

Abstract
This report provides in vivo evidence for the posttranslational control of the acetyl coenzyme A (Ac-CoA) synthetase (AcsA) enzyme of Bacillus subtilis by the acuA and acuC gene products. In addition, both in vivo and in vitro data presented support the conclusion that the yhdZ gene of B. subtilis encodes a NAD+-dependent protein deacetylase homologous to the yeast Sir2 protein (also known as sirtuin). On the basis of this new information, a change in gene nomenclature, from yhdZ to srtN (for sirtuin), is proposed to reflect the activity associated with the YdhZ protein. In vivo control of B. subtilis AcsA function required the combined activities of AcuC and SrtN. Inactivation of acuC or srtN resulted in slower growth and cell yield under low-acetate conditions than those of the wild-type strain, and the acuC srtN strain grew under low-acetate conditions as poorly as the acsA strain. Our interpretation of the latter result was that both deacetylases (AcuC and SrtN) are needed to maintain AcsA as active (i.e., deacetylated) so the cell can grow with low concentrations of acetate. Growth of an acuA acuC srtN strain on acetate was improved over that of the acuA+ acuC srtN strain, indicating that the AcuA acetyltransferase enzyme modifies (i.e., inactivates) AcsA in vivo, a result consistent with previously reported in vitro evidence that AcsA is a substrate of AcuA.
Acetate is abundant in soil, the natural habitat of the gram-positive bacterium Bacillus subtilis. This short-chain fatty acid accumulates in soil and other environments because it is the product of many fermentative processes (7). Many prokaryotes use acetate as a source of carbon and energy. The first step in the utilization of acetate is its activation into acetyl coenzyme A (Ac-CoA). Once Ac-CoA is made, it is used to synthesize lipids, can enter the tricarboxylic acid cycle to generate precursors of amino acids, or can be oxidized to generate reducing power via diverse pathways (9, 15, 26, 57).
In B. subtilis, like in many other prokaryotes, acetate is excreted into the environment until the compound from which it is derived (e.g., glucose) is depleted. At that point, the acetate switch (55) is flipped, and acetate recapture is initiated (1, 10, 35).
Two pathways are central to acetate catabolism in prokaryotes. Both of these pathways activate acetate into Ac-CoA, but they do it via different intermediates and in response to different concentrations of acetate in the environment. The pathway relevant to this work is the one illustrated in reactions 1 and 2 below, both of which are catalyzed by the Ac-CoA synthetase (AcsA) EC 6.2.1.1 (49).
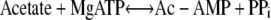 |
(1) |
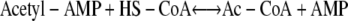 |
(2) |
The second pathway is comprised of the acetate kinase (AckA; EC 2.7.2.1) and the phosphotransacetylase (Pta; EC 2.3.1.8) enzymes (6). AckA catalyzes reaction 3, while Pta catalyzes reaction 4.
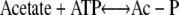 |
(3) |
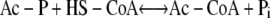 |
(4) |
Although both pathways are reversible, hydrolysis of pyrophosphate generated by reaction 1 prevents the AcsA from converting Ac-CoA to acetate. The role of these pathways is to carefully maintain a physiological balance of Ac-CoA and free CoA (13, 14, 55).
In B. subtilis, the Pta/AckA pathway appears to be a carbon overflow pathway used for the excretion of acetate (21). In contrast, the AcsA enzyme is preferentially used to capture acetate from the environment (53). The interplay of these acetate utilization pathways is carefully controlled at the transcription level by the global regulators CcpA and CodY. The latter represses acsA while activating ackA expression (34, 43, 44, 46).
Previous work from our laboratory identified some of the functions required for the posttranslational control of the B. subtilis AcsA enzyme in vitro. Enzymes encoded by the acuA and acuC genes were shown to inactivate and reactivate the Ac-CoA-forming activity of AcsA (17). In B. subtilis, the acuA and acuC loci are adjacent to the acsA gene. A gene of unknown function, acuB, is located between acuA and acuC (16, 17). In this report, we provide evidence that the AcsA enzyme of B. subtilis is posttranslationally regulated in vivo, and we identify an additional protein deacetylase enzyme involved in the control of the activity of AcsA.
At present, expression of acuABC genes appears to be responsive to conditions relevant to spore germination and outgrowth and during anaerobic respiration to nitrate (8). The data showing AcuA involvement in spore germination are relevant because they suggest that posttranslational modification is a mechanism used by the cell to control central metabolic functions other than AcsA.
The regulation of expression of the acuABC genes is complicated by the fact that the operon is under the control of the global regulatory protein CcpA, which, as mentioned above, also regulates the expression of the acsA gene. The tight regulation at the transcriptional level of the acsA gene and the effective posttranslational control of AcsA activity reflect on the importance of maintaining CoA homeostasis in this bacterium.
The posttranslational control system that regulates the AcsA enzyme of B. subtilis appears to be a variation of the one first described for Salmonella enterica, whose Acs enzyme is under acetylation control (48). In S. enterica, a protein acetyltransferase (Pat) enzyme homologous to the Gcn-5 protein of Saccharomyces cerevisiae (30, 50) uses Ac-CoA to acetylate a lysyl residue in the active site of Acs, which stops the first step of Ac-CoA formation (48) (reaction 1). The cognate deacetylase in S. enterica is a class III protein deacetylase that uses NAD+ as a substrate (not as a coenzyme), yielding O-acetyl-ADP-ribose (O-AADPR) and nicotinamide as by-products (25, 29, 39, 40, 51). Class III histone deacetylases are called sirtuins because they are homologous to the founding member of this family of proteins, the SIR2 protein of Saccharomyces cerevisiae (5, 36). In the gram-negative enterobacterium S. enterica, sirtuin-type deacetylase activity is associated with the CobB protein (45, 48, 52).
