from cross-linked EcoRI fragments by PCR and normalized to the DNA concentration input and to the transgene copy number difference, with the pseudogene ψβ assigned a value of 1 (Fig. 6). As a control for nonspecific cross-linking, we measured interactions between the LCR and exon 15 of the murine Pkd1 gene. No product was detected, which confirmed the specificity of interactions detected within the locus (data not shown). At e12.5, the LCR showed little to no association with ɛ for both β-YAC and PBGDAγ-YAC transgenic mice. However, the LCR showed a strong association with the entire fetal domain, from Gγ to PBGDAγ, including the intergenic region between the two fetal genes, for PBGDAγ-YAC transgenic mice that was significantly higher than the interaction measured for the β-YAC locus (P < 0.002) (Fig. 6A). In contrast to interactions observed for the PBGDAγ-YAC locus, we detected a strong LCR association with the entire adult domain, from the δ to β globin genes, in β-YAC mice (P < 0.002) (Fig. 6A). At the adult stage, the interaction of the LCR and the fetal domain was still significantly higher in PBGDAγ-YAC mice than in β-YAC mice (P < 0.03), supporting the presence of a sustained interaction in erythroid cells throughout development. Additionally, in PBGDAγ-YAC mice, the LCR is in close proximity to the entire adult globin domain, with no significant difference from the wild-type locus (P = 0.3), including the δ-globin gene, despite a virtual absence of transcriptional activity. Interestingly, the LCR also showed a strong association with 3′HS for both β-YAC and PBGDAγ-YAC transgenic mice at the adult stage only, comparable to that observed for the adult globin domain, but it was not detected at e12.5 for β-YAC transgenic mice.
FIG. 6.

Spatial proximity of the LCR to the different regions of the β-globin locus by 3C. A schematic linear representation of the human β-globin locus is shown above each graph. Individual hypersensitive sites (arrows), globin genes (gray boxes), the ψβ pseudogene (open box), and individual fragments below the locus (gray lines) indicate the regions analyzed, and corresponding fragment quantification (gray shadow) is depicted in the histogram. (A) Relative cross-linking frequencies observed at e12.5 between the LCR EcoRI fragment (HS2 to HS4) and the rest of the locus (ɛ, Gγ, inter-Gγ-Aγ, Aγ or PBGDAγ, ψβ, δ, inter-δ-β, β, and 3′HS). Fetal liver cells were from the transgenic β-YAC line and two PBGDAγ-YAC lines (PY367 and PY465) (n = 3 pools with 3 or 4 livers/pool). Values of relative proximity (mean ± SD for each line) were determined for each EcoRI fragment/region (gray shadow) and normalized to the ψβ region, arbitrarily set at 1. The P values were determined by ANOVA (P < 0.002 for the fetal region [Gγ, inter-Gγ-Aγ, and Aγ or PBGDAγ] for line PY367 or PY465 and P < 0.002 for the adult region [δ, inter-δ-β, and β] for β-YAC mice). Representative examples of PCR-amplified ligation products are shown below the graph for evaluation of cross-linking frequencies. (B) Relative cross-linking frequencies observed in adult erythroid cells between the LCR EcoRI fragment and the rest of the locus. Adult bone marrow Ter119+ cells (n = 3/line) were analyzed from two PBGDAγ-YAC lines (PY367 and PY465) and the wild-type β-YAC line. The P values were determined by ANOVA (P < 0.03 for the fetal region [Gγ, inter-Gγ-Aγ, and Aγ or PBGDAγ] for line PY367 or PY465, but values were comparable for the adult region [δ, inter-δ-β, β, and 3′HS] for line PY367, PY465, or β-YAC). Representative examples of PCR-amplified ligation products are shown below the graph for evaluation of cross-linking frequencies.
DISCUSSION
Our study provides new evidence on the hemoglobin switching mechanism by functional characterization of the fetal stage-specific promoter of the Aγ-globin gene. Replacement of the Aγ-globin promoter by the erythroid-specific and stage-independent PBGD promoter in a YAC containing the human β-globin locus results in a strong deferred fetal-to-adult globin switch that occurs near birth instead of midgestation, as normally observed in transgenic mice and at the mouse β-globin locus. This pattern is reminiscent of human globin gene switching. Importantly, the induced critical upregulation of unmodified fetal Gγ-gene expression from the PBGDAγ-YAC locus in definitive erythropoiesis inversely influences the adult β-globin genes and reveals a coordinate control mechanism for the fetal genes. The spatial conformation of the PBGDAγ-YAC locus in the fetus displayed parallel LCR interactions with both genes of the fetal domain, at the exclusion of the adult domain. These results led us to propose a model in which a gene pair forms a bigenic conformational subdomain through contact interactions with the LCR for coordinated expression regulation.
