

Abstract
Circadian clocks keep time via gene expression feedback loops that are controlled by time-of-day-specific changes in the synthesis, activity, and degradation of transcription factors. Within the Drosophila melanogaster circadian clock, DOUBLETIME (DBT) kinase is necessary for the phosphorylation of PERIOD (PER), a transcriptional repressor, and CLOCK (CLK), a transcriptional activator, as CLK-dependent transcription is being repressed. PER- and DBT-containing protein complexes feed back to repress CLK-dependent transcription, but how DBT promotes PER and CLK phosphorylation and how PER and CLK phosphorylation contributes to transcriptional repression have not been defined. Here, we show that DBT catalytic activity is not required for CLK phosphorylation or transcriptional repression and that PER phosphorylation is dispensable for repressing CLK-dependent transcription. These results support a model in which DBT plays a novel noncatalytic role in recruiting additional kinases that phosphorylate CLK, thereby repressing transcription. A similar mechanism likely operates in mammals, given the conserved activities of PER, DBT, and CLK orthologs.
A vast array of animal, plant, and microbial species display daily rhythms of metabolism, physiology, and behavior. These rhythms are controlled by endogenous circadian clocks that are set by ∼24-h environmental cycles but persist in the absence of environmental cues. Studies with several phylogenetically diverse model systems have revealed that the circadian timekeeping mechanism is composed of conserved transcriptional feedback loops (reviewed in reference 4). In Drosophila melanogaster, these feedback loops are initiated by two basic-helix-loop-helix-PER-ARNT-SIM transcription factors, CLOCK (CLK) and CYCLE (CYC). CLK-CYC heterodimers bind E-box elements during the late day and early evening to activate period (per) and timeless (tim) transcription (2, 8, 12, 35). The PERIOD (PER) protein then gradually accumulates during the night as a heterodimer with TIMELESS (TIM) to inhibit CLK-CYC activity (8). The gradual accumulation of PER, an important determinant of circadian period, is controlled by DOUBLETIME (DBT)-dependent phosphorylation (19, 30), which targets PER for degradation in the 26S proteasome (6, 11, 21, 27), and TIM binding, which protects PER from DBT-dependent degradation (11, 19, 21, 30, 31, 34). Once TIM undergoes light- or clock-induced degradation around dawn, PER is degraded and another cycle of CLK-CYC-mediated transcription is initiated.
Although transcriptional repression by PER (or PER-TIM) is essential for feedback loop function, how PER functions to inhibit CLK-CYC-dependent transcription is not well understood. During the circadian cycle, the accumulation of phosphorylated PER in the nucleus is coincident with hyperphosphorylation of CLK, release of CLK-CYC from E-boxes, and transcriptional repression (41). Given that PER remains bound to DBT when it enters the nucleus and binds CLK (20, 41), it is possible that DBT is also responsible for CLK phosphorylation and transcriptional repression. In support of this possibility, a PER mutant unable to bind DBT (perΔ) eliminates both hyperphosphorylation of CLK and repression of CLK-CYC-dependent transcription (17, 29). Nevertheless, PERΔ does enter the nucleus and bind CLK, which suggests that PER binding to CLK is not sufficient to remove CLK-CYC from E-boxes and repress transcription (17). These results suggest that DBT is necessary for CLK hyperphosphorylation and transcriptional repression by PER-containing complexes.
If DBT is required for CLK hyperphosphorylation, then CLK should also be hypophosphorylated in the absence of DBT. Although dbt null mutants are not viable as adults (19, 30), a dbt mutation that severely compromises kinase catalytic activity, dbtar, produces viable adults that lack circadian clock function (33). Surprisingly, CLK is hyperphosphorylated rather than hypophosphorylated in dbtar flies (41). To reconcile the different phosphorylation states of CLK in perΔ and dbtar flies, we propose that DBT plays a noncatalytic role in recruiting other kinases into PER repression complexes. Kinases recruited into this complex by DBT phosphorylate CLK, thereby releasing CLK-CYC from E-boxes and repressing transcription.
Here, we demonstrate that entry of DBT into the PER complex is required, but DBT catalytic activity is dispensable for CLK hyperphosphorylation and transcriptional repression. CLK is always hyperphosphorylated when CLK-CYC transcription is repressed, but surprisingly, PER hyperphosphorylation is not required for CLK hyperphosphorylation or repression of CLK-CYC-mediated transcription. These results strongly support a novel noncatalytic role for DBT in recruiting other kinases that phosphorylate CLK and promote transcriptional repression, indicating that PER phosphorylation by DBT is not a prerequisite for transcriptional repression.
MATERIALS AND METHODS
Fly stocks.
The w1118 strain served as a wild-type (WT) control for clock function. The UAS-dbtK/R (dbtK/R is the dbt mutant lacking kinase catalytic activity due to a K38R substitution), perΔ (per01, w1118;; perΔ-HAHIS/+), and tim-Gal4 transgenic strains were described previously (10, 17, 26). The UAS-dbtK/R line UAS-KR22, the perΔ line perΔF21, and the tim-Gal4 line 62 were used. The dbtar mutant employed in this study has the genotype dbtar/dbtP, where dbtP is a homozygous lethal P element insert that is apparently dbt null (19, 30). We used dbtar/dbtP flies because they are healthier than homozygous dbtar flies in our hands and they have the lowest level of DBT catalytic activity as viable adults. dbtar/dbtP flies were produced by crossing dbtar/TM3 flies to dbtP/TM3 flies. The perΔ genotype employed here (per01, w1118;; perΔ/dbtP) was generated to keep the genetic background comparable to that of the dbtar genotype (dbtar/dbtP) by crossing per01, w1118;; dbtp/TM3 flies with per01, w1118;; perΔ/MKRS flies. The per01, w1118;; perΔ, dbtar/dbtP double mutants, referred to as perΔ, dbtar in the text, were produced by recombining the perΔ transgene onto a third chromosome containing dbtar. Recombinant third chromosomes bearing the perΔ insert were identified as dbtar by their loss of a FokI restriction site caused by the dbtar mutation and were verified by DNA sequencing. The per01, w1118;; perΔ, dbtar/TM6B flies were crossed to per01, w1118;; dbtP/TM3 flies to generate per01, w1118;; perΔ, dbtar/dbtP double-mutant flies. UAS-dbtK/R/tim-Gal4; dbtar/dbtP flies, referred to as dbtK/R; dbtar flies in the text, were generated by crossing dbtK/R/CyO; dbtar/dbtP flies with tim-Gal4/CyO; dbtar/dbtP flies. The dbtK/R/CyO; dbtar/dbtP and tim-Gal4/CyO; dbtar/dbtP flies were used as dbtar no-driver and dbtar no-responder controls, respectively.