In this paper, we provide in vitro and in vivo evidence that the YhdZ protein of B. subtilis has sirtuin-like activity; that is, YhdZ has NAD+-dependent protein deacetylase activity. To avoid confusion generated by the use of a gene name that reflects unknown function (i.e., yhdZ), hereinafter we refer to yhdZ as srtN (for sirtuin). We present evidence that indicates that the posttranslational modification system controlling the B. subtilis AcsA enzyme is more complex than the one in S. enterica, because two deacetylases appear to be involved in the deacetylation of AcsAAc.
MATERIALS AND METHODS
Bacterial strains, media, and growth conditions.
Table 1 lists genotypes of strains of S. enterica serovar Typhimurium LT2 and B. subtilis SMY used in these studies. S. enterica strains were grown on no-carbon essential medium (2), supplemented with MgSO4 (1 mM) and l-methionine (0.5 mM). B. subtilis strains were grown on a modified Spizizen's minimal medium (47). The latter contained distilled water (90 ml), 10× Bacillus salts (10 ml; stock [wt/100 ml of water], sodium citrate [1 g], KH2PO4 [6 g], K2HPO4 [14 g], ammonium sulfate [2 g], MgSO4 [17 mg]), filter-sterilized trace elements (100 μl; stock [wt/100 ml of water], CaCl2 [5.5 mg]; FeCl2 [14 mg], MnCl2·4H2O [1 mg], ZnCl2 [2 mg], CuCl2·2H2O [0.4 mg], CoCl2·6H2O [0.6 mg], Na2MoO4 [0.6 mg], 1 M HCl [200 μl]), and glutamic acid (2 mg). A low concentration of acetate (10 mM for S. enterica, 30 mM for B. subtilis) or propionate (30 mM) was used as the sole carbon and energy source for cells grown in minimal medium. l-(+)-Arabinose (250 μM) or isopropyl-β-d-thiogalactoside (IPTG; 400 μM) was used to induce expression of plasmid-borne genes. Lysogenic broth (LB) was used as rich medium (3, 4). When used, antibiotics were present in the medium at the following concentrations: ampicillin, 100 μg/ml; erthythromycin, 1 μg/ml; chloramphenicol, 5 μg/ml; spectinomycin, 100 μg/ml; and kanamycin, 10 μg/ml. All chemicals were purchased from Sigma.
TABLE 1.
Strains and plasmids
| Strain or plasmid | Relevant genotype | Source or referencea |
|---|---|---|
| S. enterica serovar Typhimurium strains | ||
| LT2 | Wild type | |
| ER2566 | F− λ−fhuA2 [lon] ompT lacZ::T7 gene1 gal sulA11 Δ(mcrC-mrr)114::IS10 R(mcr-73::miniTn10-Tets)2 | New England BioLabs |
| R(zgb-210::Tn10) (Tets) endA1 [dcm] | ||
| LT2 derivative | ||
| TR6583 | metE205 ara-9 | K. Sanderson via J. Roth |
| TR6583 derivatives | ||
| JE4718 | cobB1176::Tn10d(tet+) pta102::MudI1734 | |
| JE7724 | cobB1176::Tn10d(tet+) pta102::MudI1734/pBAD30 | |
| JE7725 | cobB1176::Tn10d(tet+) pta102::MudI1734/pCOBB8 | |
| JE7726 | cobB1176::Tn10d(tet+) pta102::MudI1734/pSRTN1 | |
| B. subtilis strain | ||
| JE8607 | SMY (prototroph) | |
| SMY derivatives | ||
| JE8610 | acuC1::spc+ | |
| JE8611 | srtN4::spc+ | |
| JE9089 | acuC1::spc+srtN5::erm+ | |
| JE11111 | acuC1::spc+srtN5::erm+/pSRTN3 | |
| JE11281 | acuC1::spc+srtN5::erm+acuA2::kan+ | |
| Plasmids | ||
| pBAD30 | Expression vector, ParaBADbla+ | 11 |
| pHP13 | E. coli-B. subtilis shuttle vector, cat+ | A. L. Sonenshein |
| pDG646 | Antibiotic cassette harboring vector for B. subtilis, replicates in E. coli, bla+ in E. coli, erm+ in B. subtilis | BGSC |
| pDG783 | Antibiotic cassette harboring vector for B. subtilis, replicates in E. coli, bla+ in E. coli, kan+ in B. subtilis | BGSC |
| pDG1726 | Antibiotic cassette harboring vector for B. subtilis, replicates in E. coli, bla+ in E. coli, spc+ in B. subtilis | BGSC |
| pMUTIN4 | Pspac promoter harboring vector for B. subtilis, replicates in E. coli, bla+ | BGSC |
| pTYB12 | Purification vector, N-terminal self-cleavable intein tag, bla+ | New England BioLabs |
| pACUA2 | B. subtilis acuA+ cloned into pTYB12 bla+ | |
| pCOBB8 | S. enterica cobB+ cloned into pBAD30 | |
| pSRTN1 | B. subtilis srtN+ cloned into pBAD30 bla+ | |
| pSRTN2 | B. subtilis srtN+ cloned into pTYB12 bla+ | |
| pSRTN3 | B. subtilis srtN+ cloned into pHP13 | |
| Under the control of the Pspac promoter, cat+ |
Unless otherwise stated, strains were constructed during the course of this work. BGSC, Bacillus Genetic Stock Center.