The globin expression pattern of the PBGDAγ-YAC locus was markedly altered during development/adulthood, with the exception of the ɛ-globin gene. The proper expression pattern of the ɛ-globin gene in the PBGDAγ-YAC mice demonstrates its regulatory independence from the other genes of the locus and supports an autonomous control mechanism. However, expression of the modified fetal Aγ-globin gene in the locus at similar levels from embryonic through adult stages significantly impacted the expression of the other fetal and adult genes. Indeed, the unmodified fetal Gγ-globin gene, normally silenced in fetal definitive erythropoiesis, in contrast was induced, despite the presence of repressive signals/factors that bind in the vicinity of this gene (31, 34, 41). Interestingly, this particular expression pattern of the fetal Gγ-globin gene almost mimics human embryonic-fetal switching in the mouse. At birth/late gestation, the important repression, although not complete, of the fetal Gγ gene pointed to the existence of distinct neonatal silencing or of competitive elements in these late developmental stages and suggests the occurrence of two uncoupled negative regulatory mechanisms for the fetal genes. These negative fetal expression patterns likely result in part from an autonomous silencing mechanism, but for a gene pair (65). The sustained fetal Gγ-globin gene expression pattern is unlikely to be caused by the PBGD promoter itself (at a distance of 2.5 kb), but rather by the expression of the Aγ-globin gene. Such coordinated fetal regulation has previously been observed in Aγ(mutDR)-YAC mice with costimulation of Aγ and Gγ expression (41) and in humans, by Aγ-HPFH mutants associated with adult activation of Gγ expression (22, 45) or by an Aγ promoter deletion associated with a neonatal decrease in expression of Gγ (12). In parallel with this delayed silencing/repression of the fetal gene pair in the PBGDAγ-YAC locus, the markedly slower induction kinetics of adult β-globin gene expression led to neonatal fetal-to-adult globin switching, strengthening a reciprocal or antagonistic role with the active fetal genes. While the adult β-globin gene never fully reached the typical maximal expression of the wild-type locus, the severely reduced expression of δ-globin throughout life suggests that δ-globin transcriptional potential as well could not overcome the active fetal genes. Similar inverse relationships of low δ-globin expression (or HbA2) and high γ-globin expression (or HbF) in erythrocytes have been reported for individuals with different HPFH mutations, thalassemia, or stress erythropoiesis (62), thereby providing further evidence for a model of competition between the fetal and adult gene pairs.
The altered globin expression pattern at the fetal stage for the modified YAC was associated with differences in chromatin epigenetic status for the two fetal promoters as well as with direct DNA modification. The chromatin signature of histone modifications (H3 and H4 acetylation) at the β-globin promoter for the modified and wild-type globin YAC locus highlighted an open chromatin conformation but displayed no strict concordance with gene expression levels. These data suggest that chromatin organization at these promoters may be a major predeterminant in the transcriptional regulation of the globin locus but is not sufficient to control transcriptional levels of activation (7, 64). It is possible, however, that the globin transcription status could correlate with a particular distribution of histone modifications in other gene regions or with intergenic transcription (24, 40, 46). Since the structure of an open chromatin state within the globin promoters occurs independently of gene transcriptional activation (4, 30, 48), it raises questions about critical epigenetic switches and chromatin regulators. Independently of histone modifications, the decreased levels of CpG methylation within the proximal promoters and/or the coding sequences of the globin genes correlated with a transcriptionally active state, as proposed from MEL cell experiments (18). Interestingly, the difference in CpG demethylation levels for modified and wild-type YAC loci was more pronounced for fetal than adult genes. This observation at e12.5 may result from a more dynamic de novo methylation process than that of DNA demethylation, which would mainly affect the wild-type YAC locus (37, 61). While our analysis does not directly address whether DNA methylation status is a consequence or a regulator of the transcriptional activity of the locus, it would be consistent with the reported evidence that γ-globin promoter methylation promotes γ silencing (23, 29, 52).