Western blotting.
For preparing fly head extract, flies were entrained in a 12-h light/12-h dark (LD) incubator for at least 3 days and collected at the indicated time points. Isolated frozen fly heads were homogenized in radioimmunoprecipitation assay (RIPA) buffer (20 mM Tris at pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.05 mM EGTA, 10% glycerol, 1% Triton X-100, 0.4% sodium deoxycholate, 0.1% SDS [sodium dodecyl sulfate]) containing 0.5 mM PMSF (phenylmethylsulfonyl fluoride), 10 μg/ml aprotinin, 10 μg/ml leupeptin, 2 μg/ml pepstatin A, 1 mM Na3VO4, and 1 mM NaF (hereafter referred to as the protease inhibitor mixture). This homogenate was sonicated 5 to 10 times for 10 s each time, using a Misonix XL2000 model sonicator at a setting of 3 and then centrifuged at 20,000 × g for 10 min. The supernatant was collected as RIPA S extract, and protein concentration was determined by the Bradford assay. Equal amounts of RIPA S extract were run, transferred, and probed with antibodies as follows: guinea pig anti-CLK (GP-47), 1:2,000 (15); guinea pig anti-PER (GP-73), 1:3,000 (37); rat anti-TIM (TR3), 1:2,000 (37); rabbit anti-DBT, 1:4,000 (26); and mouse anti-beta-actin (Abcom), 1:2,000. Horseradish peroxidase-conjugated secondary antibodies (Sigma) against guinea pig, rat, rabbit, and mouse were diluted 1:1,000. Immunoblots were visualized using ECL plus (Amersham) reagent.
Quantitative real-time RT-PCR.
Total RNA was isolated from frozen fly heads, using Trizol (Invitrogen), and treated with a Turbo DNase DNA-free kit (Ambion) to eliminate genomic DNA contamination. DNA-free total RNA (1.0 μg) was reverse transcribed using oligo(dT)12-28 primers (Invitrogen) and Superscript II (Invitrogen). The reverse transcription (RT) product was amplified and analyzed with an Applied Biosystems model 7500 Fast real-time PCR system using Power SYBR green PCR Master Mix (Applied Biosystems) and gene-specific primers. All primer pairs were designed to span an exon-intron boundary to prevent genomic DNA amplification. The gene-specific primer pairs used for rp49 were 5′-TACAGGCCCAAGATCGTGAA-3′ and 5′-GCACTCTGTTGTCGATACCC-3′; for vri were 5′-ATGAACAACGTCCGGCTATC-3′ and 5′-CTGCGGACTTATGGATCCTC-3′; for per were 5′-TGATGGGCGACTACAACTCC-3′ and 5′-GTCGCTATTCCCATTGCTGT-3′; and for tim were 5′-GGTGGCATCTGTGTACGAAA-3′ and 5′-GATCTCGGTTCGCTCAAGTC-3′. For each sample, RNA quantity was determined by the standard curve for each gene that was analyzed. The RNA quantity of vri, per, or tim was normalized to that of rp49. For each data series, the rp49-normalized values were further normalized to levels in dbtP/+ (Fig. 1) and w1118 (see Fig. 4) at zeitgeber time 2 (ZT 2) (during LD cycles, lights on is referred to as ZT 0 and lights off is referred to as ZT 12) to yield relative mRNA levels.
FIG. 1.

Comparison of CLK phosphorylation and CLK-CYC transcription levels in dbtar flies with those in perΔ flies. (A) Western blot of CLK, PER, DBT, and β-actin levels in dbtar/dbtP (dbtar), +/dbtP (WT control), per01;; perΔ/dbtP (perΔ), and ClkJrk flies collected at ZT 2 or ZT 14. DBT, hypophosphorylated (Hypo) and hyperphosphorylated (Hyper) CLK and PER, and β-actin bands are marked. β-Actin was used as a loading control. (B and C) Quantitative real-time RT-PCR was used to measure tim (B) and vri (C) mRNA levels in heads from WT control, dbtar, and perΔ flies as designated above. The relative mRNA levels were quantified as described in Materials and Methods. Data were plotted as the means ± standard error of the means (n = 3).
FIG. 4.

DBT is in a complex with PER, TIM, and CLK in dbtar but not perΔ flies. EB3-S extracts from w1118 (WT), dbtar/dbtP (dbtar), and per01;; perΔ/+ (perΔ) fly heads collected at the indicated time points were subjected to coimmunoprecipitations (Co-IP) using a rabbit DBT antiserum (anti-DBT) or normal rabbit serum (RS). Western blots containing immunoprecipitates and input extracts (Input) were probed with CLK, PER, and TIM antibodies, respectively. DBT and hypophosphorylated (Hypo) and hyperphosphorylated (Hyper) CLK and PER bands are marked. Ctrl is a constant nonspecific band detected by PER antiserum as a loading control.
IP.