Phage P22-mediated transduction of S. enterica genes.
Lysates of the high-transducing strain HT105/1 int-201 (41, 42) of bacteriophage P22 were used as donors to move chromosomal markers into specific genetic backgrounds. Protocols for P22-mediated transductions are described elsewhere (11).
Mobilization of plasmids.
Plasmids were introduced into S. enterica or B. subtilis strains by transformation or electroporation (23, 33, 37).
Construction of B. subtilis strains.
We replaced the acuC gene of B. subtilis strain SMY with a spectinomycin resistance cassette from plasmid pDG1726 (Bacillus Genetic Stock Center) using described PCR protocols (54). We used the same method to inactivate the srtN gene with either a spectinomycin or an erythromycin resistance cassette from plasmids pDG1726 or pDG646, respectively. The acuA gene was inactivated by the insertion of a kanamycin resistance gene, which was cut from plasmid pDG783 using restriction enzyme EcoRI (Promega). The fragment containing the kanamycin resistance marker was purified, blunted with DNA polymerase I Klenow fragments (Fermentas), and repurified with a commercially available kit (Qiagen). Plasmid pACUA2 was cut with enzyme SacI, which was cut only once and within the acuA gene. The linearized plasmid was purified and blunted with DNA polymerase I Klenow fragments. The kanamycin resistance marker was ligated into the linearized pACUA2 plasmid and transformed into E. coli strain DH5α, selecting for kanamycin resistance on rich medium containing kanamycin. Plasmid DNA was recovered from the kan+ transformants, and the acuA::kan+ construct was amplified and purified. The resulting 2.1-kb PCR fragment was transformed into strain JE9089 (B. subtilis acuC1::spc+ srtN5::erm+), selecting for kanamycin resistance. The presence of the acuA::kan+ insertion in the chromosome of the resulting strain was confirmed by PCR analysis.
PCR.
All amplifications used TripleMaster polymerase (Eppendorf) and were performed using an Eppendorf Mastercycler gradient PCR thermocycler (Brinkmann Instruments). Primers were purchased from Integrated DNA Technologies (see Table S1 in the supplemental material).
Cloning of B. subtilis genes.
Template DNA was obtained from B. subtilis strain SMY. For complementation studies with S. enterica, the srtN gene was cloned into the EcoRI and HindIII sites of plasmid pBAD30 (22).
For complementation studies with B. subtilis, the srtN gene was amplified from genomic DNA purified from B. subtilis SMY with primers that had NotI and SalI restrictions sites at the 5′ and 3′ ends of the gene, respectively. The srtN PCR product, which did not include its promoter, was purified and cut with NotI and SalI enzymes. The B. subtilis⇔E. coli shuttle plasmid pHP13 (a gift from A. L. Sonenshein) was cut with the HindIII and SalI enzymes and purified. The low-level, constitutive promoter Pspac drove expression of the srtN gene. Pspac was cut from plasmid pMUTIN4, using HindIII and SalI restriction enzymes. We sequentially cloned Pspac and srtN+ into plasmid pHP13, resulting in plasmid pSRTN3 (srtN+). For a scheme of the construction of plasmid pSRTN3, see Fig. S3 in the supplemental material. For the purpose of protein purification, the srtN+ gene was cloned into the NdeI and PstI sites of plasmid pTYB12 (New England BioLabs).
Protein overproduction and isolation. (i) SrtN.
SrtN protein was overproduced in E. coli strain ER2566 harboring plasmid pSRTN2 (pTYB12 srtN+). Cells were grown with shaking (180 rpm) at 30°C to an optical density at 650 nm of 0.4, at which point IPTG was added to a final concentration of 400 μM to induce expression of the srtN+ gene. After induction, cultures were incubated with shaking (180 rpm) at 18°C for 16 h. Cells were broken using a French press (Spectronic Unicam) under 1.26 kPa of pressure. Cell debris was removed by centrifugation at 12,000 × g for 45 min at 4°C in an Avanti J-25I centrifuge (Beckman-Coulter); clarified, crude cell extract was filtered using a 45-μm filter syringe. Crude cell extract was loaded onto a 5-ml chitin affinity chromatography column (New England BioLabs), which was developed per the manufacturer's instructions. Purified SrtN protein was dialyzed against HEPES buffer (0.05 M, pH 7.5) containing KCl (0.1 M) and glycerol (25%, vol/vol).
(ii) AcsA and GST-AcuA.
AcsA and glutathione S-transferase (GST)-AcuA proteins were purified using previously described protocols (16, 17). All proteins were drop frozen in liquid nitrogen and stored at −80°C until used.
(iii) PncA.
Nicotinamidase (PncA) was purified as described previously (18) and was a gift from A. Tucker.
Protein purity analysis.
