

Abstract
The Escherichia coli polynucleotide phosphorylase (PNPase; encoded by pnp), a phosphorolytic exoribonuclease, posttranscriptionally regulates its own expression at the level of mRNA stability and translation. Its primary transcript is very efficiently processed by RNase III, an endonuclease that makes a staggered double-strand cleavage about in the middle of a long stem-loop in the 5′-untranslated region. The processed pnp mRNA is then rapidly degraded in a PNPase-dependent manner. Two non-mutually exclusive models have been proposed to explain PNPase autogenous regulation. The earlier one suggested that PNPase impedes translation of the RNase III-processed pnp mRNA, thus exposing the transcript to degradative pathways. More recently, this has been replaced by the current model, which maintains that PNPase would simply degrade the promoter proximal small RNA generated by the RNase III endonucleolytic cleavage, thus destroying the double-stranded structure at the 5′ end that otherwise stabilizes the pnp mRNA. In our opinion, however, the first model was not completely ruled out. Moreover, the RNA decay pathway acting upon the pnp mRNA after disruption of the 5′ double-stranded structure remained to be determined. Here we provide additional support to the current model and show that the RNase III-processed pnp mRNA devoid of the double-stranded structure at its 5′ end is not translatable and is degraded by RNase E in a PNPase-independent manner. Thus, the role of PNPase in autoregulation is simply to remove, in concert with RNase III, the 5′ fragment of the cleaved structure that both allows translation and prevents the RNase E-mediated PNPase-independent degradation of the pnp transcript.
Posttranscriptional regulation at the level of mRNA stability and translatability is acknowledged as an important step in gene expression control (for reviews, see references 8 and 25). An interesting system for the study of the interplay between mRNA translation and decay may be the regulation of pnp gene expression. The pnp gene encodes polynucleotide phosphorylase (PNPase), one of the major players in Escherichia coli mRNA degradation. The enzyme is widely conserved among members of the class Bacteria, and homologous genes have been found in plants and human cells (reviewed in references 3, 29, and 51). In E. coli, PNPase is a homotrimeric protein of a 711-amino-acid polypeptide with a structural core, which contains the catalytic domain, and two C-terminal RNA-binding domains, KH and S1 (5, 38). The three subunits associate via trimerization interfaces of the core domain, forming a central channel (55). In vitro, PNPase catalyzes the processive 3′-to-5′ phosphorolytic degradation of RNA, the reverse polymerization reaction, and the exchange reaction between free phosphate and the β-phosphate of ribonucleoside diphosphates (7, 19, 59). PNPase can also bind RNA via its two RNA-binding domains (34, 47, 54). In vivo, the enzyme has been shown to be implicated in both degradation and polyadenylation of RNAs (13, 36, 37) and may be found as a component of a multiprotein machine, the RNA degradosome, together with the endonuclease RNase E, the DEAD-box RNA helicase RhlB, and enolase (6, 35, 42).
The pnp gene is located downstream of rpsO, which codes for the ribosomal protein S15 (40). Transcription of pnp can originate from two different promoters, rpsOp (also referred to as P1), upstream of rpsO, and pnp-p (P2), located between rpsO and pnp (41). Both primary transcripts are processed by RNase III, which cleaves the RNAs 37 (site 1) and 75 (site 2) nucleotides downstream of the pnp-p transcription start point within a long hairpin structure in the 5′-untranslated region (5′-UTR) (39, 43, 46, 58). This originates a monocistronic mRNA 2.25 kb long, which extends from RNase III site 2 to the pnp-t Rho-independent terminator, and longer transcripts terminating downstream (39, 57, 62). Transcripts originating at rpsOp are cleaved, in addition, by RNase E in the rpsO-pnp intergenic region, and the RpsO-encoding mRNAs undergo independent processing and degradation (21, 44-46; see Fig. S1 in the supplemental material).
PNPase posttranscriptionally regulates its own expression by controlling the stability of the RNase III-processed mRNA, as the processed transcript, in the presence of PNPase, becomes very unstable (48, 49). Two models have been proposed by C. Portier and collaborators to mechanistically explain PNPase autogenous regulation. The former one, proposed in 1994 (49), postulates that PNPase can bind determinants in the 5′-UTR of pnp mRNA; this would inhibit translation, thus promoting degradation of the transcript (39, 48, 49). The current model proposed in 2001 maintains that the duplex formed by the 37-nucleotide (nt)-long RNA (RNA37) upstream of the proximal RNase III cleavage site 1 and the new 5′-monophosphate end of the mature pnp mRNA protects the transcript from degradation. PNPase would trigger pnp mRNA decay by degrading the small RNA37 from the short 3′ overhang (23).
In our view, the first model has not been completely ruled out by the second one, as the issue of translational repression was not further addressed by Jarrige et al. (23). It thus remains possible that these two non-mutually exclusive mechanisms operate in concert to autoregulate PNPase expression.
In this work, through the analysis of mutants in the leader region, we provide further support to the 2001 model and provide evidence that the putative translational control by PNPase does not play a role in autoregulation. Moreover, we show that the native pnp mRNA with its long 5′-UTR is stable and translatable, whereas the processed transcript devoid of its 5′ double-strand structure is poorly, if at all, translated and is rapidly degraded by RNase E, the major endoribonuclease implicated in the degradation of mRNAs.
MATERIALS AND METHODS
Bacterial strains and plasmids.