For immunoprecipitation (IP) assays, EB3-S extract was prepared by homogenizing frozen fly heads in EB3 buffer (10 mM HEPES at pH 7.5, 5 mM Tris at pH 7.5, 50 mM KCl, 1 mM EDTA, 0.05 mM EGTA, 10% glycerol, and 0.1% Triton X-100) containing protease inhibitor mixture, sonicated as described for preparing RIPA S extracts, and centrifuged at 20,000 × g for 10 min, and the supernatant was collected as EB3-S extract. To 1 mg of the EB3-S extract, 2.5 μl of DBT antiserum was added, and the mixture was incubated at 4°C overnight. Immune complexes were collected by incubating 35 μl of 50% protein A-Sepharose beads (Amersham) at 4°C for 2 h and then washing the beads three times with EB3 buffer. The immunoprecipitates were eluted by boiling in 15 μl of 2× loading buffer and then run in parallel with 50 μg of EB3-S extracts as input. Immunoblots were processed as described previously for Western blotting.
ChIP.
Chromatin IP (ChIP) assays were performed as described previously (41), with modifications. Briefly, frozen fly heads were homogenized and cross-linked in ∼5 volumes of HX buffer (50 mM HEPES at pH 8.0, 140 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.4% Igpel CA-630, 0.2% Triton X-100, 1% formaldehyde, 1 mM PMSF, 1 mM Na3VO4, and 1 mM NaF) for 10 min at 25°C. Glycine was added to a final concentration of 0.125 M to stop the cross-linking reaction. The homogenates were then filtered with 100-μm nylon mesh. The nuclei were harvested by centrifugation at 800 × g for 5 min and washed three times with XN wash buffer (20 mM Tris at pH 7.5, 150 mM NaCl, 1 mM EDTA, and 0.5 mM EGTA) containing protease inhibitor mixture (see “Western blotting” above). The cross-linked nuclei were suspended in RIPA buffer containing protease inhibitor mixture and sonicated 15 times for 10 s each time using a Misonix model XL2000 sonicator at a setting of 3. After centrifugation at 25,000 × g for 10 min, the supernatant was collected as XN extract and quantified by the Bradford assay. An aliquot of 50 μg of XN extract was stored at −80°C as input. For IP, 500 μg of XN extract was diluted in 4 volumes of IP buffer (20 mM Tris at pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100, and 0.01% SDS) containing protease inhibitor mixture. The diluted XN extract was precleared as described previously (41) and then immunoreacted overnight by the addition of 3 μl of anti-PER (GP-73) and salmon sperm DNA to a final concentration of 0.1 μg/μl. Immune complexes were recovered, and DNA was purified as described previously (41).
Quantitative real-time PCR was used to measure E-box-containing DNA fragments. The suspended DNA samples were further diluted at 1:40 for ChIP DNA and at 1:600 for input DNA for real-time PCR. The circadian regulatory sequence (CRS) containing a fragment upstream of per and an E-box-containing fragment upstream of cry were amplified and quantified as described above in “Quantitative real-time RT-PCR” using the primer pair 5′-CCAGTGCCAGTGCGAGTTC-3′ and 5′-GATGCCAAGTGTCAATCCAAGC-3′ for the CRS E-box; and the primer pair 5′-CCCCTTATAATCCTGGTTTTGG-3′ and 5′-TGCCTAGACAACGACAACGAC-3′ for the cry E-box. The cry E-box is not bound by CLK-CYC (data not shown) and was used as a measure of background PER binding. The percentage of total DNA was calculated by the following formula: the percentage of total DNA = (CRS quantity from ChIP × 40)/(CRS quantity from input × 10 × 600) × 100% − (cry E-box quantity from ChIP × 40)/(cry E-box quantity from input ×10 × 600) × 100%.
RESULTS
PER-DBT binding, but not DBT catalytic activity, is required for CLK hyperphosphorylation and transcriptional repression.
We first confirmed PER and CLK phosphorylation levels in perΔ and dbtar flies (see Materials and Methods for complete genotypes). The small deletion within the PERΔ protein prohibits DBT binding and eliminates PER and CLK phosphorylation and transcriptional repression, even though PERΔ is able to bind CLK and TIM (16, 17). In contrast, catalytically compromised DBTar protein shows high levels of PER and CLK phosphorylation, though its effect on transcription was not tested (33, 41). As expected, phosphorylation of PER and CLK is strongly skewed toward hyperphosphorylated forms in dbtar flies and is almost entirely hypophosphorylated in perΔ flies (Fig. 1A). Moreover, although DBTar is catalytically compromised (18, 33), it accumulates to levels that are similar to those of WT DBT (Fig. 1A), as previously seen in tim01; dbtar flies (7). Since PER and CLK are hypophosphorylated when CLK-CYC-mediated transcription is activated and are hyperphosphorylated when CLK-CYC-mediated transcription is repressed (41), we determined the tim and vri mRNA levels in perΔ and dbtar flies. Both tim and vri mRNA levels are high in perΔ flies (Fig. 1B and C), consistent with increased CLK-CYC-mediated transcription of per when PER and CLK are hypophosphorylated in WT and perΔ flies (17, 41). In contrast, tim and vri mRNAs were at low levels in dbtar flies (Fig. 1B and C), thereby strengthening the link between PER and CLK hyperphosphorylation and repression of CLK-CYC-mediated transcription. A similar decrease in tim, vri, and per mRNA levels was detected in tim01; dbtar flies (7) and was likely due to severely reduced DBT catalytic activity.