Protein purity was assessed using Fotodyne's FOTO/Eclipse electronic documentation and analysis system, including software packages FOTO/Analyst PC Image v5.0 and TotalLab 1D gel analysis v2003 from NonLinear Dynamics, Ltd. This analysis was performed using sodium dodecyl sulfate (SDS)-polyacrylamide gels (27) stained with Coomassie blue (38).
Determination of the molecular mass of the SrtN protein.
The molecular mass of the SrtN protein was determined using fast protein liquid chromatography on a Superdex-200 column (Amersham) equilibrated and developed with 50 mM sodium phosphate buffer, pH 7.4, containing 0.15 M NaCl at a flow rate of 0.5 ml/min. A standard curve was generated using proteins of known molecular mass. For this purpose, we used gel filtration standard mixture (Bio-Rad Laboratories). The compensation volume was 0.5 ml, and the empty loop volume was 0.2 ml. Protein elution was monitored at 215 and 280 nm over a period of 60 min. A plot of molecular mass (kilodaltons) versus retention time (min) was generated using the above-mentioned protein standards. The standard curve was used to determine the approximate molecular mass and oligomeric state of active SrtN enzyme.
SrtN (sirtuin)-dependent protein deacetylation assay.
The GST-AcuA-dependent acetylation of the AcsA reaction was performed with a volume of 100 μl under described conditions (17). [14C,C-1]Ac-CoA (Moravek Biochemicals) was present in the reaction mixture at a specific radioactivity of 47 mCi/mmol; the concentration of Ac-CoA in the reaction mixture was 20 μM. After a 2-h incubation period at 37°C, GST-AcuA protein was removed from the reaction using 20 μl of a 50% (vol/vol) slurry of GST·Mag beads (Novagen) equilibrated with HEPES buffer (0.05 M, pH 7.5) containing tris(2-caboxyethyl)phosphine hydrochloride (TCEP; 200 μM). Incubation and removal of the magnetic beads was performed per the manufacturer's instructions. After removal of the beads, 185 pmol of purified SrtN deacetylase and 1 mM NAD+ were added to the sample. In some experiments, PncA nicotinamidase (128 pmol) was added to the reaction mixture to relieve SrtN inhibition by nicotinamide. Samples were incubated at 37°C for 2 h, and the reaction was stopped by precipitation with trichloroacetic acid to a final concentration to 5% (wt/vol). Reaction products were analyzed by SDS-polyacrylamide gel electrophoresis and phosphorimaging analysis.
RESULTS AND DISCUSSION
Although control of AcsA activity by acetylation has been reported, the supporting data were obtained in vitro or by heterologous complementation of S. enterica strains by B. subtilis genes provided in trans, but experiments with the appropriate B. subtilis strains were not performed (17).
Here, we present results from experiments performed with strains of B. subtilis with an altered AcsA posttranslational modification system. The data indicate that the AcuA protein is the acetyltransferase that modifies and inactivates AcsA, and AcuC is one of two deacetylases that reactivates AcsAAc. The other deacetylase is encoded by yhdZ, which we show in vivo and in vitro to be a class III NAD+-dependent sirtuin deacetylase. To reflect the sirtuin protein deacetylase activity associated with the YhdZ protein, we propose a change in gene nomenclature, from yhdZ to srtN.
SrtN compensates for the lack of CobB sirtuin during growth of a S. enterica cobB strain under low-acetate or -propionate conditions.
To obtain evidence that SrtN was a bona fide sirtuin, we performed heterologous complementation experiments using S. enterica strain JE4718 (cobB pta). We introduced plasmid pSRTN1 (srtN+) into strain JE4718, resulting in strain JE7726. Inactivation of the pta gene in JE7726 was needed to force the conversion of acetate or propionate to Ac-CoA or propionyl-CoA via the Acs enzyme. Because the lack of cobB function blocks deacetylation (hence activation) of AcsAc, growth of strain JE7726 on acetate or propionate would occur only if B. subtilis SrtN deacetylated AcsAc.
Arabinose-induced expression of the B. subtilis srtN+ gene restored growth of strain JE7726 on either acetate (Fig. 1B) or propionate (Fig. 1D). A S. enterica cobB strain, which could not deacetylate AcsAc (48), exhibited a partial defect in growth on acetate (Fig. 1A and B). As a positive control, we introduced plasmid pCOBB8 (cobB+) into strain JE4718. As expected, the expression of cobB+ restored growth on acetate in the presence or absence of arabinose (Fig. 1B). Expression of the B. subtilis srtN+ gene restored growth of the S. enterica cobB strain (Fig. 1A and B). But, unlike cobB+, higher expression of srtN+ was needed to support growth of the S. enterica cobB strain on acetate (Fig. 1C and D). It is unclear whether the need for induction of srtN+ reflects the stability of SrtN in S. enterica or differences in the interactions of SrtN with the S. enterica Acs protein. Regardless of the reason, the data indicate that SrtN can substitute for CobB during growth of a S. enterica cobB strain under low-acetate or -propionate conditions, supporting the conclusion that the B. subtilis SrtN protein is a sirtuin.
FIG. 1.

