

Abstract
The ubiquitin-proteasome system has a central role in the degradation of intracellular proteins and regulates a variety of functions. Viruses belonging to several different families utilize or modulate the system for their advantage. Here we showed that the proteasome inhibitors MG132 and epoxomicin blocked a postentry step in vaccinia virus (VACV) replication. When proteasome inhibitors were added after virus attachment, early gene expression was prolonged and the expression of intermediate and late genes was almost undetectable. By varying the time of the removal and addition of MG132, the adverse effect of the proteasome inhibitors was narrowly focused on events occurring 2 to 4 h after infection, the time of the onset of viral DNA synthesis. Further analyses confirmed that genome replication was inhibited by both MG132 and epoxomicin, which would account for the effect on intermediate and late gene expression. The virus-induced replication of a transfected plasmid was also inhibited, indicating that the block was not at the step of viral DNA uncoating. UBEI-41, an inhibitor of the ubiquitin-activating enzyme E1, also prevented late gene expression, supporting the role of the ubiquitin-proteasome system in VACV replication. Neither the overexpression of ubiquitin nor the addition of an autophagy inhibitor was able to counter the inhibitory effects of MG132. Further studies of the role of the ubiquitin-proteasome system for VACV replication may provide new insights into virus-host interactions and suggest potential antipoxviral drugs.
The ubiquitin-proteasome system has a central role in the degradation of intracellular proteins and regulates a variety of functions (22). Proteins to be degraded are modified by the addition of multiple copies of the 76-amino-acid ubiquitin through the sequential activities of an activating enzyme (E1), a conjugating enzyme (E2), and a ligase (E3) (4, 12). The degradation is mediated by the 26S proteasome, a large multiprotein complex containing trypsin-, chymotrypsin-, and post-glutamyl peptidyl hydrolytic-like protease activities. In addition, ubiquitylation has nondegradative roles in DNA repair, transcriptional regulation, signal transduction, endocytosis, and intracellular trafficking (48). Viruses belonging to several families utilize or modulate the ubiquitin-proteasome system (2, 13). The inhibition of proteasomal degradation prevents the entry of influenza virus (23) and mouse hepatitis virus (54); the early postentry steps of minute virus of mice (44) and herpes simplex virus (7); and the genome replication or expression of human coxsackie 3B virus (27), adenovirus (5), cytomegalovirus (20), infectious bursal disease virus (26), and vesicular stomatitis virus (40). In some cases the effects may be secondary to the activation of a cellular stress response and signaling pathway (24, 40, 52). Proteasomal inhibitors have an indirect effect on retroviruses and rhabdoviruses by depleting free ubiquitin needed to modify proteins for budding (16).
Vaccinia virus (VACV), the representative member of the poxvirus family, replicates entirely in the cytoplasm and encodes nearly 200 proteins with roles in entry, transcription, DNA replication, virion assembly, spread, and host interactions (36). Several recent studies indicate that poxviruses modulate the ubiquitin pathway (17, 29, 31, 45, 50), but there have been no reports regarding the effects of proteasome inhibitors on replication. VACV has been used extensively as a vector for recombinant gene expression and in that capacity as a tool for immunological studies (34). While analyzing the effects of proteasome inhibitors on antigen presentation, we noted that these drugs severely reduced reporter gene expression by VACV. Here, we show that proteasome inhibitors interfere with VACV replication at a postentry step. Early gene expression occurred, whereas viral DNA replication and subsequent intermediate and late gene expression were severely inhibited.
MATERIALS AND METHODS
Cells, virus strains, and chemicals.
HeLa and BS-C-1 cells were maintained in minimum essential medium containing Earle's salts supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin (Quality Biological, Gaithersburg, MD). VACV Western Reserve (WR) and recombinant viruses vJ2R-CAT (28) and vV5-D4 (10) were propagated as described previously. MG132 (carbobenzoxy-l-leucyl-l-leucyl-l-leucinal) and the α′,β′-epoxyketone-containing natural product epoxomicin were obtained from EMD Biosciences (Gibbstown, NJ) and dissolved in dimethyl sulfoxide (DMSO) at concentrations of 20 mM and 1 mM, respectively. UBEI-41 {4[4-(5-nitro-furan-2-ylmethylene)-3,4-dioxo-pyrazolidin-1-yl]-benzoic acid ethyl ester} in DMSO was obtained from Biogenova (Frederick, Maryland). Hydroxyurea (HU) and 3-methyladenine (3-MA) were obtained from Sigma-Aldrich (St. Louis, MO) and dissolved in water at concentrations of 0.5 M and 0.2 M, respectively.
Construction of recombinant viruses.
We used a previously described homologous recombination and plaque selection method (3) to insert the chloramphenicol acetyltransferase (CAT) open reading frame adjacent to the early-stage promoter of the J2R (thymidine kinase) gene intermediate-stage promoter of the VACV G8R gene, or late promoter of the F17R gene into an intergenic site downstream of the F13L gene of VACV. The promoter-CAT sequences were amplified by PCR (Accuprime Pfx; Invitrogen, Carlsbad, CA), digested with EcoRI and BamHI (New England Biologicals, Ipswich, MA), and inserted into the compatible site in the transfer plasmid pRB21 (3). Plasmids containing CAT were transfected with Lipofectamine 2000 (Invitrogen) into BS-C-1 cells that had been infected with the small-plaque-forming recipient virus vRB12 (3). The cells were harvested 24 h after infection, lysed by repeated freezing and thawing, and diluted to infect BS-C-1 cells. Recombinant viruses that formed large plaques were clonally purified.
Infection protocol.
Unless otherwise indicated, HeLa cells were incubated with recombinant VACV in minimum essential medium containing 2.5% fetal bovine serum at 4°C. After 1 h, the cells were washed and incubated in fresh medium with drugs or an equivalent amount of the solvent at 37°C.
Cytotoxicity assay.
Cell viability was determined using a CytoTox-ONE homogenous membrane integrity assay kit (Promega, Madison, WI). HeLa cells were cultured in 48-well tissue culture dishes in the presence of DMSO, 10 μM MG132, or 2 μM epoxomicin. The lactate dehydrogenase release from cells was quantified using fluorescent substrate at excitation and emission wavelengths of 560 and 590 nm, respectively. The percentages of cytotoxicity were calculated based on a lactate dehydrogenase release from the lysed cells of 100%.
Visualization of intracellular viral cores.