In WT flies, PER-DBT complexes bind to CLK-CYC, promote CLK hyperphosphorylation, and release CLK-CYC from E-boxes (23, 41). In perΔ mutants, PERΔ forms a complex with CLK-CYC but it neither promotes CLK hyperphosphorylation nor removes CLK-CYC from E-boxes (17). Consequently, PERΔ-CLK-CYC complexes are expected to remain associated with E-boxes. ChIP assays using PER antiserum show that PER is always associated with the per CRS E-box (12, 13) in perΔ flies, but not in per+-rescued control flies (Fig. 2). ChIP experiments using antihemagglutinin (anti-HA) antibody to probe PER-HA interactions with the per CRS E-box in perΔ and per+-rescued control flies produced similar results (data not shown). Thus, even though PER is bound to CLK-CYC in perΔ flies, CLK is not phosphorylated and CLK-CYC is not released from E-boxes if DBT cannot enter the PER-CLK-CYC complex. Although PER levels in perΔ flies are much higher than those in WT flies, PER levels in WT flies are already saturated with respect to CLK (3), which argues that PER interactions with the CRS E-box are not due simply to high PER levels in perΔ flies. The inability of PER to immunoprecipitate E-box complexes in WT flies suggests that PER binding rapidly removes CLK-CYC from the E-box.
FIG. 2.
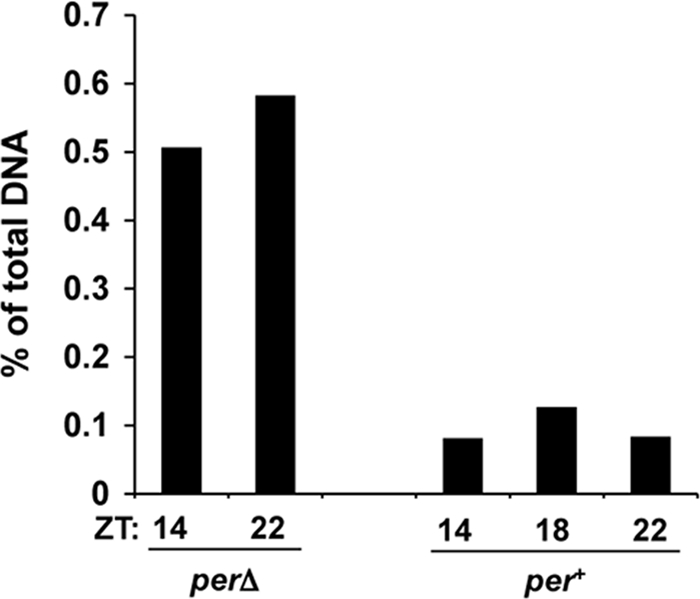
DBT is required for PER to remove CLK-CYC from E-boxes. ChIP assays were carried out to probe PER binding to the per CRS E-box in perΔ and per+ control flies at the indicated times, using PER (GP-73) antiserum. The percentage of total CRS E-box DNA bound by PER was calculated as described in Materials and Methods.
DBTar binds PER to promote CLK phosphorylation.
We previously showed that CLK hyperphosphorylation is PER dependent (41). If DBT binds PER to recruit other kinases that mediate CLK hyperphosphorylation, DBTar-dependent hyperphosphorylation of CLK should also require PER-DBTar binding. Alternatively, mutant DBTar protein may bypass the requirement for PER-DBTar binding to mediate CLK hyperphosphorylation. To determine whether hyperphosphorylation of CLK requires PER-DBTar binding, we generated perΔ, dbtar double mutants (described in Materials and Methods) and examined PER and CLK phosphorylation. PER and CLK remain hypophosphorylated in perΔ, dbtar flies, which phenocopies the perΔ rather than the dbtar mutant (Fig. 3). These results indicate that DBTar binds PER to bring about PER and CLK hyperphosphorylation and thus support a noncatalytic role for DBT.
FIG. 3.
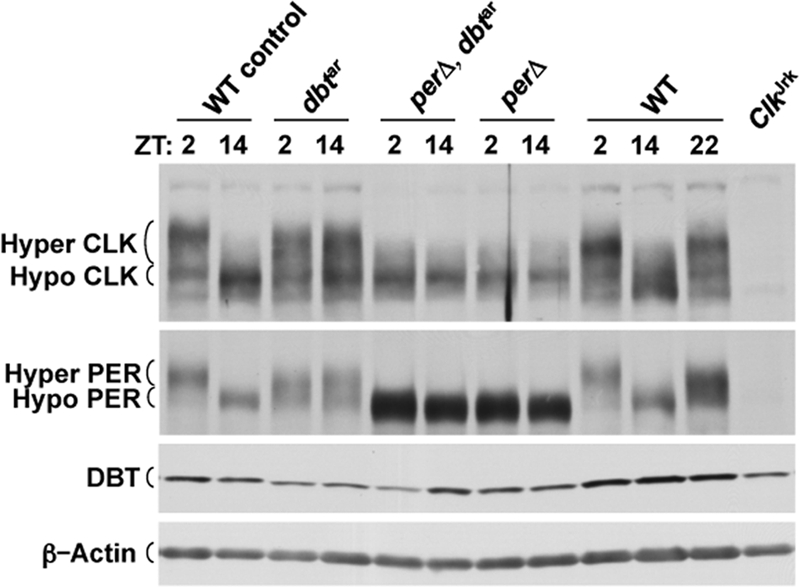
DBTar promotes CLK hyperphosphorylation (Hyper) via interactions with PER complexes. Western blot shows CLK, PER, DBT, and β-actin levels in the heads of +/dbtP (WT control), dbtar/dbtP (dbtar), per01;; perΔ, dbtar/dbtP (perΔ, dbtar), per01;; perΔ/dbtP (perΔ), w1118 (WT), and ClkJrk flies collected at ZT 2 or ZT 14. DBT, hypophosphorylated (Hypo) and hyperphosphorylated CLK and PER, and β-actin bands are marked. β-Actin was used as a loading control.