B. subtilis SrtN compensates for the lack of CobB during growth of a S. enterica cobB strain under low-acetate or -propionate conditions. Growth behavior of B. subtilis strains in 10 mM acetate (A and B) or 30 mM propionate (C and D). In panels B and D, the expression of plasmid-borne cobB+ and srtN+ was by the inclusion l+-arabinose (250 μM) in the medium. Open squares, JE4175 (wild type); open diamonds, JE7724 (cobB pta/vector); filled circles, JE7725 (cobB pta/pCOBBB8 cobB+); filled inverted triangles, JE7726 (cobB pta/pSRTN1 srtN+). On the graph, pcobB+ is pCOBB8, and psrtN+ is pSRTN1. The growth experiments were performed at least three times, and each experiment was done in triplicate. The data presented here have error bars showing variability; however, in most cases, they are smaller than the symbol. WT, wild type.
NAD+-dependent protein deacetylase (sirtuin) activity associated with SrtN.
The end-to-end homology shared by the SrtN and the S. enterica CobB proteins (see Fig. S1 in the supplemental material) also suggested SrtN was a sirtuin. We tested this idea in vitro using purified SrtN (>95% homogeneous), AcsA (>95% homogeneous), and GST-AcuA (>95% homogeneous) proteins. Radiolabeled AcsAAc protein was generated by incubation with AcuA and [14C,C-1]Ac-CoA. SrtN was added to the reaction mixture after removal of GST-AcuA, and the amount of label associated with AcsA was quantified by phosphorimaging. The data showed that the SrtN enzyme deacetylated about 50% of AcsAAc protein in the mixture and that deacetylation required NAD+ as a cosubstrate (Fig. 2, lane 3), as do other sirtuins (24, 28, 45). In addition, SrtN was inhibited by nicotinamide, another hallmark of sirtuins (Fig. 2, lane 4) (12, 19, 32); nicotinamide is the by-product of the SrtN reaction. The addition of nicotinamidase (PncA) increased SrtN activity to almost 72% (Fig. 2, lane 5), consistent with SrtN being a sirtuin. This amount of deacetylation was comparable to that of the AcuC protein deacetylase (17). Finally, like other sirtuins, B. subtilis SrtN was active as a monomer (see Fig. S2 in the supplemental material) (31, 58).
FIG. 2.

SrtN-dependent deacetylation of B. subtilis AcsAAc. The acetyltransferase reaction was performed as described in Materials and Methods. The acetyltransferase reaction mixture contained HEPES buffer (0.05 M, pH 7.5) and TCEP (200 μM) as a disulfide bond-reducing agent, GST-AcuA (3 μg [130 pmol]), and AcsA, (10 μg [170 pmol]). Amounts of protein added per 100-μl reaction mixture were as follows: for SrtN, 5 μg (185 pmol); and for PncA, 3 μg (128 pmol). SrtN-dependent AcsAAc deacetylation was assessed after removal of GST-AcuA from the reaction mixture. Deacetylation reaction mixtures were incubated at 37°C for 2 h and were stopped by precipitation with trichloroacetic acid. Reaction products were analyzed by SDS-polyacrylamide gel electrophoresis (PAGE), and quantification of AcsAAc in was performed by phosphorimaging analysis. Footnotes: 1, concentration in the reaction mixture; 2, digital light units (DLU) reflecting the amount of AscAAc; 3, normalized to DLU measured in the absence of SrtN. NMD, no measurable deacetylation.
In B. subtilis, SrtN and AcuC protein deacetylases are required for efficient growth of B. subtilis under low-acetate conditions.
Analysis of the functionality of AcsA in vivo necessitates the use of low concentrations of acetate (30 mM). Higher concentrations of acetate would be metabolized by the Pta/AckA pathway, obscuring the analysis of AcsA activity as a function of its acetylation state. Attempts to eliminate the contribution of the Pta/Ack pathway were not successful. Although we were able to construct pta acuC and pta srtN strains, all efforts to construct the pta acuC srtN strain failed, suggesting that pta acuC srtN is a synthetic lethal combination.
A disadvantage to using 30 mM acetate as the sole source of carbon and energy is that the cell yield is low, and differences in the growth rate are not dramatic. However, with the appropriate controls, one can discern the effect caused by the absence of the AcuC and SrtN functions. We found that the use of viable counts was the best approach to analyze the roles of AcuA, AcuC, and SrtN function during growth under low-acetate conditions. Relative to the wild-type strain, a strain lacking acuC displayed a reproducible and statistically significant growth defect under low-acetate conditions (Fig. 3). The observed growth defect was substantially more pronounced in the acuC srtN strain (Fig. 3), suggesting that the srtN and acuC functions were needed to maintain AcsA as active (i.e., deacetylated). If this idea were correct, one would expect the growth behavior of the srtN acuC strain to be similar to that of the acsA strain, and it was (Fig. 3). Expression of the srtN+ in trans under the control of the low-level constitutive Pspac promoter restored growth of the srtN acuC strain on acetate to the level seen in the acuC strain. As expected, the rate of growth of the srtN acuC/psrtN+ culture was similar to that of the srtN acuC+ culture and slower than that of the wild-type strain (Fig. 3). Based on these data, we concluded that efficient growth of B. subtilis on acetate required both AcuC and SrtN deacetylase activities.
FIG. 3.

Growth of Bacillus subtilis strains on 30 mM acetate. The effects of the acetylation/deacetylation mutations on cell growth were measured by the change in cell number and the growth rate. For a statistical analysis of these growth data, please see Table S2 in the supplemental material. WT, wild type.