HeLa cells were attached overnight to glass coverslips and then inoculated with purified VACV (5 PFU per cell) for 1 h at 4°C. Unbound virions were removed by washing, and the cells were incubated for 3 h at 37°C in medium with or without 300 μg per ml cycloheximide and either DMSO or 10 μM MG132. The cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) and then permeabilized with PBS containing 0.1% Triton X-100. The cells were blocked with 10% fetal bovine serum and incubated with mouse monoclonal antibody (MAb) 7D11, which specifically reacts with the L1 MV membrane protein (51), and anti-A4 rabbit polyclonal antibody (8). After 1 h, the cells were washed with PBS and incubated with Alexa Fluor 594 anti-mouse immunoglobulin G (IgG) and Alexa Fluor 488 anti-rabbit IgG and stained with diamidino-2-phenylindole dihydrochloride (5 μg per ml). Images were collected with a Leica laser-scanning confocal microscope as 0.35-μm optical stacks. Viral cores (diameter, 1 μm) that stained with anti-A4 antibody and cells that stained with diamidino-2-phenylindole dihydrochloride were counted using Imaris 6.1.5 software (Bitplane Scientific Solutions) with the same quality and intensity standard deviation values for all the samples.
Pulse-labeling proteins with [35S]methionine and [35S]cysteine.
HeLa cell monolayers were incubated with VACV (7.5 PFU per cell) for 1 h at 4°C, washed three times, and incubated in medium containing DMSO or 10 μM MG132. At various times, the cells were washed with cysteine- and methionine-free medium and incubated for 5 min with the latter containing 300 μCi of a mixture of [35S]methionine and [35S]cysteine (Perkin Elmer, Waltham, MA) per ml with DMSO or 10 μM MG132. The cells were harvested in PBS, lysed with sodium dodecyl sulfate (SDS), and analyzed by electrophoresis on a 4 to 12% polyacrylamide gel. After drying, the radioactive bands were detected using a phosphor screen with a Typhoon Variable mode imager (GE Healthcare, Piscataway, NJ).
Western blotting.
Cells were lysed by heating at 100°C for 6 min in 1× NuPAGE sample loading buffer containing 1× reducing agent and lithium dodecyl sulfate (Invitrogen). The proteins were resolved by electrophoresis on 4 to 12% Novex NuPAGE polyacrylamide gels (Invitrogen) and transferred to nitrocellulose membranes using mini iBlot gel transfer stacks (Invitrogen). The membrane was blocked with 5% nonfat milk in PBS containing 0.05% Tween 20. For multiplex fluorescent detection, the membrane was incubated with mouse anti-V5 (Invitrogen) and rabbit anti-A3 antibodies for 1 h. The membrane was washed and further incubated for 1 h with the corresponding fluorescent secondary antibodies donkey anti-mouse IRDye 680 and donkey anti-rabbit IRDye 800, respectively. Fluorescent proteins were detected using the LI-COR Odyssey infrared imager (LI-COR Biosciences, Lincoln, NE). For the chemiluminescent Western blot detection, the membrane was incubated with a primary antibody for 1 h at room temperature, washed, and further incubated with secondary antibody conjugated with horseradish peroxidase (Pierce, Rockford, IL). The blot was washed and developed using Dura or Femto chemiluminescent substrate (Pierce). To reprobe the blots with different antibodies, the membrane was stripped at 55°C for 20 min using restore buffer (Pierce).
CAT assay.
CAT activity was determined using a FAST CAT green (dideoxy)chloramphenicol acetyltransferase assay kit (Molecular Probes, Eugene, OR). The infected cell pellets from a 24-well culture plate were resuspended in 100 μl of 250 mM Tris (pH 7.4) and lysed by three freeze-thaw cycles, and the soluble fraction was obtained by centrifugation at 12,000 rpm for 5 min at 4°C. A volume of 20 μl of soluble material was mixed with 10 μl of substrate reagent A (Molecular Probes CAT assay kit). After 5 min at 37°C, 10 μl of 9 mM acetyl coenzyme A was added and further incubated for 20 to 45 min at 37°C. The reaction was stopped by the addition of ice-cold ethyl acetate solution. The organic phase was collected after centrifugation at 12,000 rpm for 3 min and dried using a Savant SpeedVac concentrator (Thermo Scientific, Waltham, MA). The pellet was resuspended in 10 μl of ethyl acetate, 2 μl was spotted onto a silica gel plate, and the chromatogram was developed in a presaturated chamber containing a chloroform:methanol solvent at an 85:15 ratio. The plate was examined under UV light, and the percent conversion of the substrate was calculated by measuring the intensities of the substrate and product according to the manufacturer's instructions.
Analysis of VACV genome replication.
Infected HeLa cells in 24-well dishes were washed in PBS, suspended in 10× SSC (0.15 M NaCl and 0.015 M sodium citrate pH 7.0 for 1× solution) containing 1 M ammonium acetate, and lysed by freezing and thawing. The lysate was digested with proteinase K (Sigma-Aldrich), sonicated, and denatured by incubation with 0.4 M NaOH at 100°C (15). The total DNA was spotted onto a Hybond-N+ (Amersham, Piscataway, NJ) nylon membrane under vacuum conditions, washed with 10× SSC, and denatured by soaking in 0.5 M NaOH, 1.5 M NaCl, and the pH was lowered with 1.5 M NaCl, 1 M Tris HCl (pH 8.0). Finally, the blot was washed with 5× SSC and hybridized using QuiK-Hyb (Stratagene) solution according to the manufacturer's instructions. VACV DNA was detected using a probe against F17R DNA radioactively labeled with Ready-To-Go DNA labeling beads (-dTCP; GE Healthcare, Waukesha, WI). The blot was developed using a phosphor screen in Typhoon Variable mode imager.
Analysis of plasmid replication.
Total DNA was isolated from infected cells using a QIAmp blood mini kit (Qiagen, Valencia, CA). DNA (2 μg) was digested with EcoRI and DpnI enzymes, resolved on a 0.8% agarose gel, and transferred to a Hybond-N+ nylon membrane. A radioactively labeled plasmid probe was generated using Ready-To-Go DNA labeling beads. Hybridization was performed using Quik-Hyb and developed by Typhoon Variable mode imager.
RESULTS
Effects of proteasome inhibitors on VACV replication.
The peptide aldehyde MG132 and the α′,β′-epoxyketone epoxomicin are widely used as proteasome inhibitors at concentrations of approximately 10 μM (25, 30). Both drugs inhibit the three major activities of the proteasome, albeit at various rates. VACV plaque formation was reduced by MG132 and epoxomicin in a concentration-dependent manner (Fig. 1A and B). The numbers of plaques were reduced by 50 to 80% at 1.25 μM MG132 and 0.25 μM epoxomicin, and no plaques were observed at concentrations above 5 and 2 μM, respectively. Note that in all experiments, whether specifically stated or not, the no-drug controls received the same amount of DMSO solvent as the experimental groups. The similar effects of the two different inhibitors supported a specific role for the proteasome in VACV replication.
FIG. 1.