The spatial conformation between the LCR and the different genes of the PBGDAγ-YAC and β-YAC loci provided novel insights into the regulatory globin switching mechanism. The strong LCR affinity for the PBGD promoter determined by 3C at e12.5 suggested that the PBGD promoter could form a functional complex. Evidence for this interaction was obtained by Aγ-globin expression at a comparable range to that for β-YAC mice at e10.5. This interaction was sustained in adulthood by similar LCR physical proximity to the PBGD promoter within the fetal subdomain. In addition, the high cross-linking frequency of the intergenic Gγ-Aγ region with the LCR in modified YAC mice parallels fetal gene expression and indicates that it could play a potential mechanistic role. At e12.5, the contrast of LCR's close proximity to the highly transcribed γ-globin genes and the lack of LCR interaction with the minimally expressed β-globin in the modified YAC compared with the wild-type YAC strongly supports a competition model between the fetal and adult genes for interaction with LCR. The LCR exclusion from the β-globin region in the modified YAC was abrogated in adulthood and likely resulted from a late gain or loss of activators/repressors at either the LCR-adult or LCR-fetal genes, leading to increased interaction of the LCR for the adult β-globin genes, to the detriment of the γ genes. An interesting finding was the equivalent frequency and strong proximity of the LCR to the δ-globin fragment in adulthood for both the wild-type and modified YACs. Considering that the modified YAC had a very low δ-globin expression level, this suggests that this region might act as an anchor point for the LCR to stabilize expression at the adult stage rather than favoring δ-globin gene expression. This highly reproducible δ-globin interaction in the vicinity of the reported binding region of the PYR/Ikaros chromatin remodeling complex (32, 42) may coincide with the Corfu deletion that results in HPFH and a decrease in β-globin expression (11, 33). Alternatively, it may implicate a region closer to the δ-globin gene (21). Noticeably, close LCR proximity to the δ-β intergenic region concordant with adult β-globin gene expression resembles the pattern observed for LCR proximity to the fetal intergenic region when the γ-globin genes are active at the fetal stage. This systematic analysis of LCR interaction within the globin locus in correlation with gene expression sheds light on the hemoglobin switch model.
Together, these data led us to hypothesize a model of “bigenic conformational subdomain” regulation. An important consequence of the LCR interaction with PBGDAγ was the LCR proximity to the Gγ gene as well as direct gene activation, which reveals a novel expression pattern. These results suggested that both γ promoters can interact cooperatively, possibly within different regions of the LCR. Evidence for LCR cointeraction with Gγ and Aγ from e12.5 in the modified YAC is provided by the following two complementary characteristics: (i) the substantial or even higher Gγ expression levels than those of PBGDAγ in development and (ii) the LCR's absence of interaction/affinity for the inactive Gγ at e12.5 in wild-type β-YAC mice infers that the Gγ gene in PBGDAγ-YAC mice would not have the potential to either alternate or compete with PBGDAγ for LCR interaction in a monogenic or one-gene-at-a-time mechanism. Our data argue that the LCR can recruit and cooperate with two genes or a gene pair in a bigenic conformation and are compatible with alternating γ- or β-globin gene transcription, as shown by Wijgerde et al. (63). Hence, this study favors a bigenic conformation mechanism for hemoglobin switching where the Gγ and Aγ genes cooperate to form the fetal γ-genic subdomain and the δ and β genes form the adult β-genic subdomain. Additional support for a gene pair simultaneously binding the LCR was also provided by similar LCR proximities to the fetal Gγ-intergenic region-PBGDAγ region in a modified YAC and to the adult δ-intergenic region-β region in the wild-type YAC. This model also corroborates previous experiments reporting LCR dual and/or concomitant promoter activation in human and murine β-globin loci (8, 28). An individual bigenic subdomain could become intertwined within the LCR complex in a niche or hub to stabilize the open chromatin for concurrent gene activation of a corresponding subdomain (Fig. 7). Interaction of the LCR with each subdomain likely consists of major anchor points within the transcriptionally active genes and/or intergenic regions that could serve to position the LCR in the most stable and sterically appropriate configuration for efficient activation by the transcriptional machinery. At the developmental switching stages, dynamic alternate transcription between the fetal and adult bigenic conformations is supported from the coexpression of the fetal and adult genes in adult red cells with the modified YAC, since no distinct cell populations expressing either γ or β were detected. Moreover, silencing of the fetal bigenic subdomain likely depends on at least two uncoupled negative regulatory mechanisms, i.e., mutual autonomous silencing and repression by competition with the adult bigenic subdomain. Our results for impaired fetal silencing at e12.5 in the modified YAC, despite the presence of all factors sufficient for adult gene activation, show a preferential fetal over adult bigenic conformation that is in accordance with the critical role played by gene order for temporal activation of the most proximal globin genes by the LCR (2, 25, 26, 55, 65). Interestingly, the comparable negative regulation pattern for native Gγ in the modified YAC in the late fetal/neonatal stage to the reported gradual fetal autonomous silencing pattern in the absence of adult gene competition (14, 54) suggests a progressive mechanism for the γ-globin genes in mutual autonomous silencing. Since adult bigenic subdomain activation in the wild-type locus is of rapid onset and kinetics in transgenic mice, we speculate that a competition mechanism in the early switching stage appears to be a major contributor to the switch from the fetal to adult globin bigenic conformation and would persist into adulthood. The existence of different anchor points in bigenic conformational subdomains of the globin locus that compete for LCR interactions could then be modulated to stimulate the fetal bigenic pair and thereby antagonize adult subdomain activation. Hence, targeting specific physical interactions within a subdomain should open novel avenues for therapy of β-thalassemia and sickle cell anemia.