We also predicted that inactivation of acuA in the acuC srtN strain would result in an unmodified (active) AcsA protein and that growth would improve. As predicted, growth of the acuC srtN acuA strain on acetate did improve, but it did not match that of the wild-type strain. The fact that inactivation of acuA did not restore wild-type growth of the acuA acuC srtN strain under low-acetate conditions suggests that the absence of AcuC and/or SrtN may affect the function of yet-to-be-identified proteins.
In summary, the posttranslational modification system that controls the activity of the Ac-CoA synthetase enzyme in the gram-positive bacterium B. subtilis is more complex than the previously studied one in the gram-negative bacterium S. enterica. Notable differences between the two systems include the following. (i) The genes encoding the protein acetyltransferase and one deacetylase enzymes may be cotranscribed in B. subtilis, but not in S. enterica. (ii) The B. subtilis acuABC genes are under transcriptional control of a global regulator responsive to the quality of the carbon source available to the cell (20, 21). At present, the transcriptional control of the S. enterica pat and cobB genes have not been reported. (iii) Unlike S. enterica, B. subtilis uses two different types of deacetylases to control AcsA activity, one whose activity depends on NAD+ (SrtN) and another whose activity does not (AcuC) (Fig. 4). Bioinformatics searches for other protein deacetylases in B. subtilis failed to identify any orthologues of acuC and srtN, suggesting that if there are other protein deacetylases in this bacterium, they may belong to a new class of this family of enzymes (56).
FIG. 4.

Schematic of the control of AcsA by the acetylation/deacetylation system of B. subtilis. While the data show that AcuA is the only acetyltransferase that can acetylate (deactivate) AcsA, the deacetylation (reactivation) step can be catalyzed by the SrtN or AcuC deacetylases. The physiological conditions under which each of the deacetylases is preferentially active are presently unknown. CoASH, free CoA.
It is not obvious why B. subtilis would require redundancy in the deacetylases that control AcsA activity. The obvious difference between AcuC and SrtN functions is that the latter requires NAD+, while the former does not use NAD+ as cosubstrate (17). We speculate that the involvement of AcuC and SrtN in AcsAAc reactivation may occur in response to different indicators of the energy charge of the cell. AcuC-dependent activation of AcsAc may be triggered in response to a low Ac-CoA:CoA ratio, allowing the cell to replenish Ac-CoA to levels needed for survival. The involvement of SrtN may be triggered when the NAD+:NADH ratio becomes high, a signal of high energy charge in the cell. Under such physiological conditions (high-energy and low-Ac-CoA conditions), the cell would need to meet the demand for Ac-CoA; thus, it would use SrtN to activate AcsAAc. These ideas are currently under investigation.
Supplementary Material
Acknowledgments
This work was supported by PHS grant R01 GM62203 to J.C.E.-S.
We are thankful to A. L. Sonenshein (Tufts University) for advice and materials.
Footnotes
Published ahead of print on 9 January 2009.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Ali, N. O., J. Bignon, G. Rapoport, and M. Debarbouille. 2001. Regulation of the acetoin catabolic pathway is controlled by sigma L in Bacillus subtilis. J. Bacteriol. 1832497-2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berkowitz, D., J. M. Hushon, H. J. Whitfield, Jr., J. Roth, and B. N. Ames. 1968. Procedure for identifying nonsense mutations. J. Bacteriol. 96215-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bertani, G. 2004. Lysogeny at mid-twentieth century: P1, P2, and other experimental systems. J. Bacteriol. 186595-600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bertani, G. 1951. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J. Bacteriol. 62293-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brachmann, C. B., J. M. Sherman, S. E. Devine, E. E. Cameron, L. Pillus, and J. D. Boeke. 1995. The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression, and chromosome stability. Genes Dev. 92888-2902. [DOI] [PubMed] [Google Scholar]
- 6.Brown, T. D., M. C. Jones-Mortimer, and H. L. Kornberg. 1977. The enzymic interconversion of acetate and acetyl-coenzyme A in Escherichia coli. J. Gen. Microbiol. 102327-336. [DOI] [PubMed] [Google Scholar]
- 7.Buckel, W. 1999. Anaerobic energy metabolism, p. 278-326. In J. W. Lengler, G. Drews, and H. G. Chlegel (ed.), Biology of the procaryotes. Thieme, Sttutgart, Germany.
- 8.Clements, L. D., U. N. Streips, and B. S. Miller. 2002. Differential proteomic analysis of Bacillus subtilis nitrate respiration and fermentation in defined medium. Proteomics 21724-1734. [DOI] [PubMed] [Google Scholar]
- 9.Cronan, J., Jr., and D. LaPorte. November 2006, posting date. Chapter 3.5.2, Tricarboxylic acid cycle and glyoxylate bypass. In R. Curtiss III et al. (ed.), EcoSal—Escherichia coli and Salmonella: cellular and molecular biology. ASM Press, Washington, DC. http://www.ecosal.org.