Proteasome inhibitors prevent VACV replication. (A, B) Plaque formation. BS-C-1 monolayers were incubated with VACV at 4°C. After 1 h, the virus inoculum was removed and replaced with medium containing 0.5% methylcellulose with or without the indicated concentrations of MG132 or epoxomicin. At 24 h after infection, the cells were stained with crystal violet and the plaques counted. (C) Virus yields. HeLa cells were incubated with VACV (3 PFU/cell) for 1 h at 4°C. The inoculum was removed and the cells were washed and incubated with fresh medium in the presence of DMSO, MG132, or epoxomicin. At the indicated times, the cells were harvested and lysed by freezing and thawing and the virus titers determined by plaque assay in BS-C-1 cells without proteasome inhibitors. (D) Cytotoxicity assay. HeLa cells were incubated with medium containing DMSO, 10 μM MG132, or 2 μM epoxomicin for either 12 or 24 h and assayed for the release of lactate dehydrogenase.
To distinguish between the inhibition of virus replication and spread, we carried out one-step growth experiments. MG132 or epoxomicin was added after virus attachment at 4°C, and virus yields were determined by plaque assay at intervals of time after increasing the temperature to 37°C. In the absence of drug, an increase in infectivity was detected by 10 h and reached a plateau at 20 h (Fig. 1C). In contrast, no increase in infectivity was detected at any time in the presence of 10 μM MG132 or 2 μM epoxomicin. At those drug concentrations, there was no evidence of enhanced cytotoxicity, as measured by the release of lactate dehydrogenase (Fig. 1D). The latter result is consistent with other reports that cell viability and growth are generally not affected for at least 20 h by proteasome inhibitors (25).
Effect of proteasome inhibitors on viral core entry and uncoating.
The initial steps in infection involve the binding of VACV to the cell, membrane fusion, and the penetration of the core into the cytoplasm (37). The core mediates early gene expression (21, 38), and the DNA is then uncoated by a process that requires protein synthesis (19). We adapted a confocal microscopic procedure developed to analyze the binding and entry of VACV. In the original procedure, cells were fixed, permeabilized, and incubated with antibodies to membrane and core proteins 30 min after infection (47). Importantly, the fixation procedure prevents antibody from reacting with the core of intact virions, and therefore, intracellular cores are specifically detected by their punctate fluorescence. In our adaptation, the cells were infected for 3 h in the presence or absence of cycloheximide, a protein synthesis inhibitor that prevents DNA uncoating and stabilizes cores, prior to antibody staining. In the experiment for which the results are summarized in Fig. 2, we detected approximately 6.5 cores per cell after treatment with cycloheximide and MG132 or DMSO (Fig. 2), indicating that proteasomal activity was not needed for virus entry. In the absence of cycloheximide, the number of cores was reduced to 1.5 and 3.9 in the presence of DMSO and MG132, respectively (Fig. 2). Based on the difference between the number of cores in the presence and absence of cycloheximide, one could infer that 77% and 40% of the cores were uncoated under the two conditions. In a repeat experiment, these values were 56% and 45% for DMSO- and MG132-treated cells, respectively. Using DNase sensitivity as a measure of uncoating, Joklik (18) had reported that 50% of the virus particles were uncoated in 2 to 3 h and that this level reached a maximum of 60%. Although uncoating may be reduced somewhat or delayed in the presence of MG132, the effect seemed insufficient to account for the complete inhibition of virus replication.
FIG. 2.

Core entry and uncoating in the presence of MG132. HeLa cells on glass coverslips were inoculated for 1 h at 4°C with purified VACV (5 PFU per cell) and then incubated for 3 h at 37°C. After fixation and permeabilization, the cells were stained with mouse MAb 7D11, which specifically reacts with the L1 MV membrane protein, and a rabbit polyclonal antibody specific for the A4 MV core protein, followed by Alexa Fluor 594 anti-mouse IgG and Alexa Fluor 488 anti-rabbit IgG. Cell nuclei were stained with diamidino-2-phenylindole dihydrochloride. An average of 275 cells in each group were counted, and the numbers of cores per cell were plotted.
Effect of MG132 on viral gene expression.
VACV gene expression is transcriptionally regulated in a cascade fashion (1). The enzymes and factors required for early gene expression are packaged within the infectious particle, allowing viral mRNA synthesis within minutes after entry. In contrast, the expression of intermediate and late genes is delayed and dependent on the de novo synthesis of stage-specific transcription factors and viral DNA. Pulse-labeling with radioactive amino acids followed by SDS-polyacrylamide gel electrophoresis provides a method of analyzing newly made proteins during the course of infection. In the experiment for which the results are shown in Fig. 3A, cells were washed with methionine- and cysteine-deficient medium at the indicated times between 0 and 12 h after infection and then incubated with [35S]methionine and [35S]cysteine for 5 min. In the absence of MG132, the shutoff of cell protein synthesis and the appearance of viral late proteins were detected at 6 h (Fig. 3A). Viral early proteins are difficult to detect unless very high multiplicities of virus are used (35) because the majority of the label is incorporated into the cellular proteins during the first few hours. In the presence of MG132, viral late proteins were not detected and host cell protein synthesis continued (Fig. 3A). The lower incorporation of radioactivity in the presence of MG132 may be an artifact due to the very rapid amino acid depletion that occurs in the presence of proteosome inhibitors when the medium contains reduced amounts of amino acids (46).
FIG. 3.

Effects of proteasome inhibitor on VACV protein synthesis. (A) Amino acid labeling. HeLa cells were inoculated with VACV (7.5 PFU per cell) at 4°C and then incubated at 37°C in medium containing DMSO or 10 μM MG132. At the indicated times, the cells were washed in medium lacking methionine and cysteine and incubated for 5 min with [35S]methionine and [35S]cysteine. Cells were lysed with SDS and analyzed by electrophoresis on a 4 to 12% polyacrylamide gel. After drying, radioactive bands were detected using a phosphor screen. Bands representing VACV late proteins are marked by dots. M represents mock-infected cells that were labeled for 5 min at 12 h after DMSO or MG132 treatment. (B, C) Western blotting of representative early and late proteins. HeLa cells were infected with vV5-D4 in the presence of DMSO or 10 μM MG132. At the indicated times, the cells were harvested and lysed and the proteins were resolved by polyacrylamide gel electrophoresis. The proteins were transferred to a nitrocellulose membrane and probed for D4 protein using mouse MAb to V5 tag and polyclonal rabbit antibody to A3 followed by fluorescently labeled secondary antibodies. In all panels, the positions of the marker proteins are shown on the left.
Western blotting allows the specific detection of viral proteins without the need for shutting off host protein synthesis and avoids potential artifacts due to amino acid labeling. By using a multiplex fluorescent antibody detection procedure, we were able to analyze the production of the replication factor uracil DNA glycosylase, an early protein encoded by the D4R gene, and a major core protein encoded by the late A3L gene in the same blot. The recombinant virus vV5-D4, which encodes D4 with a V5 epitope tag under its natural promoter, was used to infect cells in the presence of DMSO or MG132. vV5-D4 was detected at 2 h and reached a maximum at 6 h in the DMSO-treated, infected control cells (Fig. 3B). When MG132 was added immediately following virus adsorption, vV5-D4 synthesis continued to higher than normal levels by 15 h (Fig. 3B). In the DMSO-treated cells, the A3 core protein accumulated to high levels between 8 and 24 h. In contrast, the A3 protein was not detected in MG132-treated cells (Fig. 3C) unless the blot was greatly overexposed using chemiluminescence detection (data not shown). The continued increase of vV5-D4 at late times (Fig. 3B) while A3 synthesis was inhibited (Fig. 3C) indicated that the latter was not due to general effects on protein synthesis. The ability of the infected cells to synthesize early proteins in the presence of MG132 was also demonstrated by flow cytometry using a MAb to the E3 protein (data not shown), which does not react on Western blots (55).