FIG. 7.
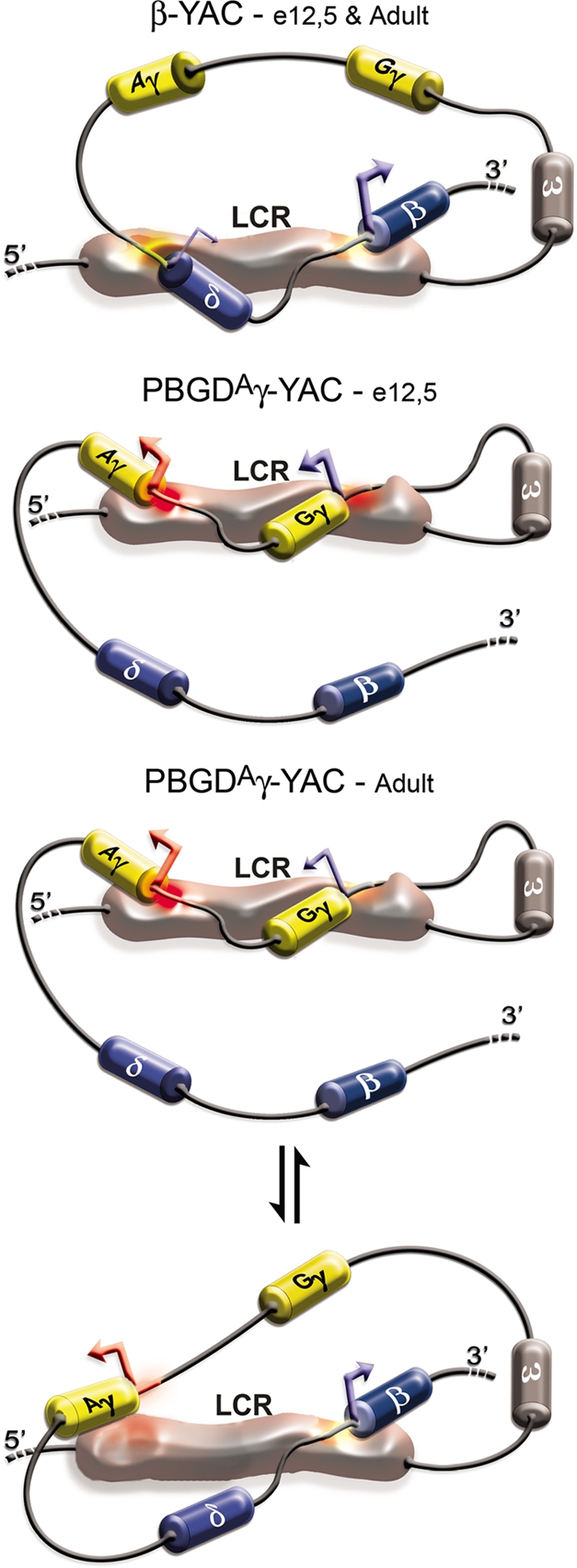
Schematic model of LCR interactions with active globin conformational subdomains at e12.5 and adult stages. In the YAC-β-globin locus (top), the LCR at the e12.5 and adult stages has affinity for the adult bigenic subdomain, which may be stabilized by anchor points at δ-globin/δ upstream region and/or the δ-β intergenic region for proper interaction with the adult δ- and β-globin genes in a niche. In PBGDAγ-YAC mice at e12.5 (middle), the LCR is recruited to the fetal bigenic subdomain. Interaction of the LCR with the PBGD promoter upstream of the Aγ-globin gene could also serve as an anchor point, favoring activation of the unmodified Gγ-globin gene and preventing activation of adult β-globin genes by steric exclusion from the niche. At the adult stage (bottom), the higher expression levels argue that the LCR shuttles preferentially between the fetal conformational subdomain and the PBGDAγ and adult β-globin genes, and occasionally to the natural adult δ- and β-globin bigenic conformational subdomain (not illustrated).
Our results show that γ-globin promoters are critical regulators of the fetal-to-adult hemoglobin switching onset and kinetics in definitive erythropoiesis. Importantly, this study provides evidence for a bigenic conformational model where a gene pair intertwined with the LCR forms an open chromatin niche for coactivation.