The bacteria and plasmids are listed in Tables 1 and 2, respectively. The mutant pnp alleles created in this work (Table 3) were obtained either directly by PCR or by conventional in vitro recombination in an intermediate plasmid as detailed below. A linear mutant DNA fragment (either as a purified PCR product or recovered from the intermediate plasmid) was transferred by electroporation into the recipient strain harboring pKD46, a replication-thermosensitive plasmid expressing bla (Ampr) and the λ Red recombination system, as described previously (9). Coordinates of the pnp locus are given, throughout, relative to the pnp-p transcription start point (23), conventionally defined as +1, corresponding to coordinate G7947 in NCBI accession no. ECAE000397 and to coordinate 3309346 in the complete E. coli sequence (NCBI accession no. U00096.2) on the complementary strand. pGEX104, the parental plasmid in which all the mutations in the pnp 5′ and 3′ regions have been constructed, harbors a DNA fragment delimited by EcoRI-KpnI that contains the pnp regions −201 to +249 and 2239 to 2589 separated by a cat cassette (Δpnp-871::cat allele). In Δpnp-871::cat, an amplicon obtained by PCR of plasmid pKD13 with primers FG1701 and FG1702 replaces the 248-2238 pnp region. The mutations in the 5′ and 3′ regions have been obtained by replacing, respectively, the EcoRI-DraIII and XbaI-KpnI regions of the pGEX104 pnp insert with equivalent DNA fragments harboring the specific mutations obtained by three-step PCR (30) using appropriate oligonucleotides (Table 4). Construction of the primary single and double pnp mutants is outlined in Fig. 1. To obtain the 5′-end mutants in both the leader and the coding regions, C-5705/pKD46, which contains the rpsOt::aph (Kanr) allele (an insertion of the aph cassette at −60) was transformed with the entire EcoRI-KpnI mutant inserts. Camr transformants were screened for the loss of the rpsOt::aph marker. FLP-mediated excision of resistance cassettes from the mutants transformed with pCP20 was performed as described previously (9). The deletion of the pnp-p terminator was obtained by selecting Camr recombinants of C-1a/pKD46 transformed with the pGEX1010 insert, which harbors the Δpnp-1010t deletion (positions 2309 to 2328) of the pnp Rho-independent terminator obtained by three-step PCR (30) using oligonucleotides FG1802 and FG1803. Camr transformants were screened by PCR to detect pnp-t deletion. The presence of each mutation was confirmed by sequencing suitable DNA fragments obtained by PCR.
TABLE 1.
Genotypes of the bacterial strains used in this study
| Strain | Genotype | Source or reference |
|---|---|---|
| BZ31 zce-726::Tn10 | his ΔtrpE5 mukB106 smbB-rne-131 zce-726::Tn10 (λ) | Kindly provided by M. Dreyfus |
| C-1a | E. coli C, prototrophic | 52 |
| C-5684 | Δrnc38::kan | By P1*SK7622 transduction into C-1a (this work) |
| C-5686 | Δrne-131∼Tn10 | By P1*BZ31 zce-726::Tn10 transduction into C-1a (this work) |
| C-5705 | rpsOt::aph | From C-1a/pKD46 by λ Red-mediated recombination with DNA fragment obtained by PCR of pKD13 with primers FG1701 and FG1702; aph cassette inserted at −60 from pnp-p into rpsO transcription terminator (this work) |
| C-5729 | Δpnp-871::cat | From C-5705/pKD46 by λ Red-mediated recombination with pGEX104 insert (this work) |
| C-5731 | Δpnp-871 | From C-5729/pCP20 by FLP-mediated excision of cat cassette (this work) |
| C-5801 | ΔpnpL1001 Δpnp-871::cat | From C-5705/pKD46 by λ Red-mediated recombination with pGEX1001 insert (this work) |
| C-5802 | ΔpnpL1002 Δpnp-871::cat | From C-5705/pKD46 by λ Red-mediated recombination with pGEX1002 insert (this work) |
| C-5803 | ΔpnpL1003 Δpnp-871::cat | From C-5705/pKD46 by λ Red-mediated recombination with pGEX1003 insert (this work) |
| C-5804 | ΔpnpL1004 Δpnp-871::cat | From C-5705/pKD46 by λ Red-mediated recombination with pGEX1004 insert (this work) |
| C-5805 | ΔpnpL1005 Δpnp-871::cat | From C-5705/pKD46 by λ Red-mediated recombination with pGEX1005 insert (this work) |
| C-5806 | ΔpnpL1006 Δpnp-871::cat | From C-5705/pKD46 by λ Red-mediated recombination with pGEX1006 insert (this work) |
| C-5807 | pnpL1007 Δpnp-871::cat | From C-5705/pKD46 by λ Red-mediated recombination with pGEX1007 insert (this work) |
| C-5808 | pnpL1008 Δpnp-871::cat | From C-5705/pKD46 by λ Red-mediated recombination with pGEX1008 insert (this work) |
| C-5809 | pnpL1009 Δpnp-871::cat | From C-5705/pKD46 by λ Red-mediated recombination with pGEX1009 insert (this work) |
| C-5810 | Δpnp-1010t Δpnp-871::cat | From C-5705/pKD46 by λ Red-mediated recombination with pGEX1010 insert (this work) |
| C-5811 | ΔpnpL1011 Δpnp-871::cat | From C-5705/pKD46 by λ Red-mediated recombination with pGEX1011 insert (this work) |
| C-5812 | ΔpnpL1012 Δpnp-871::cat | From C-5705/pKD46 by λ Red-mediated recombination with pGEX1012 insert (this work) |