- 10.Cruz Ramos, H., T. Hoffmann, M. Marino, H. Nedjari, E. Presecan-Siedel, O. Dreesen, P. Glaser, and D. Jahn. 2000. Fermentative metabolism of Bacillus subtilis: physiology and regulation of gene expression. J. Bacteriol. 1823072-3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis, R. W., D. Botstein, and J. R. Roth. 1980. A manual for genetic engineering: advanced bacterial genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 12.Denu, J. M. 2005. Vitamin B3 and sirtuin function. Trends Biochem. Sci. 30479-483. [DOI] [PubMed] [Google Scholar]
- 13.El-Mansi, M. 2005. Free CoA-mediated regulation of intermediary and central metabolism: an hypothesis which accounts for the excretion of alpha-ketoglutarate during aerobic growth of Escherichia coli on acetate. Res. Microbiol. 156874-879. [DOI] [PubMed] [Google Scholar]
- 14.El-Mansi, M., A. J. Cozzone, J. Shiloach, and B. J. Eikmanns. 2006. Control of carbon flux through enzymes of central and intermediary metabolism during growth of Escherichia coli on acetate. Curr. Opin. Microbiol. 9173-179. [DOI] [PubMed] [Google Scholar]
- 15.Erb, T. J., I. A. Berg, V. Brecht, M. Muller, G. Fuchs, and B. E. Alber. 2007. Synthesis of C5-dicarboxylic acids from C2-units involving crotonyl-CoA carboxylase/reductase: the ethylmalonyl-CoA pathway. Proc. Natl. Acad. Sci. USA 10410631-10636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gardner, J. G., and J. C. Escalante-Semerena. 2008. Biochemical and mutational analyses of AcuA, the acetyltransferase enzyme that controls the activity of the acetyl coenzyme a synthetase (AcsA) in Bacillus subtilis. J. Bacteriol. 1905132-5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gardner, J. G., F. J. Grundy, T. M. Henkin, and J. C. Escalante-Semerena. 2006. Control of acetyl-coenzyme A synthetase (AcsA) activity by acetylation/deacetylation without NAD+ involvement in Bacillus subtilis. J. Bacteriol. 1885460-5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garrity, J., J. G. Gardner, W. Hawse, C. Wolberger, and J. C. Escalante-Semerena. 2007. N-lysine propionylation controls the activity of propionyl-CoA synthetase. J. Biol. Chem. 28230239-30245. [DOI] [PubMed] [Google Scholar]
- 19.Grubisha, O., B. C. Smith, and J. M. Denu. 2005. Small molecule regulation of Sir2 protein deacetylases. FEBS J. 2724607-4616. [DOI] [PubMed] [Google Scholar]
- 20.Grundy, F. J., A. J. Turinsky, and T. M. Henkin. 1994. Catabolite regulation of Bacillus subtilis acetate and acetoin utilization genes by CcpA. J. Bacteriol. 1764527-4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grundy, F. J., D. A. Waters, T. Y. Takova, and T. M. Henkin. 1993. Identification of genes involved in utilization of acetate and acetoin in Bacillus subtilis. Mol. Microbiol. 10259-271. [DOI] [PubMed] [Google Scholar]
- 22.Guzman, L. M., D. Belin, M. J. Carson, and J. Beckwith. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 1774121-4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harwood, C. R., and S. M. Cutting. 1990. Molecular biological methods for Bacillus. John Wiley & Sons, New York, NY.
- 24.Imai, S., C. M. Armstrong, M. Kaeberlein, and L. Guarente. 2000. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403795-800. [DOI] [PubMed] [Google Scholar]
- 25.Jackson, M. D., and J. M. Denu. 2002. Structural identification of 2′- and 3′-O-acetyl-ADP-ribose as novel metabolites derived from the Sir2 family of beta-NAD+-dependent histone/protein deacetylases. J. Biol. Chem. 27718535-18544. [DOI] [PubMed] [Google Scholar]
- 26.Kornberg, H. L. 1966. The role and control of the glyoxylate cycle in Escherichia coli. Biochem. J. 991-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227680-685. [DOI] [PubMed] [Google Scholar]
- 28.Landry, J., J. T. Slama, and R. Sternglanz. 2000. Role of NAD+ in the deacetylase activity of the SIR2-like proteins. Biochem. Biophys. Res. Commun. 278685-690. [DOI] [PubMed] [Google Scholar]
- 29.Lee, S., L. Tong, and J. M. Denu. 2008. Quantification of endogenous sirtuin metabolite O-acetyl-ADP-ribose. Anal. Biochem. 383174-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marmorstein, R., and S. Y. Roth. 2001. Histone acetyltransferases: function, structure, and catalysis. Curr. Opin. Genet. Dev. 111555-1561. [DOI] [PubMed] [Google Scholar]
- 31.Min, J., J. Landry, R. Sternglanz, and R. M. Xu. 2001. Crystal structure of a SIR2 homolog-NAD complex. Cell 105269-279. [DOI] [PubMed] [Google Scholar]
- 32.Neugebauer, R. C., U. Uchiechowska, R. Meier, H. Hruby, V. Valkov, E. Verdin, W. Sippl, and M. Jung. 2008. Structure-activity studies on splitomicin derivatives as sirtuin inhibitors and computational prediction of binding mode. J. Med. Chem. 511203-1213. [DOI] [PubMed] [Google Scholar]