Supplementary Material
Acknowledgments
This study was supported by the CIHR/IRSC (M.T.); H.B. was a recipient of a CIHR-IG and of FRSQ studentships.
We thank W. Lemsaddek for help and support and Eric Milot and James Ellis for interesting and helpful discussions.
Footnotes
Published ahead of print on 29 December 2008.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Anderson, K. P., J. A. Lloyd, E. Ponce, S. C. Crable, J. C. Neumann, and J. B. Lingrel. 1993. Regulated expression of the human β-globin gene in transgenic mice requires an upstream globin or nonglobin promoter. Mol. Biol. Cell 41077-1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bauchwitz, R., and F. Costantini. 2000. Developmentally distinct effects on human ɛ-, γ- and δ-globin levels caused by the absence or altered position of the human β-globin gene in YAC transgenic mice. Hum. Mol. Genet. 9561-574. [DOI] [PubMed] [Google Scholar]
- 3.Beauchemin, H., M. J. Blouin, and M. Trudel. 2004. Differential regulatory and compensatory responses in hematopoiesis/erythropoiesis in α- and β-globin hemizygous mice. J. Biol. Chem. 27919471-19480. [DOI] [PubMed] [Google Scholar]
- 4.Bender, M. A., M. Bulger, J. Close, and M. Groudine. 2000. β-Globin gene switching and DNase I sensitivity of the endogenous β-globin locus in mice do not require the locus control region. Mol. Cell 5387-393. [DOI] [PubMed] [Google Scholar]
- 5.Blouin, M.-J., H. Beauchemin, A. Wright, M. DePaepe, M. Sorette, A.-M. Bleau, B. Nakamoto, C.-N. Ou, G. Stamatoyannopoulos, and M. Trudel. 2000. Genetic correction of sickle cell disease: insights using transgenic mouse models. Nat. Med. 6177-182. [DOI] [PubMed] [Google Scholar]
- 6.Blouin, M. J., and M. Trudel. 1997. Characterization of the hematopoietic precursors in sickle cell disease of SAD transgenic mouse model. Blood 9022-23. [Google Scholar]
- 7.Bottardi, S., A. Aumont, F. Grosveld, and E. Milot. 2003. Developmental stage-specific epigenetic control of human β-globin gene expression is potentiated in hematopoietic progenitor cells prior to their transcriptional activation. Blood 1023989-3997. [DOI] [PubMed] [Google Scholar]
- 8.Bresnick, E. H., and G. Felsenfeld. 1994. Dual promoter activation by the human β-globin locus control region. Proc. Natl. Acad. Sci. USA 911314-1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bungert, J., U. Davé, K.-C. Lim, K. H. Lieuw, J. A. Shavit, Q. Liu, and J. D. Engel. 1995. Synergistic regulation of human β-globin gene switching by locus control region elements HS3 and HS4. Genes Dev. 93083-3096. [DOI] [PubMed] [Google Scholar]
- 10.Bungert, J., K. Tanimoto, S. Patel, Q. Liu, M. Fear, and J. D. Engel. 1999. Hypersensitive site 2 specifies a unique function within the human β-globin locus control region to stimulate globin gene transcription. Mol. Cell. Biol. 193062-3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chakalova, L., C. S. Osborne, Y. F. Dai, B. Goyenechea, A. Metaxotou-Mavromati, A. Kattamis, C. Kattamis, and P. Fraser. 2005. The Corfu δβ thalassemia deletion disrupts γ-globin gene silencing and reveals post-transcriptional regulation of HbF expression. Blood 1052154-2160. [DOI] [PubMed] [Google Scholar]
- 12.Coleman, M. B., J. G. R. Adams, M. H. Steinberg, and W. P. Winter. 1994. A four base pair deletion 5′ to the AγT gene is associated not only with decreased expression of the AγT-globin gene, but also of the Gγ-globin gene in cis. Am. J. Hematol. 47307-311. [DOI] [PubMed] [Google Scholar]
- 13.Dekker, J., K. Rippe, M. Dekker, and N. Kleckner. 2002. Capturing chromosome conformation. Science 2951306-1311. [DOI] [PubMed] [Google Scholar]
- 14.Dillon, N., and F. Grosveld. 1991. Human γ-globin genes silenced independently of other genes in the β-globin locus. Nature 350252-254. [DOI] [PubMed] [Google Scholar]
- 15.Dostie, J., T. A. Richmond, R. A. Arnaout, R. R. Selzer, W. L. Lee, T. A. Honan, E. D. Rubio, A. Krumm, J. Lamb, C. Nusbaum, R. D. Green, and J. Dekker. 2006. Chromosome conformation capture carbon copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 161299-1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eleouet, J. F., and P. H. Romeo. 1993. CCACC-binding or simian-virus-40-protein-1-binding proteins cooperate with human GATA-1 to direct erythroid-specific transcription and to mediate 5′ hypersensitive site 2 sensitivity of a TATA-less promoter. Eur. J. Biochem. 212763-770. [DOI] [PubMed] [Google Scholar]