To further investigate the effects of proteasome inhibitors on the expression of early, intermediate, and late genes, recombinant viruses expressing the same reporter gene regulated by different promoters were constructed. CAT was used as the reporter because previous studies had shown that MG132 did not affect its expression (9). The early, intermediate, and late promoters were derived from the J2R (thymidine kinase), G8R (late transcription factor), and F17R (11K core protein) genes, respectively. The recombinant viruses vpJ2R-CAT, vpG8R-CAT, and vpF17R-CAT were adsorbed to cells at 4°C, and then various concentrations of MG132 were added to the medium. CAT expression from the early promoter, measured at 6 h after raising the temperature to 37°C, was reduced slightly at 2.5 μM MG132 but remained constant until the drug concentration reached 10 μM, after which another small decline was noted (Fig. 4A). In contrast, CAT expressed at 8 h from intermediate (Fig. 4B) and late (Fig. 4C) promoters exhibited a profound dose-dependent decrease with increasing levels of MG132. Furthermore, the inhibition of CAT synthesis regulated by the late promoter could not be overcome by increasing the multiplicity of the virus infection (Fig. 4D).
FIG. 4.

Effect of MG132 on gene expression regulated by early, intermediate, and late promoters. (A) Early promoter. HeLa cells were inoculated with vpJ2R-CAT for 1 h at 4°C and then with the indicated concentrations of MG132 at 37°C for 6 h. Cells were harvested and CAT activity was determined. CAT activity is indicated as the percentage of substrate conversion normalized to the activity with no inhibitor. (B, C) Intermediate and late promoters. The protocol was similar to that described for panel A, except that cells were infected with vpG8R-CAT or vpF17R-CAT and that activity was determined 8 h after infection. (D) Effect of virus multiplicity. HeLa cells were infected with vpF17R-CAT virus with the indicated virus multiplicity (PFU/cell) as described for panel C, and CAT activity was determined.
Effects of the pretreatment of cells with MG132.
Since early gene expression precedes intermediate and late gene expression, it was possible that the specificity of the drugs was only apparent and actually reflected the length of time needed to establish an adverse cellular effect. To evaluate this, HeLa cells were divided into two groups. One group was incubated with 10 μM MG132 for 6 h prior to infection. The cells in both groups were then infected with vpJ2R-CAT or vpF17R-CAT in the presence or absence of MG132. Early-promoter-regulated CAT activity in the vpJ2R-CAT-infected cells was relatively unaffected by MG132 even when MG132 treatment started 6 h before infection and continued after infection (Fig. 5A), indicating that the length of the treatment was not critical. On the other hand, late-promoter-regulated CAT activity in cells infected with vpF17R-CAT was reduced by MG132 whether the drug treatment started 6 h before or at the time of infection (Fig. 5A). Furthermore, the action of MG132 was not toxic on the cells since a 6-h treatment prior to infection had no adverse effect on vpF17R-CAT expression when the drug was removed from one culture at the time of infection (Fig. 5A). Thus, the stage specificity of the promoter, not the duration of the MG132 treatment, was important.
FIG. 5.

Duration and reversibility of proteasome inhibition on VACV gene expression. (A) Effect of MG132 pretreatment. HeLa cells were divided into groups that were infected with vpJ2R-CAT or vpF17R-CAT and harvested 8 h later. Group 1 was infected in the presence of DMSO; group 2 was infected in the presence of MG132; group 3 was pretreated with MG132 for 6 h, and then the drug was replaced with DMSO at the start of infection; and group 4 was pretreated with MG132 for 6 h, and the addition of MG132 was continued during infection. After harvesting, CAT activity was determined. CAT activity is indicated as the percentage of substrate conversion normalized to the activity with no inhibitor. Error bars are shown. (B) Removal of MG132 after infection. HeLa cells were infected with vpF17R-CAT in the presence or absence of 10 μM MG132. The drug was removed at 1, 2, 4, and 6 h after infection, and in each case, the cells were incubated for an additional 6 h without drug so that CAT activity was determined at 7, 8, 10, and 12 h, respectively, after infection. hpi, hours postinfection. (C) Removal of epoxomicin after infection. The protocol was identical to that described for panel B except that 2 μM epoxomicin was used instead of MG132.
Determination of the critical time for MG132 inhibition.
As described above, MG132 exerted no lingering adverse effect when present for 6 h and removed at the time of virus infection. In the next experiments, MG132 was added following virus adsorption and removed at later times to determine the critical period of drug activity. HeLa cells were infected with vpF17R-CAT in the presence or absence of 10 μM MG132. At the indicated times after infection, the cells were washed and incubated with medium lacking inhibitor for another 6 h, and CAT activity was determined. Thus, when the drug was removed at 1, 2, 4, and 6 h, CAT activity was determined at 7, 8, 10, and 12 h after infection, respectively. MG132 had little or no effect on CAT activity when the drug was removed 1 or 2 h after infection (Fig. 5B). However, when MG132 was removed 4 or 6 h after infection, CAT activity was substantially reduced (Fig. 5B). This result contrasts with the reversibility of MG132 when it was present for 6 h prior to infection (Fig. 5A) and suggests that a step in virus replication between 2 and 4 to 6 h is irreversibly blocked. When an experiment similar to that depicted in Fig. 5B was performed with epoxomicin, an irreversible inhibitor of the proteasome, CAT activity was reduced even if the drug was removed 1 or 2 h after infection (Fig. 5C).
These experiments suggested that the critical time for proteasome inhibition occurred later than 2 h after infection. To establish this more directly, the time-of-addition experiments were carried out. HeLa cells were infected with vpF17R-CAT, and MG132 was added at times (0 to 6 h) preceding significant late gene expression under relatively low (3 PFU/cell) multiplicity conditions. CAT expression, measured at 9 h after infection, was almost as severely reduced when MG132 was added at 2 h as at zero time (Fig. 6A), clearly showing that the drug effect occurs postentry. However, CAT expression was much less inhibited when MG132 was added at 4 or 6 h, indicating that the critical period for proteasome inhibition was between 2 and 4 h.
FIG. 6.