The dependence of PER and CLK hyperphosphorylation on PER-DBTar binding implies that DBTar is present in a complex with PER and CLK-CYC. To determine if this is the case, DBT antiserum was used to immunoprecipitate head extracts from dbtar, WT, and perΔ flies collected at different times of day. DBT coimmunoprecipitated PER and CLK only when PER and CLK were hyperphosphorylated, as determined at ZT 2 and ZT 14 in dbtar flies and at ZT 22 and ZT 2 in WT flies (Fig. 4). However, DBT did not coimmunoprecipitate PER and CLK when PER and CLK were hypophosphorylated at ZT 2 and ZT 14 in perΔ flies and at ZT 14 in WT flies (Fig. 4). These data demonstrate that DBTar is indeed in a complex with PER and CLK in dbtar flies and confirm previous results showing that PER-DBT-CLK-CYC complexes form in phase with PER and CLK hyperphosphorylation and transcriptional repression in WT flies but lack DBT in perΔ flies (17, 41). Taken together with the PER-DBTar binding required for DBTar-dependent PER and CLK hyperphosphorylation (Fig. 3), these results suggest that DBT recruits or DBT and PER jointly recruit other kinases that phosphorylate PER and CLK to repress transcription.
Blocking DBT catalytic activity in clock cells prevents PER phosphorylation but not CLK hyperphosphorylation and CLK-CYC transcriptional repression.
Although results from dbtar flies strongly support a noncatalytic role for DBT in PER and CLK phosphorylation and transcriptional repression, it is possible that DBTar retains a modified kinase activity that is still capable of phosphorylating PER and CLK with altered specificity (33), consistent with recent results showing low levels of casein phosphorylation by DBTar (18). Phosphorylation of different target sites can alter protein activity, localization, and/or degradation. For instance, phosphorylation of Ser 659 on mPER2 promotes mPER2 stabilization, whereas phosphorylation of other sites by CK1δ/ɛ promotes mPER2 degradation (40). Likewise, DBT phosphorylates S47 on PER to promote degradation (6), whereas other PER phosphorylation sites are predicted to control subcellular localization (1, 25, 38). To determine whether PER and CLK phosphorylation in dbtar flies is due to other kinases or to altered DBTar target site specificity, we employed a dominant negative form of DBT (DBTK38R) that lacks kinase catalytic activity due to a K38R substitution (26). Since dbt null mutants die during development (19, 30), DBTK38R (hereafter referred to as DBTK/R) is expressed in a tissue-specific manner using the Gal4/UAS system, thereby avoiding tissues that give rise to developmental lethality. Overexpressing DBTK/R in vitro and in oscillator cells from WT flies antagonizes PER hyperphosphorylation, thus demonstrating that DBT mediates the vast majority of PER phosphorylation (26). PER is hypophosphorylated in flies that overexpress DBTK/R in oscillator cells (26), which contrasts with PER hyperphosphorylation in dbtar flies (Fig. 1 and 3). This difference in PER phosphorylation implies that DBTar retains reduced and/or off-target catalytic activity that phosphorylates PER.
To produce the greatest reduction in DBT catalytic activity, tim-Gal4 and UAS-dbtK/R transgenes were used to overexpress DBTK/R in oscillator cells from dbtar flies (referred to as dbtK/R; dbtar flies). In dbtK/R; dbtar flies, the high levels of DBTK/R compared to that of DBTar suggest that DBTK/R will effectively block DBTar catalytic activity (Fig. 5A). Indeed, PER levels are constitutively high and are almost completely hypophosphorylated in dbtK/R; dbtar flies (Fig. 5A, compare lanes 3 and 4 to lanes 1 and 2, 5 and 6, or 11 and 12). In contrast, CLK is constitutively hyperphosphorylated in dbtK/R; dbtar flies (Fig. 5A, compare lanes 3 and 4 to lanes 1 and 2, 5 and 6, or 11 and 12), which further supports our argument that DBT catalytic activity is not necessary for CLK hyperphosphorylation. Given that PER hypophosphorylation is associated with CLK-CYC transcriptional activation and that CLK hyperphosphorylation is associated with CLK-CYC transcriptional repression, we wanted to determine the level of CLK-CYC-dependent transcription in dbtK/R; dbtar flies. The levels of per and vri mRNA are near that of the WT trough in dbtK/R; dbtar fly heads (Fig. 5B and C), thus reinforcing the link between CLK hyperphosphorylation and repression of CLK-CYC-dependent transcription and severing the association between PER hypophosphorylation and activation of CLK-CYC-dependent transcription.
FIG. 5.

Blocking DBT catalytic activity in oscillator cells prevents PER phosphorylation but not CLK hyperphosphorylation and CLK-CYC transcriptional repression. (A) Western blot of CLK, PER, DBT, and β-actin levels in w1118 (WT), UAS-dbtK/R/tim-Gal4; dbtar/dbtP (dbtK/R; dbtar), UAS-dbtK/R/+; dbtar/dbtP (dbtar no driver), tim-Gal4/+; dbtar/dbtP (dbtar no responder), and ClkJrk flies collected at the indicated times. DBT, DBTK/R, hypophosphorylated (Hypo) and hyperphosphorylated (Hyper) CLK and PER, and β-actin bands are marked. β-Actin was used as a loading control. (B and C) Quantitative real-time RT-PCR was used to measure per (B) and vri (C) mRNA levels in heads from WT, dbtK/R; dbtar, dbtar no driver, and dbtar no responder flies as designated above. The relative mRNA levels were quantified as described in Materials and Methods. The data were plotted as the means ± standard error of the means (n = 3).
DISCUSSION
Nonphosphorylated PER accumulates to high levels in homozygous dbtP null mutant larvae and dbtK/R adults due to the lack of DBT catalytic activity (26, 30). Despite the high levels of PER in dbtP larvae, CLK-CYC-dependent transcription is not repressed by monomeric PER in LD cycles (30), consistent with results for perΔ flies and the requirement that DBT recruit additional kinases that phosphorylate CLK. The high levels of nonphosphorylated PER that accumulate in dbtK/R; dbtar flies indicate that DBT catalytic activity has been eliminated. Nevertheless, CLK is hyperphosphorylated in dbtK/R; dbtar flies, thus demonstrating that DBT catalytic activity is dispensable for CLK hyperphosphorylation. Conversely, DBT catalytic activity is required for PER hyperphosphorylation. The presence of phosphorylated PER in dbtar flies implies that DBTar retains catalytic activity but that this activity is defective since it leads only to the partial degradation of PER (Fig. 1 and 3 to 5). The remaining phosphorylated PER apparently forms a PER repression complex sufficient to sustain transcriptional repression.