- 17.Fang, X., P. Xiang, W. Yin, G. Stamatoyannopoulos, and Q. Li. 2007. Cooperativeness of the higher chromatin structure of the β-globin locus revealed by the deletion mutations of the DNase I hypersensitive site 3 of the LCR. J. Mol. Biol. 36531-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng, Y. Q., R. Desprat, H. Fu, E. Olivier, C. M. Lin, A. Lobell, S. N. Gowda, M. I. Aladjem, and E. E. Bouhassira. 2006. DNA methylation supports intrinsic epigenetic memory in mammalian cells. PLoS Genet. 2e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forget, B. G. 1998. Molecular basis of hereditary persistence of fetal hemoglobin. Ann. N. Y. Acad. Sci. 85038-44. [DOI] [PubMed] [Google Scholar]
- 20.Fraser, S. T., J. Isern, and M. H. Baron. 2007. Maturation and enucleation of primitive erythroblasts during mouse embryogenesis is accompanied by changes in cell-surface antigen expression. Blood 109343-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gaensler, K. M., Z. Zhang, C. Lin, S. Yang, K. Hardt, and L. Flebbe-Rehwaldt. 2003. Sequences in the Aγ-delta intergenic region are not required for stage-specific regulation of the human β-globin gene locus. Proc. Natl. Acad. Sci. USA 1003374-3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilman, J. G., N. Mishima, X. J. Wen, T. A. Stoming, J. Lobel, and T. H. Huisman. 1988. Distal CCAAT box deletion in the Aγ globin gene of two black adolescents with elevated fetal Aγ globin. Nucleic Acids Res. 1610635-10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goren, A., G. Simchen, E. Fibach, P. E. Szabo, K. Tanimoto, L. Chakalova, G. P. Pfeifer, P. J. Fraser, J. D. Engel, and H. Cedar. 2006. Fine tuning of globin gene expression by DNA methylation. PLoS ONE 1e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gribnau, J., K. Diderich, S. Priuzina, R. Calzolari, and P. Fraser. 2000. Intergenic transcription and developmental remodelling of chromatin subdomains in the human β-globin locus. Mol. Cell 5377-386. [DOI] [PubMed] [Google Scholar]
- 25.Hanscombe, O., D. Whyatt, P. Fraser, N. Yannoutsos, D. Greaves, N. Dillon, and F. Grosveld. 1991. Importance of globin gene order for correct developmental expression. Genes Dev. 51387-1394. [DOI] [PubMed] [Google Scholar]
- 26.Harju, S., P. A. Navas, G. Stamatoyannopoulos, and K. R. Peterson. 2005. Genome architecture of the human β-globin locus affects developmental regulation of gene expression. Mol. Cell. Biol. 258765-8778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hodge, D., E. Coghill, J. Keys, T. Maguire, B. Hartmann, A. McDowall, M. J. Weiss, S. Grimmond, and A. Perkins. 2006. A global role for EKLF in definitive and primitive erythropoiesis. Blood 1073359-3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu, X., M. Bulger, J. N. Roach, S. K. Eszterhas, E. Olivier, E. E. Bouhassira, M. Groudine, and S. Fiering. 2004. Promoters of the murine embryonic β-like globin genes Ey and βh1 do not compete for interaction with the β-globin locus control region. Proc. Natl. Acad. Sci. USA 1001111-1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jane, S. M., D. L. Gumucio, P. A. Ney, J. M. Cunningham, and A. W. Nienhuis. 1993. Methylation-enhanced binding of Sp1 to the stage selector element of the human γ-globin gene promoter may regulate developmental specificity of expression. Mol. Cell. Biol. 133272-3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson, K. D., J. A. Grass, C. Park, H. Im, K. Choi, and E. H. Bresnick. 2003. Highly restricted localization of RNA polymerase II within a locus control region of a tissue-specific chromatin domain. Mol. Cell. Biol. 236484-6493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Katsube, T., and Y. Fukumaki. 1995. A role for the distal CCAAT box of the γ-globin gene in Hb switching. J. Biochem. 11768-76. [DOI] [PubMed] [Google Scholar]