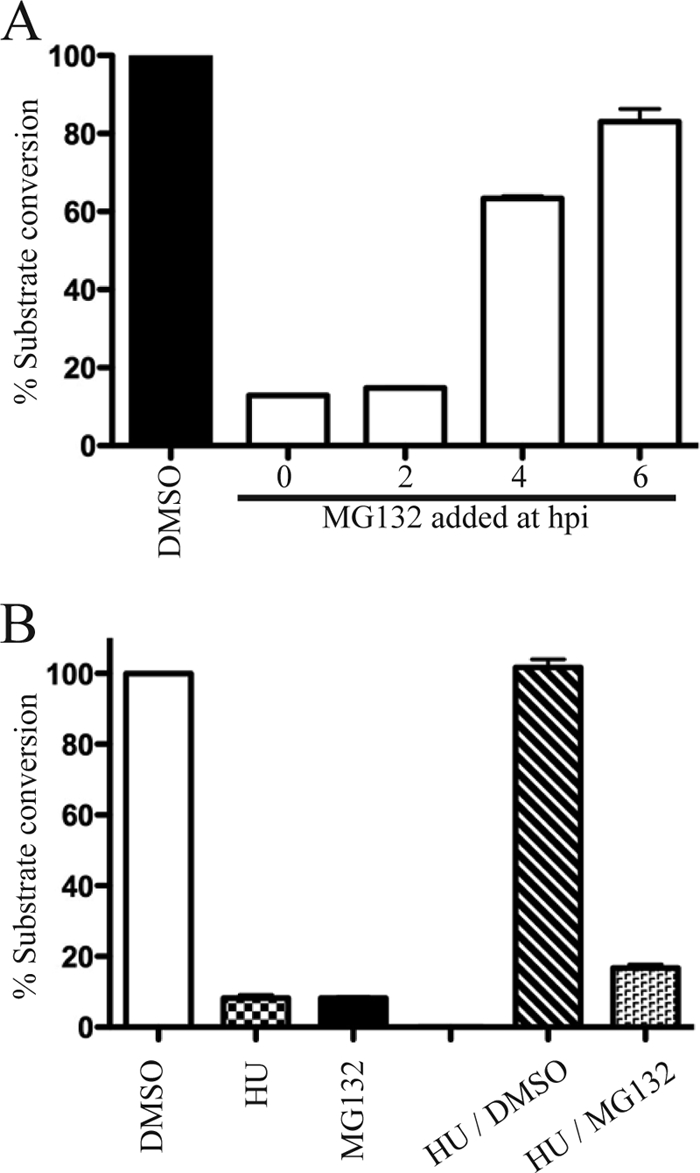
Effect of time of MG132 addition on late gene expression. (A) HeLa cells were inoculated with vpF17R-CAT for 1 h at 4°C. After virus adsorption, the cells were incubated at 37°C and 10 μM MG132 was added at the indicated hours postinfection (hpi). The cells were harvested 9 h after infection, and CAT activity was measured. CAT activity is indicated as the percentage of substrate conversion normalized to the activity with no inhibitor. (B) The first three lanes show CAT activity from HeLa cell cultures that were infected with vpF17R-CAT for 6 h in the presence of DMSO, HU, or MG132. In the last two lanes, cultures were infected with vpF17R-CAT for 3 h in the presence of HU. The HU was then removed and replaced with DMSO or MG132. After an additional 6 h, the cells were harvested and CAT activity was determined.
A variation of the above experiment was carried out using 5 mM HU, which prevents DNA synthesis but not virus entry or early gene expression (42). Some cultures were infected with vpF17R-CAT in the presence of DMSO, HU, or MG132 to confirm the effects of the inhibitors on CAT expression. As expected, both HU and MG132 were inhibitory (Fig. 6B). Other cultures were infected with vpF17R-CAT for 3 h in the presence of HU in order to allow early gene expression. Then, the HU was removed and replaced with DMSO or MG132, and the incubation continued for an additional 6 h. The reversibility of the HU treatment was shown by CAT activity in the DMSO sample (Fig. 6B). Importantly, MG132 still inhibited CAT activity even though early gene expression had been allowed to proceed for 3 h in the presence of HU (Fig. 6B).
Effect of MG132 on VACV genome and plasmid replication.
Experiments to determine whether MG132 prevents viral DNA replication were carried out next. The total DNA from infected cells was immobilized on a membrane, which was then incubated with a radioactively labeled viral DNA probe. In the control untreated cells, viral DNA increased from 3 to 24 h (Fig. 7A and B). The specificity of the viral DNA detection was confirmed by the absence of DNA synthesis in the presence of AraC (data not shown). In parallel incubations, MG132 and epoxomicin severely reduced viral DNA synthesis (Fig. 7A and B). In a variation of this experiment, cells were allowed to express early proteins for 3 h in the presence of HU. The HU was then removed and replaced with either DMSO or MG132, as in the protocol for which the results are depicted in Fig. 6B. In the absence of MG132, viral DNA was detected at 4 h after the removal of HU and increased at 6 and 8 h. In contrast, no viral DNA was detected even at 8 h in the presence of MG132 (Fig. 7C). Thus, the effect of MG132 on viral genome replication could account for the block in intermediate and viral late protein synthesis.
FIG. 7.

Effect of proteasome inhibitors on VACV genome and plasmid replication. (A, B) Effects of MG132 and epoxomicin on genome replication. HeLa cells were infected with VACV and incubated in the presence of DMSO, 10 μM MG132, or 2 μM epoxomicin. The level of viral DNA was determined at the indicated hours postinfection by slot blot analysis with a radioactive viral DNA probe. The intensities of the radioactive bands were quantified using ImageQuant software and plotted. (C) Effect on genome replication of MG132 addition following early gene expression. HeLa cells infected with vpF17R-CAT were treated with HU for 3 h to allow early gene expression. At the end of this time, the HU was removed and the cells were resuspended in medium containing MG132 or DMSO. At 2, 4, 6, and 8 h after the removal of HU, DNA was extracted from the cells and subjected to slot blot analysis. (D) Effect of MG132 on plasmid replication. HeLa cells were transfected with pUC19. After 24 h, the cells were infected with VACV in the absence (−) or presence (+) of 10 μM MG132. Mock-infected cells (Mock) and infected cells (vWR) were harvested at the indicated times, and the total DNA was digested with BamHI and DpnI and resolved by agarose gel electrophoresis. DNA fragments were transferred to a nylon membrane and probed for plasmid-specific sequences.
We considered the possibility that viral DNA replication failed to occur because the proteasome inhibitor prevented the complete protein uncoating of the genome within the virus core and access by the newly synthesized viral replication proteins. To test this model, we took advantage of previous observations that any circular DNA can be replicated in cells infected with poxviruses (6, 32). Plasmid replication occurs in cytoplasmic viral factories and requires all viral proteins known to be necessary for VACV genome replication, making it a surrogate for the latter (10). HeLa cells were transfected with pUC19 and then either mock infected or infected with VACV with or without the addition of MG132. DNA was isolated from cells at various times and digested with DpnI to specifically cleave the methylated input DNA and with BamHI to resolve newly replicated unmethylated concatenated DNA. DNA was subjected to gel electrophoresis, transferred to a membrane, and detected by hybridization with a specific probe. In the infected untreated cells, Dpn-resistant plasmid DNA increased from 3 to 12 h, whereas no Dpn-resistant DNA was detected in infected cells that had been treated with MG132 (Fig. 7D). Thus, proteasome function was required for VACV-mediated DNA replication of a transfected “naked” DNA template as well as viral genomic DNA.