The CLK phosphorylation state parallels transcriptional activity in the WT, per mutant, and dbt mutant genotypes; CLK is hyperphosphorylated when CLK-CYC-dependent transcription is repressed and is hypophosphorylated when CLK-CYC-dependent transcription is activated (41) (Fig. 1 and 5). In contrast, results with dbtK/R; dbtar flies demonstrate that PER hyperphosphorylation is not a prerequisite for transcriptional repression and strengthen the argument that DBT plays a noncatalytic role in PER complexes to mediate transcriptional repression. The loss of CLK-CYC activity in dbtK/R; dbtar flies suggests that CLK is phosphorylated at the same sites that repress transcriptional activity in WT flies, thus implying that the same kinases mediate CLK phosphorylation in the WT and in the dbtK/R; dbtar mutant strains. Furthermore, the 270-amino-acid CLK-CYC interaction domain of PER, which contains the DBT binding region (17, 29), is sufficient to inhibit CLK-CYC transcription in cell culture (5), consistent with a requirement for DBT, but not PER phosphorylation, in repressing CLK-CYC transcription. Likewise, eliminating DBT expression via RNA interference in cell culture gives rise to high levels of nonphosphorylated PER that are unable to repress CLK-CYC-dependent transcription (28), further supporting a requirement for DBT in repressing CLK-CYC-mediated transcription.
Our results demonstrate that DBT catalytic activity is not necessary for phosphorylating CLK or repressing CLK-CYC-mediated transcription, but how DBT carries out these activities is not known. We speculate that DBT forms a physical bridge to recruit other kinases that phosphorylate CLK and repress CLK-CYC-dependent transcription (Fig. 6). During the early evening, PER destabilization by DBT phosphorylation and stabilization by TIM binding result in the gradual accumulation of PER (11, 19, 21, 30, 31, 34). PER phosphorylation by CK2 then promotes the nuclear localization of PER-DBT and PER-DBT-TIM complexes (1, 25, 38), where continued phosphorylation by DBT produces hyperphosphorylated PER (6, 30). As PER-DBT-TIM complexes accumulate in the nucleus late at night, they bind CLK-CYC via interactions between PER and CLK (3, 22, 23). Once the PER-DBT-TIM-CLK-CYC complex is formed, DBT recruits other kinases that hyperphosphorylate CLK, thereby releasing CLK-CYC from E-boxes and repressing transcription (39, 41). Although DBT may recruit other kinases via direct binding (Fig. 6), it is also possible that entry of DBT into the PER repression complex could promote binding of other kinases to PER and/or CLK, which then directly phosphorylate CLK. CLK phosphorylation coincides with transcriptional repression, which implies that CLK phosphorylation represses transcription. However, experimental support for this possibility awaits identification of CLK phosphorylation sites and/or the kinases that phosphorylate CLK. After dawn, light or clock-dependent degradation of TIM permits SLIMB binding of phosphorylated PER S47 (6), which targets PER for degradation in the 26S proteasome (11, 21, 27). Phosphorylation of CLK by DBT also triggers CLK degradation (16, 41), and by mid day, hypophosphorylated CLK derived from new synthesis and/or dephosphorylation binds E-boxes to initiate the next cycle of transcription.
FIG. 6.

Model of noncatalytic DBT function during the circadian cycle (see text for description). Transcriptional regulatory events occurring during late night (left panel), early morning (middle panel), and late day (right panel) are shown. PER, red shape; TIM, orange oval; DBT, plum shape; CLK-CYC, green ovals; DBT-bridged CLK kinases, Xs in purple oval; DBT phosphorylation, plum-colored oval with a P; DBT-bridged CLK kinase phosphorylation, purple oval with a P; PER phosphorylation by other kinases, yellow oval with a P; E-box regulatory element, blue rectangle; transcriptional repression, arrow with red X; transcriptional activation, arrow with black waveform; PER degradation, red arrow to red stippled PER; TIM degradation, orange arrow to orange stippled TIM; CLK degradation, green arrow to green stippled CLK; inhibition of CLK-CYC binding to E-boxes, black arrow.
Loss of DBT catalytic and noncatalytic activities together, or DBT catalytic activity alone, stops the oscillator at different circadian phases. The loss of PER-DBT binding in flies expressing PERΔ disables both DBT catalytic and bridging functions, thus stopping the feedback loop during the early evening when CLK is hypophosphorylated and CLK-CYC-mediated transcription is high. In contrast, loss of DBT catalytic function alone in dbtar and dbtK/R; dbtar flies stops the feedback loop during the early morning, when CLK is hyperphosphorylated and CLK-CYC-mediated transcription is low. The different phases of oscillators stopped by the loss of DBT catalytic or catalytic plus noncatalytic functions suggest that DBT catalytic activity is required for progression from transcriptional repression to activation.
The DBT bridge model may be applicable to eukaryotic clocks in general. Like CLK in Drosophila, the feedback loop activators WHITE COLLAR 1 and WHITE COLLAR 2 (WCC) in Neurospora and CLOCK (as a heterodimer with BMAL1) in mammals are transcriptionally active when they are hypophosphorylated and repressed when they are hyperphosphorylated (24, 32, 36). CK1 and CK2 mediate FREQUENCY (FRQ)-dependent hyperphosphorylation of the WCC in Neurospora Crassa to repress transcription (14), but whether these processes are mediated by catalytic activity or by bridging is not known. In mammals, CRYPTOCHROME-PERIOD (CRY-PER) complexes contain CK1ɛ and CK1δ and promote CLOCK hyperphosphorylation and transcriptional repression (24). As in Drosophila, expression of dominant negative CK1ɛ represses CLOCK-BMAL1-dependent transcription in cultured mammalian cells (9). Although the state of CLOCK phosphorylation in these cells was not determined, the strong link between CLOCK phosphorylation and transcriptional repression argues for a CK1ɛ bridging function. Additional experiments will be necessary to support or rule out a role for CK1 bridging in Neurospora and mammals.