- 32.Keys, J. R., M. R. Tallack, Y. Zhan, P. Papathanasiou, C. C. Goodnow, K. M. Gaensler, M. Crossley, J. Dekker, and A. C. Perkins. 2008. A mechanism for Ikaros regulation of human globin gene switching. Br. J. Haematol. 141398-406. [DOI] [PubMed] [Google Scholar]
- 33.Kulozik, A. E., N. Yarwood, and R. W. Jones. 1988. The Corfu δβ0 thalassemia: a small deletion acts at a distance to selectively abolish β-globin gene expression. Blood 711509. [PubMed] [Google Scholar]
- 34.Liu, L. R., Z. W. Du, H. L. Zhao, X. L. Liu, X. D. Huang, J. Shen, L. M. Ju, F. D. Fang, and J. W. Zhang. 2005. T to C substitution at −175 or −173 of the γ-globin promoter affects GATA-1 and Oct-1 binding in vitro differently but can independently reproduce the hereditary persistence of fetal hemoglobin phenotype in transgenic mice. J. Biol. Chem. 2807452-7459. [DOI] [PubMed] [Google Scholar]
- 35.Lloyd, J. A., S. S. Case, E. Ponce, and J. B. Lingrel. 1994. Positive transcriptional regulation of the human γ-globin gene. γPE is a novel nuclear factor with multiple binding sites near the gene. J. Biol. Chem. 26919385-19393. [PubMed] [Google Scholar]
- 36.Lloyd, J. A., J. M. Krakowsky, S. C. Crable, and J. B. Lingrel. 1992. Human γ- to β-globin gene switching using a mini construct in transgenic mice. Mol. Cell. Biol. 121561-1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsuo, K., J. Silke, O. Georgiev, P. Marti, N. Giovannini, and D. Rungger. 1998. An embryonic demethylation mechanism involving binding of transcription factors to replicating DNA. EMBO J. 171446-1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mignotte, V., J. F. Eleouet, N. Raich, and P.-H. Romeo. 1989. Cis- and trans-acting elements involved in the regulation of the erythroid promoter of the human porphobilinogen deaminase gene. Proc. Natl. Acad. Sci. USA 866548-6552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mignotte, V., L. Wall, E. deBoer, F. Grosveld, and P. H. Romeo. 1989. Two tissue-specific factors bind the erythroid promoter of the human porphobilinogen deaminase gene. Nucleic Acids Res. 1737-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miles, J., J. A. Mitchell, L. Chakalova, B. Goyenechea, C. S. Osborne, L. O'Neill, K. Tanimoto, J. D. Engel, and P. Fraser. 2007. Intergenic transcription, cell-cycle and the developmentally regulated epigenetic profile of the human β-globin locus. PLoS ONE 2e630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Omori, A., O. Tanabe, J. D. Engel, Y. Fukamizu, and K. Tanimoto. 2005. Adult-stage γ-globin silencing is mediated by a promoter direct repeat element. Mol. Cell. Biol. 253443-3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O'Neill, D., K. Bornschlegel, M. Flamm, M. Castle, and A. Bank. 1991. A DNA-binding factor in adult hematopoietic cells interacts with a pyrimidine-rich domain upstream from the human δ-globin gene. Proc. Natl. Acad. Sci. USA 888953-8957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patrinos, G. P., M. de Krom, E. de Boer, A. Langeveld, A. M. A. Imam, J. Strouboulis, W. de Laat, and F. G. Grosveld. 2004. Multiple interactions between regulatory regions are required to stabilize an active chromatin hub. Genes Dev. 181495-1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peterson, K. R., C. H. Clegg, Q. Li, and G. Stamatoyannopoulos. 1997. Production of transgenic mice with yeast artificial chromosomes. Trends Genet. 1361-66. [DOI] [PubMed] [Google Scholar]
- 45.Pistidda, P., L. Frogheri, L. Oggiano, L. Guiso, L. Manca, F. Dore, B. Masala, J. G. Gilman, and M. Longinotti. 1995. Fetal hemoglobin expression in compound heterozygotes for −117(G→A) Aγ HPFH and β0 39 nonsense thalassemia. Am. J. Hematol. 49264-270. [DOI] [PubMed] [Google Scholar]
- 46.Plant, K. E., S. J. Routledge, and N. J. Proudfoot. 2001. Intergenic transcription in the human β-globin gene cluster. Mol. Cell. Biol. 216507-6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Raich, N., T. Enver, B. Nakamoto, B. Josephson, T. Papayannopoulou, and G. Stamatoyannopoulos. 1990. Autonomous development control of human embryonic globin gene switching in transgenic mice. Science 2501147-1149. [DOI] [PubMed] [Google Scholar]