| C-5813 | ΔpnpL1013 Δpnp-871::cat | From C-5705/pKD46 by λ Red-mediated recombination with pGEX1013 insert (this work) |
| C-5814 | ΔpnpL1014 Δpnp-871::cat | From C-5705/pKD46 by λ Red-mediated recombination with pGEX1014 insert (this work) |
| C-5815 | ΔpnpL1015 Δpnp-871::cat | From C-5705/pKD46 by λ Red-mediated recombination with pGEX1015 insert (this work) |
| C-5851 | ΔpnpL1001 Δpnp-871 | From C-5801/pCP20 by FLP-mediated excision (this work) |
| C-5852 | ΔpnpL1002 Δpnp-871 | From C-5802/pCP20 by FLP-mediated excision (this work) |
| C-5853 | ΔpnpL1003 Δpnp-871 | From C-5803/pCP20 by FLP-mediated excision (this work) |
| C-5854 | ΔpnpL1004 Δpnp-871 | From C-5804/pCP20 by FLP-mediated excision (this work) |
| C-5855 | ΔpnpL1005 Δpnp-871 | From C-5805/pCP20 by FLP-mediated excision (this work) |
| C-5856 | ΔpnpL1006 Δpnp-871 | From C-5806/pCP20 by FLP-mediated excision (this work) |
| C-5857 | pnpL1007 Δpnp-871 | From C-5807/pCP20 by FLP-mediated excision (this work) |
| C-5858 | pnpL1008 Δpnp-871 | From C-5808/pCP20 by FLP-mediated excision (this work) |
| C-5859 | pnpL1009 Δpnp-871 | From C-5809/pCP20 by FLP-mediated excision (this work) |
| C-5860 | Δpnp-1010t Δpnp-871 | From C-5810/pCP20 by FLP-mediated excision (this work) |
| C-5861 | ΔpnpL1011 Δpnp-871 | From C-5811/pCP20 by FLP-mediated excision (this work) |
| C-5862 | ΔpnpL1012 Δpnp-871 | From C-5812/pCP20 by FLP-mediated excision (this work) |
| C-5863 | ΔpnpL1013 Δpnp-871 | From C-5813/pCP20 by FLP-mediated excision (this work) |
| C-5864 | ΔpnpL1014 Δpnp-871 | From C-5814/pCP20 by FLP-mediated excision (this work) |
| C-5865 | ΔpnpL1015 Δpnp-871 | From C-5815/pCP20 by FLP-mediated excision (this work) |
| C-5868 | rne-3071∼Tn10 | By P1*KG241 transduction into C-1a, Tetr selection, screening for TS; presence of rne-3071 confirmed by sequencing |
| C-5869 | rne+∼Tn10 | By P1*KG241 transduction into C-1a, Tetr selection, screening for TS+; presence of rne+ confirmed by sequencing |
| C-5870 | Δpnp-871 rne-3071∼Tn10 | By P1*KG241 transduction into C-5731, Tetr selection, screening for TS; presence of rne-3071 confirmed by sequencing |
| C-5871 | Δpnp-871 Tn10 | By P1*KG241 transduction into C-5731, Tetr selection, screening for TS+; presence of rne+ confirmed by sequencing |
| DH10B | F−mcrA Δ(mrr-hsdRMS-mcrBC) φ80dlacZΔM15 ΔlacX74 deoR recA1 araD139 Δ(ara-leu)7697 galU galK rpsL endA1 nupG | 32 |
| KG241 | rne-3071∼Tn10 | By P1*BZ31 into N3431, Tetr selection, screening for TS; presence of rne-3071 confirmed by sequencing |
| MRE600 | rna | 60 |
| N-3431 | lacZ43 λ−relA1 spoT1 rne-3071 | 18 |
| N-3433 | lacZ43 λ−relA1 spoT1 thi-1 | 18 |
| SK7622 | thyA715 rph-1 rncΔ38::kan | 1 (kindly provided by S. Kushner) |
TABLE 2.
Characteristics of the plasmids used in this study
| Plasmid | Relevant characteristics | Reference |
|---|---|---|
| pAZ76 | pGM743 derivative; harbors 1-380 pnp operon region under T7 promoter | This work |
| pAZ77 | pGM743 derivative; harbors 76-380 pnp operon region under T7 promoter | This work |
| pAZ101 | pGZ119HE derivative; pnp+ | 47 |
| pAZ133 | pAZ101 derivative; harbors Δpnp-833 allele that encodes KHS1-truncated PNPase | 4 |
| pAZ133L1001 | Harbors ΔpnpL1001 Δpnp-833 double mutation | This work |
| pAZ133L1002 | Harbors ΔpnpL1002 Δpnp-833 double mutation | This work |
| pCP20 | Thermosensitive replication, thermoinducible FLP (FRT-specific) recombinase; Ampr Camr | 9 |
| pGEX-1 | Cloning vector; Ampr | 53 |
| pGEX104 | pGEX-1 derivative; harbors pnpL+ Δpnp-871::cat | This work |
| pGEX1001 | pGEX104 derivative; harbors ΔpnpL1001 Δpnp-871::cat | This work |
| pGEX1002 | pGEX104 derivative; harbors ΔpnpL1002 Δpnp-871::cat | This work |
| pGEX1003 | pGEX104 derivative; harbors ΔpnpL1003 Δpnp-871::cat | This work |
| pGEX1004 | pGEX104 derivative; harbors ΔpnpL1004 Δpnp-871::cat | This work |
| pGEX1005 | pGEX104 derivative; harbors Δpnp-1005 Δpnp-871::cat | This work |
| pGEX1006 | pGEX104 derivative; harbors Δpnp-1006 Δpnp-871::cat | This work |
| pGEX1007 | pGEX104 derivative; harbors pnp-1007 Δpnp-871::cat | This work |
| pGEX1008 | pGEX104 derivative; harbors pnp-1008 Δpnp-871::cat | This work |
| pGEX1009 | pGEX104 derivative; harbors pnp-1009 Δpnp-871::cat | This work |
| pGEX1010 | pGEX104 derivative; harbors Δpnp-1010t Δpnp-871::cat | This work |
| pGEX1011 | pGEX104 derivative; harbors ΔpnpL1011 Δpnp-871::cat | This work |
| pGEX1012 | pGEX104 derivative; harbors ΔpnpL1012 Δpnp-871::cat | This work |
| pGEX1013 | pGEX104 derivative; harbors ΔpnpL1013 Δpnp-871::cat | This work |
| pGEX1014 | pGEX104 derivative; harbors ΔpnpL1014 Δpnp-871::cat | This work |
| pGEX1015 | pGEX104 derivative; harbors ΔpnpL1015 Δpnp-871::cat | This work |
| pGM743 | Promoterless pGZ119EH derivative | 62 |
| pGZ119HE | oriVColD Camr | 28 |
| pKD13 | oriRγ bla; harbors the aph (Camr) cassette flanked by FRT sites | 9 |
| pKD46 | oriR101 repA101(Ts) araC araBp-gam-bet-exo Ampr | 9 |
TABLE 3.