Acknowledgments
We thank Isaac Edery for providing PER and TIM antisera. We also thank members of the Hardin laboratory for helpful discussions.
This work was supported by NIH grants NS051280 (P.E.H.) and MH056895 (J.L.P.).
Footnotes
Published ahead of print on 12 January 2009.
REFERENCES
- 1.Akten, B., E. Jauch, G. K. Genova, E. Y. Kim, I. Edery, T. Raabe, and F. R. Jackson. 2003. A role for CK2 in the Drosophila circadian oscillator. Nat. Neurosci. 6251-257. [DOI] [PubMed] [Google Scholar]
- 2.Allada, R., N. E. White, W. V. So, J. C. Hall, and M. Rosbash. 1998. A mutant Drosophila homolog of mammalian Clock disrupts circadian rhythms and transcription of period and timeless. Cell 93791-804. [DOI] [PubMed] [Google Scholar]
- 3.Bae, K., C. Lee, P. E. Hardin, and I. Edery. 2000. dCLOCK is present in limiting amounts and likely mediates daily interactions between the dCLOCK-CYC transcription factor and the PER-TIM complex. J. Neurosci. 201746-1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bell-Pedersen, D., V. M. Cassone, D. J. Earnest, S. S. Golden, P. E. Hardin, T. L. Thomas, and M. J. Zoran. 2005. Circadian rhythms from multiple oscillators: lessons from diverse organisms. Nat. Rev. Genet. 6544-556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang, D. C., and S. M. Reppert. 2003. A novel C-terminal domain of Drosophila PERIOD inhibits dCLOCK:CYCLE-mediated transcription. Curr. Biol. 13758-762. [DOI] [PubMed] [Google Scholar]
- 6.Chiu, J. C., J. T. Vanselow, A. Kramer, and I. Edery. 2008. The phospho-occupancy of an atypical SLIMB-binding site on PERIOD that is phosphorylated by DOUBLETIME controls the pace of the clock. Genes Dev. 221758-1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cyran, S. A., G. Yiannoulos, A. M. Buchsbaum, L. Saez, M. W. Young, and J. Blau. 2005. The double-time protein kinase regulates the subcellular localization of the Drosophila clock protein period. J. Neurosci. 255430-5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Darlington, T. K., K. Wager-Smith, M. F. Ceriani, D. Staknis, N. Gekakis, T. D. Steeves, C. J. Weitz, J. S. Takahashi, and S. A. Kay. 1998. Closing the circadian loop: CLOCK-induced transcription of its own inhibitors per and tim. Science 2801599-1603. [DOI] [PubMed] [Google Scholar]
- 9.Eide, E. J., E. L. Vielhaber, W. A. Hinz, and D. M. Virshup. 2002. The circadian regulatory proteins BMAL1 and cryptochromes are substrates of casein kinase I(epsilon). J. Biol. Chem. 27717248-17254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emery, P., W. V. So, M. Kaneko, J. C. Hall, and M. Rosbash. 1998. CRY, a Drosophila clock and light-regulated cryptochrome, is a major contributor to circadian rhythm resetting and photosensitivity. Cell 95669-679. [DOI] [PubMed] [Google Scholar]
- 11.Grima, B., A. Lamouroux, E. Chelot, C. Papin, B. Limbourg-Bouchon, and F. Rouyer. 2002. The F-box protein Slimb controls the levels of clock proteins period and timeless. Nature 420178-182. [DOI] [PubMed] [Google Scholar]
- 12.Hao, H., D. L. Allen, and P. E. Hardin. 1997. A circadian enhancer mediates PER-dependent mRNA cycling in Drosophila melanogaster. Mol. Cell. Biol. 173687-3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hao, H., N. R. Glossop, L. Lyons, J. Qiu, B. Morrish, Y. Cheng, C. Helfrich-Forster, and P. Hardin. 1999. The 69 bp circadian regulatory sequence (CRS) mediates per-like developmental, spatial, and circadian expression and behavioral rescue in Drosophila. J. Neurosci. 19987-994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He, Q., J. Cha, Q. He, H. C. Lee, Y. Yang, and Y. Liu. 2006. CKI and CKII mediate the FREQUENCY-dependent phosphorylation of the WHITE COLLAR complex to close the Neurospora circadian negative feedback loop. Genes Dev. 202552-2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Houl, J. H., W. Yu, S. M. Dudek, and P. E. Hardin. 2006. Drosophila CLOCK is constitutively expressed in circadian oscillator and non-oscillator cells. J. Biol. Rhythms 2193-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim, E. Y., and I. Edery. 2006. Balance between DBT/CKI(epsilon) kinase and protein phosphatase activities regulate phosphorylation and stability of Drosophila CLOCK protein. Proc. Natl. Acad. Sci. USA 1036178-6183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim, E. Y., H. W. Ko, W. Yu, P. E. Hardin, and I. Edery. 2007. A DOUBLETIME kinase binding domain on the Drosophila PERIOD protein is essential for its hyperphosphorylation, transcriptional repression, and circadian clock function. Mol. Cell. Biol. 275014-5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kivimae, S., L. Saez, and M. W. Young. 2008. Activating PER repressor through a DBT-directed phosphorylation switch. PLoS Biol. 6e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kloss, B., J. L. Price, L. Saez, J. Blau, A. Rothenfluh, C. S. Wesley, and M. W. Young. 1998. The Drosophila clock gene double-time encodes a protein closely related to human casein kinase I(epsilon). Cell 9497-107. [DOI] [PubMed] [Google Scholar]