Inhibition of ubiquitin-activating enzyme E1 inhibits viral late protein synthesis.
Proteasomal inhibitors act at the final stage of the ubiquitin pathway. In contrast, the inhibitors of E1 act at the first step. UBEI-41, also known as PYR-41, is a specific inhibitor of E1 ubiquitination (53). At a concentration of 25 μM, UBEI-41 had no effect on early promoter-regulated CAT synthesis (Fig. 8A) but severely inhibited late promoter-regulated CAT synthesis (Fig. 8B). At this concentration of UBEI-41, E1 ubiquitylation is inhibited by 85% (53).
FIG. 8.

Effect of ubiquitin E1 inhibitor on late gene expression. HeLa cells were inoculated with vpJ2R-CAT (A) or vpF17R-CAT (B) at 4°C for 1 h. The cells were then incubated in fresh medium containing the indicated concentrations of UBEI-41 at 37°C. After 8 h, CAT activity was determined. CAT activity is indicated as the percentage of substrate conversion normalized to the activity with no inhibitor. Error bars are shown.
The effect of MG132 was not overcome by an autophagy inhibitor or by the increased expression of ubiquitin.
Previous studies had shown that MG132 could induce the formation of autophagic vesicles due to endoplasmic reticulum stress and that this could be prevented by 3-MA, an inhibitor of autophagy (11). However, when 3-MA and MG132 were added at the same time, CAT activity was similar to that obtained with MG132 alone (Fig. 9A).
FIG. 9.

Lack of an effect of 3-MA or the overexpression of ubiquitin on MG132 inhibition of late gene expression. (A) Effect of 3-MA. Following adsorption with vpF17R-CAT, HeLa cells were incubated with DMSO, 3-MA, MG132, or 3-MA plus MG132. CAT activity was determined 8 h after infection. The values were normalized to the activity with no inhibitor. (B) CAT expression after the overexpression of ubiquitin. HeLa cells were transfected with the indicated amounts of plasmid encoding hemagglutinin-tagged ubiquitin (pHA-Ub) regulated by the cytomegalovirus early promoter. After 24 h, the cells were inoculated with vpF17R-CAT and then incubated in the presence of DMSO or 10 μM MG132. CAT activity was determined 8 h after infection and normalized. (C) Modification of proteins by epitope-tagged ubiquitin. HeLa cells were transfected as described for panel B. After 24 h, the cells were treated with DMSO or 10 μM MG132 for an additional 8 h. The lysates were analyzed by Western blotting with antibody to the influenza virus epitope tag. The position of free ubiquitin is shown on the left, and the positions of the marker proteins are on the right.
In the presence of proteasome inhibitors, polyubiquitinated proteins accumulate, resulting in the reduction of free ubiquitin available for conjugation (33). In some cases, this effect of proteasome inhibitors was overcome by the transfection of a ubiquitin overexpression plasmid (41). We found that the transfection of an epitope-tagged ubiquitin overexpression plasmid did not alter the effect of MG132 on CAT expression in VACV-infected cells (Fig. 9B), even though free and conjugated ubiquitin were detected under these conditions (Fig. 9C).
DISCUSSION
We showed that the proteasome inhibitors MG132 and epoxomicin blocked a postentry step in VACV replication. In the presence of MG132, early gene expression continued to higher than normal levels by late times, whereas viral genome replication and intermediate and late gene expression were severely reduced. Events occurring at approximately 2 h after infection (+2 h) appeared critical, as late gene expression occurred when MG132 was added at −6 h and removed at zero time or added at zero time and removed at +2 h, whereas late gene expression was inhibited severely when the drug was first added at +2 h but only slightly at +4 or +6 h. The narrow 2- to 4-h window for the effect of proteasome inhibitors on VACV replication can be contrasted with evidence for effects at prolonged times during the replication of human cytomegalovirus (20). The inhibition of ubiquitin-activating enzyme E1 also reduced VACV late gene expression, further demonstrating the role of the ubiquitin-proteasome system in VACV replication. Ubiquitylation and proteasomal degradation are closely linked, since the inhibition of ubiquitylation can prevent proteasomal degradation (53) and the inhibition of proteasomal degradation reduces the pool of free ubiquitin (33). Although the increasing expression of ubiquitin by transfection did not reverse the effect of MG132, this negative result is insufficient to rule out a nonproteasomal role for ubiquitylation in VACV replication.
Based on the timing and biochemical data, we excluded virus entry and early gene expression as being directly affected by the proteasome inhibitors. Our data pointed to viral DNA replication as an early target of inhibiting proteasome activity, despite the synthesis of early proteins. This block in genome replication by proteasome inhibitors may have been due in part to interference with the complete uncoating of the core, which would prevent access of the viral DNA replication proteins to the genome template. However, this could not explain the effects of the proteasome inhibitors since VACV-induced plasmid replication was also prevented by the addition of MG132. Previous studies had demonstrated the ability of transfected plasmids to replicate in the cytoplasm of poxvirus-infected cells (6, 32). Importantly, plasmid replication requires each factor known to be required for VACV genome replication (10). Thus, the inhibition of both genome and plasmid replication suggested effects on DNA synthesis that go beyond the blockage of uncoating. A possible mechanism by which proteasome inhibitors might act on DNA replication was suggested by recent reports describing the role of barrier to integration factor (BAF). Wiebe and Traktman (49) showed that the essential role of the VACV early B1 replication protein is to phosphorylate BAF, thereby preventing its association with cytoplasmic viral DNA. We considered that by preventing BAF degradation, proteasome inhibitors might increase BAF to levels that exceeded the ability of B1 kinase to effectively inactivate it. However, the BAF level remained unchanged in proteasome inhibitor-treated cells compared to that of the control cells, and MG132 had a similar inhibitory effect on VACV late gene expression when BAF expression was knocked down by short hairpin RNA (P.S. Satheshkumar, unpublished data). The possibility remains that other still-unknown cell proteins might play a role analogous to that of BAF.
Further studies of the role of the ubiquitin-proteasome system in the replication of VACV and other poxviruses may provide new insights into virus-host interactions. In addition, proteasome inhibitors might serve as antipoxviral drugs. In support of this notion, proteasome inhibitors have been shown to prolong the survival of mice inoculated with Epstein-Barr virus-transformed B cells (56) and to protect against coxsackievirus-induced myocarditis in mice (14). Furthermore, proteasome inhibitors are being used clinically for the treatment of certain cancers (39, 43).