- 33.O'Toole, G. A., M. R. Rondon, and J. C. Escalante-Semerena. 1993. Analysis of mutants of defective in the synthesis of the nucleotide loop of cobalamin. J. Bacteriol. 1753317-3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ratnayake-Lecamwasam, M., P. Serror, K. W. Wong, and A. L. Sonenshein. 2001. Bacillus subtilis CodY represses early-stationary-phase genes by sensing GTP levels. Genes Dev. 151093-1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Renna, M. C., N. Najimudin, L. R. Winik, and S. A. Zahler. 1993. Regulation of the Bacillus subtilis alsS, alsD, and alsR genes involved in post-exponential-phase production of acetoin. J. Bacteriol. 1753863-3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rine, J., and I. Herskowitz. 1987. Four genes responsible for a position effect on expression from HML and HMR in Saccharomyces cerevisiae. Genetics 1169-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ryu, J., and R. J. Hartin. 1990. Quick transformation in Salmonella typhimurium LT2. BioTechniques 843-45. [PubMed] [Google Scholar]
- 38.Sasse, J. 1991. Detection of proteins, p. 10.6.1-10.6.8. In F. A. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.), Current protocols in molecular biology, vol. 1. Wiley Interscience, New York, NY. [Google Scholar]
- 39.Sauve, A. A., I. Celic, J. Avalos, H. Deng, J. D. Boeke, and V. L. Schramm. 2001. Chemistry of gene silencing: the mechanism of NAD+-dependent deacetylation reactions. Biochemistry 4015456-15463. [DOI] [PubMed] [Google Scholar]
- 40.Sauve, A. A., C. Wolberger, V. L. Schramm, and J. D. Boeke. 2006. The biochemistry of sirtuins. Annu. Rev. Biochem. 75435-465. [DOI] [PubMed] [Google Scholar]
- 41.Schmieger, H. 1971. A method for detection of phage mutants with altered transducing ability. Mol. Gen. Genet. 110378-381. [DOI] [PubMed] [Google Scholar]
- 42.Schmieger, H., and H. Backhaus. 1973. The origin of DNA in transducing particles in P22-mutants with increased transduction-frequencies (HT-mutants). Mol. Gen. Genet. 120181-190. [DOI] [PubMed] [Google Scholar]
- 43.Serror, P., and A. L. Sonenshein. 1996. CodY is required for nutritional repression of Bacillus subtilis genetic competence. J. Bacteriol. 1785910-5915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shivers, R. P., S. S. Dineen, and A. L. Sonenshein. 2006. Positive regulation of Bacillus subtilis ackA by CodY and CcpA: establishing a potential hierarchy in carbon flow. Mol. Microbiol. 62811-822. [DOI] [PubMed] [Google Scholar]
- 45.Smith, J. S., C. B. Brachmann, I. Celic, M. A. Kenna, S. Muhammad, V. J. Starai, J. L. Avalos, J. C. Escalante-Semerena, C. Grubmeyer, C. Wolberger, and J. D. Boeke. 2000. A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc. Natl. Acad. Sci. USA 976658-6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sonenshein, A. L. 2007. Control of key metabolic intersections in Bacillus subtilis. Nat. Rev. Microbiol. 5917-927. [DOI] [PubMed] [Google Scholar]
- 47.Spizizen, J. 1958. Transformation of biochemically deficient strains of Bacillus subtilis by deoxyribonucleate. Proc. Natl. Acad. Sci. USA 441072-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Starai, V. J., I. Celic, R. N. Cole, J. D. Boeke, and J. C. Escalante-Semerena. 2002. Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science 2982390-2392. [DOI] [PubMed] [Google Scholar]
- 49.Starai, V. J., and J. C. Escalante-Semerena. 2004. Acetyl-coenzyme A synthetase (AMP forming). Cell. Mol. Life Sci. 612020-2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sternglanz, R., and H. Schindelin. 1999. Structure and mechanism of action of the histone acetyltransferase Gcn5 and similarity to other N-acetyltransferases. Proc. Natl. Acad. Sci. USA 968807-8808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tanner, K. G., J. Landry, R. Sternglanz, and J. M. Denu. 2000. Silent information regulator family of NAD-dependent histone/protein deacetylases generates a unique product, 1-O-acetyl-ADP-ribose. Proc. Natl. Acad. Sci. USA 9714178-14182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsang, A. W., and J. C. Escalante-Semerena. 1998. CobB, a new member of the SIR2 family of eucaryotic regulatory proteins, is required to compensate for the lack of nicotinate mononucleotide:5,6-dimethylbenzimidazole phosphoribosyltransferase activity in cobT mutants during cobalamin biosynthesis in Salmonella typhimurium LT2. J. Biol. Chem. 27331788-31794. [DOI] [PubMed] [Google Scholar]
- 53.Turinsky, A. J., T. R. Moir-Blais, F. J. Grundy, and T. M. Henkin. 2000. Bacillus subtilis ccpA gene mutants specifically defective in activation of acetoin biosynthesis. J. Bacteriol. 1825611-5614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wach, A. 1996. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast 12259-265. [DOI] [PubMed] [Google Scholar]
- 55.Wolfe, A. J. 2005. The acetate switch. Microbiol. Mol. Biol. Rev. 6912-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang, X. J., and E. Seto. 2007. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 265310-5318. [DOI] [PubMed] [Google Scholar]
- 57.Zarzycki, J., A. Schlichting, N. Strychalsky, M. Muller, B. E. Alber, and G. Fuchs. 2008. Mesaconyl-coenzyme A hydratase, a new enzyme of two central carbon metabolic pathways in bacteria. J. Bacteriol. 1901366-1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao, K., X. Chai, and R. Marmorstein. 2003. Structure of the yeast Hst2 protein deacetylase in ternary complex with 2′-O-acetyl ADP ribose and histone peptide. Structure 111403-1411. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.