- 20.Kloss, B., A. Rothenfluh, M. W. Young, and L. Saez. 2001. Phosphorylation of period is influenced by cycling physical associations of double-time, period, and timeless in the Drosophila clock. Neuron 30699-706. [DOI] [PubMed] [Google Scholar]
- 21.Ko, H. W., J. Jiang, and I. Edery. 2002. Role for Slimb in the degradation of Drosophila Period protein phosphorylated by Doubletime. Nature 420673-678. [DOI] [PubMed] [Google Scholar]
- 22.Lee, C., K. Bae, and I. Edery. 1999. PER and TIM inhibit the DNA binding activity of a Drosophila CLOCK-CYC/dBMAL1 heterodimer without disrupting formation of the heterodimer: a basis for circadian transcription. Mol. Cell. Biol. 195316-5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee, C., K. Bae, and I. Edery. 1998. The Drosophila CLOCK protein undergoes daily rhythms in abundance, phosphorylation, and interactions with the PER-TIM complex. Neuron 21857-867. [DOI] [PubMed] [Google Scholar]
- 24.Lee, C., J. P. Etchegaray, F. R. Cagampang, A. S. Loudon, and S. M. Reppert. 2001. Posttranslational mechanisms regulate the mammalian circadian clock. Cell 107855-867. [DOI] [PubMed] [Google Scholar]
- 25.Lin, J. M., V. L. Kilman, K. Keegan, B. Paddock, M. Emery-Le, M. Rosbash, and R. Allada. 2002. A role for casein kinase 2alpha in the Drosophila circadian clock. Nature 420816-820. [DOI] [PubMed] [Google Scholar]
- 26.Muskus, M. J., F. Preuss, J. Y. Fan, E. S. Bjes, and J. L. Price. 2007. Drosophila DBT lacking protein kinase activity produces long-period and arrhythmic circadian behavioral and molecular rhythms. Mol. Cell. Biol. 278049-8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naidoo, N., W. Song, M. Hunter-Ensor, and A. Sehgal. 1999. A role for the proteasome in the light response of the timeless clock protein. Science 2851737-1741. [DOI] [PubMed] [Google Scholar]
- 28.Nawathean, P., and M. Rosbash. 2004. The doubletime and CKII kinases collaborate to potentiate Drosophila PER transcriptional repressor activity. Mol. Cell 13213-223. [DOI] [PubMed] [Google Scholar]
- 29.Nawathean, P., D. Stoleru, and M. Rosbash. 2007. A small conserved domain of Drosophila PERIOD is important for circadian phosphorylation, nuclear localization, and transcriptional repressor activity. Mol. Cell. Biol. 275002-5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Price, J. L., J. Blau, A. Rothenfluh, M. Abodeely, B. Kloss, and M. W. Young. 1998. double-time is a novel Drosophila clock gene that regulates PERIOD protein accumulation. Cell 9483-95. [DOI] [PubMed] [Google Scholar]
- 31.Price, J. L., M. E. Dembinska, M. W. Young, and M. Rosbash. 1995. Suppression of PERIOD protein abundance and circadian cycling by the Drosophila clock mutation timeless. EMBO J. 144044-4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ripperger, J. A., and U. Schibler. 2006. Rhythmic CLOCK-BMAL1 binding to multiple E-box motifs drives circadian Dbp transcription and chromatin transitions. Nat. Genet. 38369-374. [DOI] [PubMed] [Google Scholar]
- 33.Rothenfluh, A., M. Abodeely, and M. W. Young. 2000. Short-period mutations of per affect a double-time-dependent step in the Drosophila circadian clock. Curr. Biol. 101399-1402. [DOI] [PubMed] [Google Scholar]
- 34.Rothenfluh, A., M. W. Young, and L. Saez. 2000. A TIMELESS-independent function for PERIOD proteins in the Drosophila clock. Neuron 26505-514. [DOI] [PubMed] [Google Scholar]
- 35.Rutila, J. E., V. Suri, M. Le, W. V. So, M. Rosbash, and J. C. Hall. 1998. CYCLE is a second bHLH-PAS clock protein essential for circadian rhythmicity and transcription of Drosophila period and timeless. Cell 93805-814. [DOI] [PubMed] [Google Scholar]
- 36.Schafmeier, T., A. Haase, K. Kaldi, J. Scholz, M. Fuchs, and M. Brunner. 2005. Transcriptional feedback of Neurospora circadian clock gene by phosphorylation-dependent inactivation of its transcription factor. Cell 122235-246. [DOI] [PubMed] [Google Scholar]
- 37.Sidote, D., J. Majercak, V. Parikh, and I. Edery. 1998. Differential effects of light and heat on the Drosophila circadian clock proteins PER and TIM. Mol. Cell. Biol. 182004-2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith, E. M., J. M. Lin, R. A. Meissner, and R. Allada. 2008. Dominant-negative CK2alpha induces potent effects on circadian rhythmicity. PLoS Genet. 4e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taylor, P., and P. E. Hardin. 2008. Rhythmic E-box binding by CLK-CYC controls daily cycles in per and tim transcription and chromatin modifications. Mol. Cell. Biol. 284642-4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vanselow, K., J. T. Vanselow, P. O. Westermark, S. Reischl, B. Maier, T. Korte, A. Herrmann, H. Herzel, A. Schlosser, and A. Kramer. 2006. Differential effects of PER2 phosphorylation: molecular basis for the human familial advanced sleep phase syndrome (FASPS). Genes Dev. 202660-2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu, W., H. Zheng, J. H. Houl, B. Dauwalder, and P. E. Hardin. 2006. PER-dependent rhythms in CLK phosphorylation and E-box binding regulate circadian transcription. Genes Dev. 20723-733. [DOI] [PMC free article] [PubMed] [Google Scholar]