Acknowledgments
We thank Jon Yewdell for helpful discussions, Norman Cooper and Catherine Cotter for providing cells, Suman Das for providing the plasmid encoding hemagglutinin-tagged ubiquitin, Mariano Esteban for providing antibody to the A4 protein, Paula Traktman for a cell line expressing BAF shRNA, and Matthew Gastinger for assistance with confocal microscopic imaging and analysis.
The work was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
Published ahead of print on 7 January 2009.
REFERENCES
- 1.Baldick, C. J., Jr., and B. Moss. 1993. Characterization and temporal regulation of mRNAs encoded by vaccinia virus intermediate stage genes. J. Virol. 673515-3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Banks, L., D. Pim, and M. Thomas. 2003. Viruses and the 26S proteasome: hacking into destruction. Trends Biochem. Sci. 28452-459. [DOI] [PubMed] [Google Scholar]
- 3.Blasco, R., and B. Moss. 1995. Selection of recombinant vaccinia viruses on the basis of plaque formation. Gene 158157-162. [DOI] [PubMed] [Google Scholar]
- 4.Burckstummer, T., K. L. Bennett, A. Preradovic, G. Schutze, O. Hantschel, G. Superti-Furga, and A. Bauch. 2006. An efficient tandem affinity purification procedure for interaction proteomics in mammalian cells. Nat. Methods 31013-1019. [DOI] [PubMed] [Google Scholar]
- 5.Corbin-Lickfett, K. A., and E. Bridge. 2003. Adenovirus E4-34kDa requires active proteasomes to promote late gene expression. Virology 315234-244. [DOI] [PubMed] [Google Scholar]
- 6.DeLange, A. M., and G. McFadden. 1986. Sequence-nonspecific replication of transfected plasmid DNA in poxvirus-infected cells. Proc. Natl. Acad. Sci. USA 83614-618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delboy, M. G., D. G. Roller, and A. V. Nicola. 2008. Cellular proteasome activity facilitates herpes simplex virus entry at a postpenetration step. J. Virol. 823381-3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Demkowicz, W. E., J. S. Maa, and M. Esteban. 1992. Identification and characterization of vaccinia virus genes encoding proteins that are highly antigenic in animals and are immunodominant in vaccinated humans. J. Virol. 66386-398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deroo, B. J., and T. K. Archer. 2002. Proteasome inhibitors reduce luciferase and beta-galactosidase activity in tissue culture cells. J. Biol. Chem. 27720120-20123. [DOI] [PubMed] [Google Scholar]
- 10.De Silva, F. S., and B. Moss. 2005. Origin-independent plasmid replication occurs in vaccinia virus cytoplasmic factories and requires all five known poxvirus replication factors. Virol. J. 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ding, W. X., H. M. Ni, W. Gao, T. Yoshimori, D. B. Stolz, D. Ron, and X. M. Yin. 2007. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am. J. Pathol. 171513-524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dye, B. T., and B. A. Schulman. 2007. Structural mechanisms underlying posttranslational modification by ubiquitin-like proteins. Annu. Rev. Biophys. Biomol. Struct. 36131-150. [DOI] [PubMed] [Google Scholar]
- 13.Gao, G., and H. Luo. 2006. The ubiquitin-proteasome pathway in viral infections. Can. J. Physiol. Pharmacol. 845-14. [DOI] [PubMed] [Google Scholar]
- 14.Gao, G., J. Zhang, X. Si, J. Wong, C. Cheung, B. McManus, and H. Luo. 2008. Proteasome inhibition attenuates coxsackievirus-induced myocardial damage in mice. Am. J. Physiol. Heart Circ. Physiol. 295H401-H408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia, A. D., and B. Moss. 2001. Repression of vaccinia virus Holliday junction resolvase inhibits processing of viral DNA into unit-length genomes. J. Virol. 756460-6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harty, R. N., M. E. Brown, J. P. McGettigan, G. Wang, H. R. Jayakar, J. M. Huibregtse, M. A. Whitt, and M. J. Schnell. 2001. Rhabdoviruses and the cellular ubiquitin-proteasome system: a budding interaction. J. Virol. 7510623-10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang, J. N., Q. Huang, X. L. Zhou, M. M. Shen, A. Yen, S. X. Yu, G. Q. Dong, K. B. Qu, P. Y. Huang, E. M. Anderson, S. Daniel-Issakani, R. M. L. Buller, D. G. Payan, and H. H. Lu. 2004. The poxvirus p28 virulence factor is an E3 ubiquitin ligase. J. Biol. Chem. 27954110-54116. [DOI] [PubMed] [Google Scholar]
- 18.Joklik, W. K. 1964. The intracellular uncoating of poxvirus DNA. I. The fate of radioactively-labeled rabbitpox virus. J. Mol. Biol. 8263-276. [DOI] [PubMed] [Google Scholar]
- 19.Joklik, W. K. 1964. The intracellular uncoating of poxvirus DNA. II. The molecular basis of the uncoating process. J. Mol. Biol. 8277-288. [DOI] [PubMed] [Google Scholar]
- 20.Kaspari, M., N. Tavalai, T. Stamminger, A. Zimmermann, R. Schilf, and E. Bogner. 2008. Proteasome inhibitor MG132 blocks viral DNA replication and assembly of human cytomegalovirus. FEBS Lett. 582666-672. [DOI] [PubMed] [Google Scholar]
- 21.Kates, J. R., and B. McAuslan. 1967. Messenger RNA synthesis by a “coated” viral genome. Proc. Natl. Acad. Sci. USA 57314-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kerscher, O., R. Felberbaum, and M. Hochstrasser. 2006. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu. Rev. Cell Dev. Biol. 22159-180. [DOI] [PubMed] [Google Scholar]
- 23.Khor, R., L. J. McElroy, and G. R. Whittaker. 2003. The ubiquitin-vacuolar protein sorting system is selectively required during entry of influenza virus into host cells. Traffic 4857-868. [DOI] [PubMed] [Google Scholar]
- 24.La Frazia, S., C. Amici, and M. G. Santoro. 2006. Antiviral activity of proteasome inhibitors in herpes simplex virus-1 infection: role of nuclear factor-kappaB. Antivir. Ther. 11995-1004. [PubMed] [Google Scholar]
- 25.Lee, D. H., and A. L. Goldberg. 1998. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 8397-403. [DOI] [PubMed] [Google Scholar]