New pnp alleles constructed in this work
| Allele | Description |
|---|---|
| Δpnp-871 | Deletion 250-2238, in-frame insertion of 78-nt-long SCAR871 sequence |
| Δpnp-871::cat | Deletion 250-2238, insertion of cat cassettea |
| ΔpnpL1001 | Deletion 38-73 (5′-UTR) |
| ΔpnpL1002 | Deletion 1-75 (5′-UTR) |
| ΔpnpL1003 | Deletion 1-108 (5′-UTR) |
| ΔpnpL1004 | Deletion 1-127 (5′-UTR) |
| Δpnp-1005 | Deletion 162-167 (within 5′-coding sequence) |
| Δpnp-1006 | Deletion 178-180 (within 5′-coding sequence) |
| pnpL1007 | Transversion GG 147-148 CC (Shine-Dalgarno) |
| pnp-1008 | Transversion G159T (start codon) |
| pnp-1009 | Transversion T157A (start codon) |
| Δpnp-1010t | Deletion 2309-2328 (Rho-independent terminator) |
| ΔpnpL1011 | Deletion 4-75 (5′-UTR) |
| ΔpnpL1012 | Deletion 4-108 (5′-UTR) |
| ΔpnpL1013 | Deletion 114-145 (5′-UTR) |
| ΔpnpL1014 | Deletion 125-145 (5′-UTR) |
| ΔpnpL1015 | Deletion 132-145 (5′-UTR) |
| rpsOt::aph | Insertion of aph cassettea at −60 |
See the text.
TABLE 4.
Oligonucleotides used in this study
| Oligonucleotide no. | 5′→3′ sequence | Coordinatesa |
|---|---|---|
| FG1080/38b | CTAATACGACTCACTATAGGGGTCACGGTGTGTTGGCC | 206-190 |
| FG1081/20 | GTCGCGAGGATGCGCAGAAG | 79-98 |
| FG1626/40b | CTAATACGACTCACTATAGGGCATTAGCCGCGCGAACCTC | 37-19 |
| FG1627/21 | GAATGATCTTCCGTTGCAGAG | 1-21 |
| FG1682/41b | CTAATACGACTCACTATAGGGGTAGGGTGGAGCTGCTTCGA | 33-52 |
| FG1683/20 | ATTCCGGGGATCCGTCGACC | 1315-1334 |
| FG1701/59c | CTGCGTCGCTAATTCTTGCGAGTTTCAGAAAAGGGGCCTGGCTTTTGAAGCTCACGCTG | −101-61/ 1267-1249 |
| FG1702/60c | CCAGTGAATTGCTGCCGTCAGCTTGAAAAAAGGGGCCACTCAAGATCCCCTTATTAGAAG | −21-60/98-117 |
| FG1802/28 | CATTTGCCCTTAACCGGGCAGGACGCCT | 2299-2308/ 2329-2346 |
| FG1803/30 | CCTGCCCGGTTAAGGGCAAATGGCAACCTT | 2340-2329/ 2308-2291 |
Coordinates are from pnp-P +1 if not otherwise indicated.
Boldface letters indicate T7 promoter sequence.
Boldface letters and coordinates are from the pKD13 plasmid.
FIG. 1.

Construction of mutants in the pnp 5′ end. C-5705 (upper drawing) harboring an aph (Kanr) cassette and the plasmid pKD46, which expresses the λ Red system, was transformed with a DNA fragment (second drawing) harboring a mutant pnp 5′-end (pnpLmut, depicted by an x) and the Δpnp-871::cat allele. After λ Red-mediated recombination, chloramphenicol-resistant clones were selected and screened for the loss of Kanr and pKD46. Such clones, putatively pnpL Δpnp-871::cat (third drawing), were transformed at 30°C with the thermosensitive plasmid pCP20, which confers ampicillin resistance and harbors a thermoinducible FLP recombinase. After growth at 42°C without antibiotic selection, PCR amplicons of Cams Amps clones (bottom drawing) were then sequenced to verify the presence of the desired pnpL Δpnp-871 mutations.
Bacterial strains were grown in LD broth (17) supplemented with chloramphenicol (30 μg/ml), ampicillin (100 μg/ml), kanamycin (50 μg/ml), 0.4% glucose, or 1% l-arabinose when needed.
Northern blotting and data processing.
Bacterial cultures were grown at 37°C in LD broth supplemented with antibiotics when needed and grown up to an optical density at 600 nm (OD600) of 0.8, if not otherwise indicated. For RNA decay analysis, rifampin and nalidixic acid (final concentrations of 800 μg/ml and 60 μg/ml, respectively) were added and samples for RNA extraction were taken at different time points. RNA extraction was performed using the RNeasy (or miRNeasy, for small RNA analysis) minikit (Qiagen). Basic procedures for Northern blot analysis and synthesis of radiolabeled riboprobes by in vitro transcription with T7 RNA polymerase were previously described (4, 12). The specific riboprobes were obtained by in vitro transcription with T7 RNA polymerase of DNA fragments obtained by PCR amplification as follows. The template for the SCAR871 riboprobe, complementary to the Δpnp-871 scar, was obtained by PCR amplification of genomic C-5731 DNA with primers FG1682 and FG1683. The template for ANTI37, complementary to the small RNA37, was obtained by PCR of pAZ101 with primers FG1626 and FG1627. The template for the pnp mRNA riboprobe (PNP5, complementary to 79-206) was obtained by PCR of pAZ101 with primers FG1080 and FG1081.
Autoradiographic images and densitometric analysis of Northern blots were obtained by phosphorimaging using ImageQuant software (Molecular Dynamics). The intensities of pnp mRNA signals were normalized to the intensity of the pnpL+ signal in the absence of PNPase (relative abundance, as reported in Table 5 and in the histogram of Fig. 4). The ratio of the abundance in the presence of PNPase to the abundance in its absence thus represents the fraction of pnp mRNA escaping autogenous regulation (fraction escaping PNPase [FEP]), whereas (1-FEP) is the mRNA fraction subject to autoregulation in each strain (raw autogenous regulation index [RAI]). A normalized autogenous regulation index (AI) was then calculated as the ratio of the RAI of each mutant to the RAI obtained in pnpL+ signal. In experiments of mRNA decay analysis, mRNA half-lives were estimated by regression analysis of curves obtained by plotting the percentage of mRNA remaining (calculated as the densitometric signal at a given time after rifampin addition divided by the signal at time 0) versus time after rifampin addition.