- 48.Reik, A., A. Telling, G. Zitnik, D. Cimbora, E. Epner, and M. Groudine. 1998. The locus control region is necessary for gene expression in the human β-globin locus but not the maintenance of an open chromatin structure in erythroid cells. Mol. Cell. Biol. 185992-6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Russell, J. E., and S. A. Liebhaber. 1998. Reversal of lethal α- and β-thalassemias in mice by expression of human embryonic globins. Blood 923057-3063. [PubMed] [Google Scholar]
- 50.Ryan, T. M., C.-W. Sun, J. Ren, and T. M. Townes. 2000. Human γ-globin gene promoter element regulates human β-globin gene development specificity. Nucleic Acids Res. 282736-2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sabatino, D. E., A. P. Cline, P. G. Gallagher, L. J. Garrett, G. Stamatoyannopoulos, B. G. Forget, and D. M. Bodine. 1998. Substitution of the human β-spectrin promoter for the human Aγ-globin promoter prevents silencing of a linked human β-globin gene in transgenic mice. Mol. Cell. Biol. 186634-6640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sengupta, P. K., D. Lavelle, and J. DeSimone. 1994. Increased binding of Sp1 to the γ-globin gene promoter upon site-specific cytosine methylation. Am. J. Hematol. 46169-172. [DOI] [PubMed] [Google Scholar]
- 53.Shih, D. M., R. J. Wall, and S. G. Shapiro. 1993. A 5′ control region of the human ɛ-globin gene is sufficient for embryonic specificity in transgenic mice. J. Biol. Chem. 26830066-30071. [PubMed] [Google Scholar]
- 54.Stamatoyannopoulos, G., B. Josephson, J.-W. Zhang, and Q. Li. 1993. Developmental regulation of human γ-globin genes in transgenic mice. Mol. Cell. Biol. 137636-7644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tanimoto, K., Q. Liu, J. Bungert, and J. D. Engel. 1999. Effects of altered gene order or orientation of the locus control region on human β-globin gene expression in mice. Nature 398344-348. [DOI] [PubMed] [Google Scholar]
- 56.Thivierge, C., A. Kurbegovic, M. Couillard, R. Guillaume, O. Cote, and M. Trudel. 2006. Overexpression of PKD1 causes polycystic kidney disease. Mol. Cell. Biol. 261538-1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tolhuis, B., R. J. Palstra, E. Splinter, F. Grosveld, and W. de Latt. 2002. Looping and interaction between hypersensitive sites in the active β-globin locus. Mol. Cell 101453-1465. [DOI] [PubMed] [Google Scholar]
- 58.Trudel, M., J. Magram, L. Bruckner, and F. Costantini. 1987. Upstream Gγ-globin and downstream β-globin gene domain in human erythroid cells. Mol. Cell. Biol. 74024-4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trudel, M., J. Magram, K. Chada, R. Wilson, and F. Costantini. 1987. Expression of normal, mutant and hybrid human globin genes in transgenic mice, p. 305-321. In G. Stamatoyannopoulos and A. Nienhuis (ed.), The regulation of hemoglobin switch. Alan R. Liss, Inc., Baltimore, MD. [PubMed]
- 60.Trudel, M., N. Saadane, M.-C. Garel, J. Bardakdjian-Michau, Y. Blouquit, J.-L. Guerquin-Kern, P. Rouyer-Fessard, D. Vidaud, A. Pachnis, P.-H. Romeo, Y. Beuzard, and F. Costantini. 1991. Towards a transgenic mouse model of sickle cell disease: hemoglobin SAD. EMBO J. 103157-3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Turek-Plewa, J., and P. P. Jagodzinski. 2005. The role of mammalian DNA methyltransferases in the regulation of gene expression. Cell. Mol. Biol. Lett. 10631-647. [PubMed] [Google Scholar]
- 62.Weatherhall, D. J., and J. B. Clegg (ed.). 2001. The thalassaemia syndromes. Blackwell Science, Oxford, United Kingdom.
- 63.Wijgerde, M., F. Grosveld, and P. Fraser. 1995. Transcription complex stability and chromatin dynamics in vivo. Nature 377209-213. [DOI] [PubMed] [Google Scholar]
- 64.Yin, W., G. Barkess, X. Fang, P. Xiang, H. Cao, and G. Stamatoyannopoulos. 2007. Histone acetylation at the human β-globin locus changes with developmental age. Blood 1104101-4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yu, M., H. Han, P. Xiang, Q. Li, and G. Stamatoyannopoulos. 2006. Autonomous silencing as well as competition controls γ-globin gene expression during development. Mol. Cell. Biol. 264775-4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.