- 26.Liu, J., L. Wei, T. Jiang, L. Shi, and J. Wang. 2007. Reduction of infectious bursal disease virus replication in cultured cells by proteasome inhibitors. Virus Genes 35719-727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo, H., J. Zhang, C. Cheung, A. Suarez, B. M. McManus, and D. Yang. 2003. Proteasome inhibition reduces coxsackievirus B3 replication in murine cardiomyocytes. Am. J. Pathol. 163381-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mackett, M., G. L. Smith, and B. Moss. 1984. General method for production and selection of infectious vaccinia virus recombinants expressing foreign genes. J. Virol. 49857-864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mansouri, M., E. Bartee, K. Gouveia, B. T. H. Nerenberg, J. Barrett, L. Thomas, G. Thomas, G. McFadden, and K. Fruh. 2003. The PHD/LAP-domain protein M153R of myxomavirus is a ubiquitin ligase that induces the rapid internalization and lysosomal destruction of CD4. J. Virol. 771427-1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meng, L., R. Mohan, B. H. Kwok, M. Elofsson, N. Sin, and C. M. Crews. 1999. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc. Natl. Acad. Sci. USA 9610403-10408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mercer, A. A., S. B. Fleming, and N. Ueda. 2005. F-box-like domains are present in most poxvirus ankyrin repeat proteins. Virus Genes 31127-133. [DOI] [PubMed] [Google Scholar]
- 32.Merchlinsky, M., and B. Moss. 1988. Sequence-independent replication and sequence-specific resolution of plasmids containing the vaccinia virus concatemer junction: requirements for early and late trans-acting factors, p. 87-93. In T. Kelly and B. Stillman (ed.), Cancer cells 6/eukaryotic DNA replication. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- 33.Mimnaugh, E. G., H. Y. Chen, J. R. Davie, J. E. Celis, and L. Neckers. 1997. Rapid deubiquitination of nucleosomal histones in human tumor cells caused by proteasome inhibitors and stress response inducers: effects on replication, transcription, translation, and the cellular stress response. Biochemistry 3614418-14429. [DOI] [PubMed] [Google Scholar]
- 34.Moss, B. 1996. Genetically engineered poxviruses for recombinant gene expression, vaccination, and safety. Proc. Natl. Acad. Sci. USA 9311341-11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moss, B. 1968. Inhibition of HeLa cell protein synthesis by the vaccinia virion. J. Virol. 21028-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moss, B. 2007. Poxviridae: the viruses and their replication, p. 2905-2946. In D. M. Knipe and P. M. Howley (ed.), Fields virology, vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 37.Moss, B. 2006. Poxvirus entry and membrane fusion. Virology 34448-54. [DOI] [PubMed] [Google Scholar]
- 38.Munyon, W. H., and S. Kit. 1966. Induction of cytoplasmic ribonucleic acid synthesis in vaccinia-infected LM cells during inhibition of protein synthesis. Virology 29303-306. [DOI] [PubMed] [Google Scholar]
- 39.Nencioni, A., F. Grunebach, F. Patrone, A. Ballestrero, and P. Brossart. 2007. Proteasome inhibitors: antitumor effects and beyond. Leukemia 2130-36. [DOI] [PubMed] [Google Scholar]
- 40.Neznanov, N., E. M. Dragunsky, K. M. Chumakov, L. Neznanova, R. C. Wek, A. V. Gudkov, and A. K. Banerjee. 2008. Different effect of proteasome inhibition on vesicular stomatitis virus and poliovirus replication. PLoS ONE 3e1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patnaik, A., V. Chau, and J. W. Wills. 2000. Ubiquitin is part of the retrovirus budding machinery. Proc. Natl. Acad. Sci. USA 9713069-13074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pogo, B. G. T., and S. Dales. 1971. Biogenesis of vaccinia: separation of early stages from maturation by means of hydroxyurea. Virology 43144-151. [DOI] [PubMed] [Google Scholar]
- 43.Richardson, P. G., C. Mitsiades, T. Hideshima, and K. C. Anderson. 2006. Bortezomib: proteasome inhibition as an effective anticancer therapy. Annu. Rev. Med. 5733-47. [DOI] [PubMed] [Google Scholar]
- 44.Ros, C., and C. Kempf. 2004. The ubiquitin-proteasome machinery is essential for nuclear translocation of incoming minute virus of mice. Virology 324350-360. [DOI] [PubMed] [Google Scholar]
- 45.Sperling, K. M., A. Schwantes, B. S. Schnierle, and G. Sutter. 2008. The highly conserved orthopoxvirus 68k ankyrin-like protein is part of a cellular SCF ubiquitin ligase complex. Virology 374234-239. [DOI] [PubMed] [Google Scholar]
- 46.Vabulas, R. M., and F. U. Hartl. 2005. Protein synthesis upon acute nutrient restriction relies on proteasome function. Science 3101960-1963. [DOI] [PubMed] [Google Scholar]
- 47.Vanderplasschen, A., M. Hollinshead, and G. L. Smith. 1998. Intracellular and extracellular vaccinia virions enter cells by different mechanisms. J. Gen. Virol. 79877-887. [DOI] [PubMed] [Google Scholar]
- 48.Welchman, R. L., C. Gordon, and R. J. Mayer. 2005. Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat. Rev. Mol. Cell Biol. 6599-609. [DOI] [PubMed] [Google Scholar]
- 49.Wiebe, M. S., and P. Traktman. 2007. Poxviral B1 kinase overcomes barrier to autointegration factor, a host defense against virus replication. Cell Host Microbe 1187-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilton, B. A., S. Campbell, N. Van Buuren, R. Garneau, M. Furukawa, Y. Xiong, and M. Barry. 2008. Ectromelia virus BTB/kelch proteins, EVM150 and EVM167, interact with cullin-3-based ubiquitin ligases. Virology 37482-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wolffe, E. J., S. Vijaya, and B. Moss. 1995. A myristylated membrane protein encoded by the vaccinia virus L1R open reading frame is the target of potent neutralizing monoclonal antibodies. Virology 21153-63. [DOI] [PubMed] [Google Scholar]
- 52.Wong, J., J. Zhang, X. Si, G. Gao, and H. Luo. 2007. Inhibition of the extracellular signal-regulated kinase signaling pathway is correlated with proteasome inhibitor suppression of coxsackievirus replication. Biochem. Biophys. Res. Commun. 358903-937. [DOI] [PubMed] [Google Scholar]
- 53.Yang, Y., J. Kitagaki, R. M. Dai, Y. C. Tsai, K. L. Lorick, R. L. Ludwig, S. A. Pierre, J. P. Jensen, I. V. Davydov, P. Oberoi, C. C. Li, J. H. Kenten, J. A. Beutler, K. H. Vousden, and A. M. Weissman. 2007. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res. 679472-9481. [DOI] [PubMed] [Google Scholar]
- 54.Yu, G. Y., and M. M. Lai. 2005. The ubiquitin-proteasome system facilitates the transfer of murine coronavirus from endosome to cytoplasm during virus entry. J. Virol. 79644-648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yuwen, H., J. H. Cox, J. W. Yewdell, J. R. Bennink, and B. Moss. 1993. Nuclear localization of a double-stranded RNA-binding protein encoded by the vaccinia virus E3L gene. Virology 195732-744. [DOI] [PubMed] [Google Scholar]
- 56.Zou, P., J. Kawada, L. Pesnicak, and J. I. Cohen. 2007. Bortezomib induces apoptosis of Epstein-Barr virus (EBV)-transformed B cells and prolongs survival of mice inoculated with EBV-transformed B cells. J. Virol. 8110029-10036. [DOI] [PMC free article] [PubMed] [Google Scholar]