TABLE 5.
pnp mRNA abundance and stability in the presence and absence of PNPase
| Mutationa | Coordinate
|
Relative abundanceb
|
AIc | Half-life (min)
|
|||
|---|---|---|---|---|---|---|---|
| Start | End | −PNPase | +PNPase | −PNPase | +PNPase | ||
| None (pnpL+) | 0 | 0 | 1.00 | 0.06 | 1.00 | 4 | NDd |
| Δ1001 | 38 | 73 | 1.42 | 0.81 | 0.46 | 5 | 8 |
| Δ1002 | 1 | 75 | 0.15 | 0.11 | 0.28 | ND | ND |
| Δ1011 | 4 | 75 | 0.24 | 0.21 | 0.12 | ND | ND |
| Δ1003 | 1 | 108 | 0.57 | 0.37 | 0.37 | 4 | 8 |
| Δ1012 | 4 | 108 | 0.54 | 0.52 | 0.04 | ND | ND |
| Δ1004 | 1 | 127 | 0.37 | 0.39 | −0.06 | ND | ND |
| Δ1013 | 114 | 145 | 0.24 | 0.05 | 0.84 | 5 | ND |
| Δ1014 | 125 | 145 | 0.20 | 0.04 | 0.85 | ND | ND |
| Δ1015 | 132 | 145 | 0.20 | 0.04 | 0.86 | ND | ND |
| Δ1005 | 162 | 167 | 0.58 | 0.20 | 0.70 | ND | ND |
| Δ1006 | 178 | 180 | 0.68 | 0.18 | 0.79 | ND | ND |
| Δ1010 | 2309 | 2328 | 0.90 | 0.13 | 0.92 | ND | ND |
| 1007 | 147 | 148 | 0.53 | 0.08 | 0.92 | ND | ND |
| 1008 | 159 | 159 | 0.22 | 0.07 | 0.74 | 2 | ND |
| 1009 | 157 | 157 | 1.08 | 0.17 | 0.90 | 7 | ND |
Mutations are listed in the same order as in Fig. 2.
Ratio to pnpL+ in the absence of PNPase.
Normalized AI.
ND, not determined.
FIG. 4.

Relative abundance and stability of Δpnp-871 mRNA. Relative abundance and stability were estimated by phosphorimaging quantification of Northern blot signals as described in Materials and Methods. The intensity of all discrete signals has been summed up to evaluate the total abundance of Δpnp-871 scar-containing transcript. Data have been normalized to the signals obtained in the pnpL+ strain in the absence of PNPase. Data from the pnpL+ strain in the presence of PNPase are the average of four experiments, and the error bar is shown. The normalized AI (see text) and half-lives (t1/2), when evaluated, are also reported. nd, not determined. Cross-hatched bars, strains harboring pAZ101 expressing PNPase; open bars, strains harboring the empty vector pGZ119HE. In the wild-type C-1a strain (with pnp in a single copy), the half-life is about 1 min (62; unpublished observation).
Preparation of E. coli crude extracts, sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) of proteins and immunoassays.
E. coli crude extracts were obtained as described previously (47). Protein content was determined using Coomassie Plus protein assay reagent (Pierce). Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was performed as described previously (26) on 10% resolving gels containing 0.1% SDS. High-molecular-weight markers (Amersham Biosciences, England) were used as size references. For immunological detection of PNPase, the gels were blotted onto a nitrocellulose (Hybond ECL) sheet and incubated with polyclonal anti-PNPase antibodies (15, 50). Immunoreactive bands were revealed using the ECL Western blotting reagent (Amersham GE Healthcare).
In vitro translation assay.
Escherichia coli MRE600 cells were grown in LB medium supplemented with 0.5% glucose at 37°C up to an OD600 of 1.2, washed with 0.9% NaCl, and harvested immediately. Cell-free (S30) extracts were prepared as described previously (27), except that extracts were prepared in 10 mM Tris-HCl (pH 7.1), containing 10 mM Mg acetate, 60 mM NH4Cl, and 6 mM β-mercaptoethanol and the preincubation step was omitted.
In vitro translation assays were carried out using in vitro-transcribed pnp mRNAs and S30 extracts (normalized for their ribosome content) in a mixture containing 28 mM Tris-HCl (pH 7.7), 10 mM Mg acetate, 50 mM NH4Cl, 2 mM dithiothreitol, 2 mM ATP, 0.4 mM GTP, 10 mM phosphoenolpyruvate, 0.025 μg of pyruvate kinase/μl reaction mixture, 200 mM of each amino acid (excepting Met), 0.1 μM [35S]Met, 1 μg/μl E. coli MRE600 tRNA mixture, and 0.12 mM citrovorum (Serva). After 30 min of incubation at 37°C, samples were spotted onto 3MM paper discs in order to establish the trichloroacetic acid-insoluble radioactivity incorporated.
Full-length and 5′-deletion pnp mRNAs were obtained by in vitro runoff transcription of 80 μg/ml SmaI-linearized pAZ76 and pAZ77, respectively, 2,000 U of T7 RNA polymerase in a transcription mixture containing 40 mM Tris-HCl (pH 8.1), 3.75 mM NTPs, 22 mM MgCl2, 10 mM spermidine, 5 mM dithiothreitol, 50 μg bovine serum albumin, and 60 U RNAsin [Amersham]. After 4 h of incubation at 37°C, the mRNA was purified by LiCl precipitation followed by ethanol precipitation.
RESULTS
cis-acting elements controlling PNPase autogenous regulation: experimental plan.
To identify specific cis-acting determinants of PNPase autoregulation and discriminate between the two proposed models, we constructed several pnpL Δpnp-871 mutants and one Δpnp-1010t Δpnp-871 chromosomal double mutant. In the Δpnp-871 mutants, a 78-nt-long scar sequence replaces in frame a 1,989-bp pnp region from codons 83 to 743. Expression of the Δpnp-871 mutant allele can thus be specifically monitored by Northern blotting even in the presence of an ectopically expressed pnp+ gene using a riboprobe (SCAR871) complementary to the unique scar sequence. Δpnp-1010t is a deletion of the Rho-independent transcription terminator immediately downstream of pnp, whereas the pnpL mutants harbor deletions or point mutations in the leader (either 5′-UTR or 5′ coding) region of Δpnp-871.
The general strategy for the 5′ mutant construction is described in Materials and Methods and outlined in Fig. 1. The pnp double mutant strains (Fig. 2 and Table 1) were then transformed with either pAZ101, which expresses PNPase from its natural promoter, pnp-p, or the parental plasmid pGZ119HE. The abundance of the chromosomally encoded Δpnp-871 mRNA in the different mutants in the presence and absence of ectopically provided PNPase was then monitored by Northern blotting using a radiolabeled SCAR871 riboprobe.
FIG. 2.

Map of the pnpL Δpnp-871 mutations. The pnp 5′- and 3′-UTRs are drawn to scale; the Δpnp-871 allele, which is 225 bp long, is on an arbitrary scale. Coordinates shown above the vertical bars are nucleotides from the pnp-p transcription start point (bent arrow). The coordinates of pnp codons retained in the mutant allele are shown below the upright arrowheads. Inverted arrows indicate the large and the small stem-loops (SL1and SL2, respectively). RIII1 and RIII2 are the two RNase III cut points. Checkerboard region, in-frame 78-nt-long scar (SCAR871) in Δpnp-871 allele; lollipop, pnp-t Rho-independent terminator. The Shine-Dalgarno and start codon sequences are also shown. The bars indicate the extent of the deletions, and boldface letters represent the bases substituted in the different mutants.
An example of such analyses is shown in Fig. 3A (see Fig. S2 in the supplemental material). The intensity of pnp mRNA signals was measured by phosphorimaging, and the results were normalized to the intensity of the pnpL+ signal in the absence of PNPase (Table 5 and Fig. 4, histogram). A normalized AI was calculated as described in Materials and Methods. We assumed that AI values of ≥0.7 and <0.5 indicate autoregulation and lack thereof, respectively (see below).
FIG. 3.

An example of Northern blot analysis of abundance and stability of Δpnp-871 mRNA with different pnpL mutations. RNA extracted from E. coli strains harboring the pnpL Δpnp-871 mutations in the presence or absence of ectopically expressed PNPase, as indicated in the figure, was analyzed by Northern blotting using a radiolabeled SCAR871 riboprobe. (A) Abundance of RNA extracted from cultures grown up to OD600 of 0.8. (B) Stability of RNA extracted from cultures grown to an OD600 of 0.8 and treated with rifampin (rif) as described in Materials and Methods. Time points (min) after rifampin addition and the half-lives (t1/2) estimated by phosphorimaging quantification of the signals are indicated. The lengths of the pnpL+ Δpnp-871 transcripts (which are 1,911 bp shorter than their corresponding pnp+ mRNAs; see Fig. S1 in the supplemental material for correspondence) are indicated in kb. The signals from the mutants are in fair agreement with the mRNAs expected from the harbored deletions. Moreover, mutants affected in RNase III processing also show some minor signals deriving from rpsOp. This complex signaling profile has been verified in part by hybridization with specific radiolabeled DNA oligonucleotide probes and an rpsO-specific riboprobe. The intensity of all discrete signals has been summed up to evaluate the total abundance of Δpnp-871 scar-containing transcript. The greater stability of the lowest band in panel B could be imputed to processing of the higher-molecular-weight transcripts.
To measure mRNA half-life (Table 5, last two columns), rifampin was added to cultures so as to inhibit transcription initiation and RNA was extracted at different time points for Northern blot analysis (see Fig. 3B as an example and see Fig. S3 in the supplemental material). The signal intensities of pnp mRNA remaining were measured and normalized to the intensity detected before transcription inhibition. Half-lives could not be estimated under conditions of very low transcript abundance.
A processable pnp mRNA double-stranded 5′ stem is the unique determinant of PNPase autoregulation.
Deletion ΔpnpL1001, which removes the upper (central) part of the large stem-loop (SL1) that serves as a substrate for RNase III, abolishes the endonucleolytic processing, as assessed by Northern blotting (data not shown). The primary transcript conserves a stem-loop of the same length as the double-stranded stem generated by RNase III digestion. The former, however, cannot be degraded by PNPase as it lacks the dangling 3′ end. As shown in Table 5 and Fig. 4, the abundance and stability of the pnp transcript in the presence of PNPase were over 10-fold higher than those of the wild type, thus indicating that in the ΔpnpL1001 mutant autoregulation was abolished. Although the AI of this mutant was relatively high (0.46), in the presence of PNPase its mRNA was greatly stabilized with a half-life of 8 min. These data are in agreement with previous results with RNase III-deficient strains or point mutations that prevent RNase III processing of the stem-loop (23, 49) and show that a double-stranded stem-loop as short as the stem produced by RNase III processing is sufficient for pnp mRNA stabilization.
Deletions ΔpnpL1002 and ΔpnpL1011 completely remove the large stem-loop. In the ΔpnpL1002 mutant, the transcript 5′-end corresponds to that of the RNase III-processed pnp mRNA (starting with a U), whereas the ΔpnpL1008 mutant conserves three nucleotides (GAA) at the natural pnp-p transcription start point so as to minimize effects of the deletion on transcription initiation efficiency. The abundance of pnp mRNA in these mutants was substantially decreased in both the presence and the absence of PNPase to a level comparable to that of the wild type in the presence of PNPase, and the AI was low. Thus, removal of SL1 generates a transcript that is unstable independently of PNPase. Similar results were obtained with mutants harboring longer deletions (ΔpnpL1003, ΔpnpL1012, and ΔpnpL1004) of the pnp 5′-UTR. Although the abundance of the pnp mRNA expressed from these latter mutants was slightly higher than that of the ΔpnpL1002 and ΔpnpL1008 mutants, it did not decrease in the presence of PNPase, giving low autoregulation indexes. Thus, the instability of pnp transcripts missing the double-stranded region at the 5′-UTR appears to be independent of PNPase autogenous control.
Other mutations (ΔpnpL1013, -1014, and -1015) that removed the small stem-loop (SL2) in the pnp 5′-UTR and/or the region immediately upstream of the Shine-Dalgarno sequence decreased to some extent the pnp mRNA abundance in the absence of PNPase relative to the wild-type allele. However, PNPase further reduced the amount of pnp mRNA so that the AI remained greater than 0.7. Likewise, deletions ΔpnpL1005 and ΔpnpL1010, which remove, respectively, a 5′ region of the coding sequence previously implicated in autogenous control (49) and the Rho-independent transcription terminator, did not affect autogenous regulation.
Overall, these data suggest that removal of the double-stranded 5′-UTR, which under wild-type conditions is removed by the concerted RNase III and PNPase activities, is necessary and sufficient to destabilize pnp mRNA.
Translation is not a direct target of PNPase autoregulation.
It has been proposed that PNPase indirectly induces its own mRNA decay by binding to the pnp 5′-UTR and preventing translation (49). A prediction of this model is that mutations preventing translation would greatly destabilize pnp mRNA in a PNPase-independent manner. We therefore constructed an AGGA→ACCA double transversion in the pnp Shine-Dalgarno sequence (pnpL1007), and a TTG→TTT transversion (pnp-1008) that destroys the natural UUG start codon of pnp. As a control, TTG was replaced with the canonical (ATG; pnp-1009) start codon sequence.
In the absence of PNPase, the pnp mRNA abundance of such constructs correlated with their expected translatability. All of these constructs, however, were subject to autoregulation (AI of >0.7) as PNPase drastically reduced their pnp transcript levels. Therefore, although in the absence of PNPase pnp mRNA translation seems to affect its stability, translation inhibition is not sufficient to fully destabilize the pnp mRNA at a level comparable to that observed in the presence of PNPase. This indicates that PNPase cannot control the stability of its mRNA by simply inhibiting translation of the processed transcript and suggests that an additional stabilizing factor needs to be directly removed by PNPase.
The RNase III-processed pnp mRNA with a single-stranded 5′-UTR is degraded by RNase E in a PNPase-independent manner and is not translatable.
RNase E-dependent degradation is thought to be the major pathway for mRNA decay and has been implicated in RNase III-processed pnp mRNA destabilization (20, 23). A major RNase E cut site was previously found 178 to 180 nt within the pnp coding sequence (20). An in-frame deletion of such a site (Δpnp-1006), however, did not affect autogenous regulation (Fig. 4), suggesting that additional RNase E sites could be used for pnp mRNA decay.
We thus tested pnp mRNA abundance and stability in an rne-thermosensitive (rne-3071) (18) mutant. As shown in Fig. 5A (upper panel), a temperature shift from 30 to 44°C led to a remarkable increase of pnp mRNA abundance in the rne-3071 strain, whereas it decreased in the isogenic wild-type strain. The half-lives at 30 and 44°C in the mutant were about 2 and 10 min, respectively (Fig. 5B), whereas in the wild type at 44°C, half-life could not be adequately assessed given the low abundance of the mRNA. Interestingly, the abundance of the small RNA37 in the rne-3071 mutant at 44°C did not concomitantly increase with the mature pnp mRNA (Fig. 5A, middle panel), suggesting that at the nonpermissive temperature its turnover was not affected and, therefore, at 44°C a larger proportion of the pnp mRNA 5′-UTR was mostly single stranded.
FIG. 5.

Stabilization of pnp mRNA and PNPase expression upon RNase E inactivation. Strains C-5869 (rne+; wild type [wt]), C-5868 (rne-3071; RNase E-thermosensitive mutant [rnets]), and C-5684 (Δrnc38::kan; RNase III-null mutant [Δrnc]) were grown at 30°C to an OD600 of 0.8 and then shifted at 44°C. At different time points from the temperature shift, samples were taken for RNA and protein extraction. Northern and Western blotting were then performed as described in Materials and Methods. (A, upper panels) Northern blotting of a denaturing agarose gel electrophoresis hybridized with radiolabeled PNP5 riboprobe that reveals monocistronic (2.25, arrowhead) and polycistronic (upper bands) pnp mRNAs. (Middle panels) Northern blotting of a denaturing acrylamide gel electrophoresis hybridized with radiolabeled ANTI37 riboprobe that reveals a ladder of PNP37 polyadenylated forms. (Lower panels) Western blotting with anti-PNPase serum as described in Materials and Methods. (B) Decay of C-5686 pnp mRNA at 30 and 44°C. Rifampin (rif) was added to an aliquot of the 30°C culture at an OD600 of 0.8 and 40 min after the shift at 44°C. Samples were taken at different time points for RNA extractions, and half-lives (t1/2) were assessed as described. The strain and sample time points after either temperature shift or rifampin addition (in min) are indicated on the figure. The apparent molecular size (in kb) of the major transcripts detected is also shown (see Fig. S1 in the supplemental material). Some variability in the relative intensity of the 2.25- and 5.4-kb mRNAs has been observed with the rne mutant in different experiments.
It may be noticed that the RNA37 appears as a ladder of discrete bands (Fig. 5A). These forms derive from polyadenyl polymerase (PAP)-dependent oligoadenylation, as they disappeared in pcnB mutants (see Fig. S4, lower panel, in the supplemental material). Nevertheless, PAP does not seem to play any role in autoregulation, since this is not affected in pcnB mutants (23; see Fig. S4, upper panel, in the supplemental material).
We also tested PNPase abundance under such conditions (Fig. 5A, lower panel). Like in the wild-type strain, in the rne-3071 strain the PNPase abundance remained substantially unchanged upon temperature upshift, notwithstanding the dramatic (over fivefold) increase in pnp mRNA. This suggests that pnp mRNA with a single-stranded 5′-UTR is poorly, if at all, translated. On the contrary, in an RNase III mutant a greater protein abundance corresponded to the higher pnp mRNA level (Fig. 5A).
To investigate on the relationships between the structure of pnp mRNA and its translatability, we transformed the rne-3071 and its wild-type isogenic strain with plasmids expressing a truncated PNPase (Pnp-ΔKHS1 from the Δpnp-833 allele, a mutant that does not exhibit autoregulation) (4) under the pnp-p promoter and a wild-type or mutant 5′-UTR. This allowed us to distinguish the endogenous from the ectopically expressed pnp mRNAs and PNPases by Northern and Western blotting, respectively. The data obtained are shown in Fig. 6. In the wild-type strain, the amounts of both Pnp and Pnp-ΔKHS1 proteins only slightly decreased at 44°C relative to 30°C, although the mRNA level decreased at the higher temperature. This could be dependent on the long turnover time of PNPase. In the rne(Ts) strain, however, the increased mRNA abundance at 44°C was not accompanied by an increase in Pnp-ΔKHS1—rather, it slightly decreased. This suggests that the pnp mRNA, coordinately processed by RNase III and PNPase, is not (efficiently) translated. The highest level of Pnp-ΔKHS1, corresponding to the highest abundance of mRNA, was attained with the ΔpnpL1001 leader sequence in both wild-type and rne(Ts) strains at both temperatures. This indicates that a pnp mRNA with a nonprocessable double-stranded stem may be efficiently translated. Curiously, the abundance of the plasmid-encoded ΔpnpL1001 mRNA was lower in the rne(Ts) strain and appeared to diminish at 44°C. It might be that in the absence of RNase E-dependent pathways, other decay pathways may take over. On the other hand, both the plasmid-encoded ΔpnpL1002 Δpnp-KHS1 mRNA and ΔPnp-KHS1 were undetectable in the rne+ host and in the rne(Ts) mutant at 30°C. In the rne(Ts) strain at 44°C, however, the mRNA signal, albeit weak, was present, whereas the protein could not be detected. This suggests that the RNase III-processed mRNA missing a double-stranded stem is not only very unstable but also poorly, if at all, translated.
FIG. 6.

Expression of PNPase upon RNase E inactivation in different 5′-UTR mutants. Strains C-5869 (rne+) and C-5868 (rne-3071; RNase E-thermosensitive mutant [rnets]) were transformed with plasmids pAZ133, pAZ133L1001, and pAZ133L1002, which express a KHS1-truncated PNPase under a pnpL+, ΔpnpL1001, and ΔpnpL1002 5′-UTR, respectively. Cultures were grown at 30°C and shifted at 44°C to inactivate the thermosensitive RNase E, and samples for RNA and protein extraction were taken before (0), and at different time points (indicated in min) after the temperature shift. Northern blotting with radiolabeled PNP5 riboprobe (upper panel) and Western blotting with anti-PNPase (middle panel) and anti-ribosomal protein S1 (lower panel) antisera were then performed as described in Materials and Methods. The extra bands in the S1 panel are remnants of prior labeling with anti-PNPase antibodies.
This conclusion was supported by the in vitro translation experiment shown in Fig. 7. Incorporation of [35S]Met in an in vitro translation system was about 14-fold higher using a pnp mRNA starting from the natural pnp-p at +1 than with an mRNA starting at +76, which corresponds to the RNase III-processed pnp mRNA.
FIG. 7.
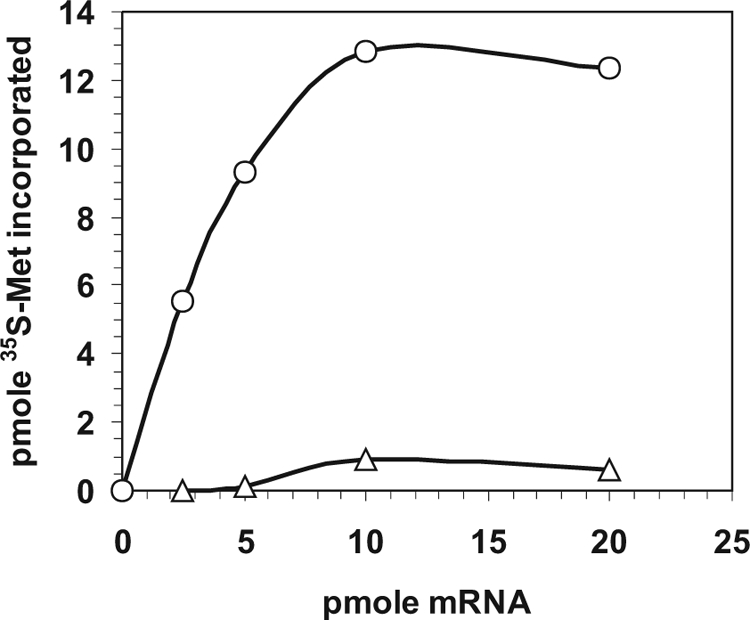
In vitro translation of full-length and 5′-end-truncated pnp mRNA. Incorporation of [35S]methionine in an in vitro translation system at increasing concentration of pnp native (circles) and 5′-truncated (triangles) mRNAs was performed as described in Materials and Methods.
From the above results, it appears that RNase E acts in concert with and downstream of the step controlled by PNPase for the destabilization of pnp mRNA (Fig. 8). To test whether the RNA degradosome could be implicated in autoregulation, we analyzed the pnp mRNA in an RNase E mutant with the C-terminal domain required for the assembly of the degradosome deleted (31). However, pnp abundance was only slightly increased and the half-life was doubled (3 min in the mutant versus 1.5 min in the wild-type strain; data not shown). This could be imputed to a lower activity of the mutant RNase E. Moreover, rhlB deletion mutants did not affect autoregulation (data not shown). Thus, the RNA degradosome appears to be marginally implicated, if at all, in PNPase autogenous regulation.
FIG. 8.
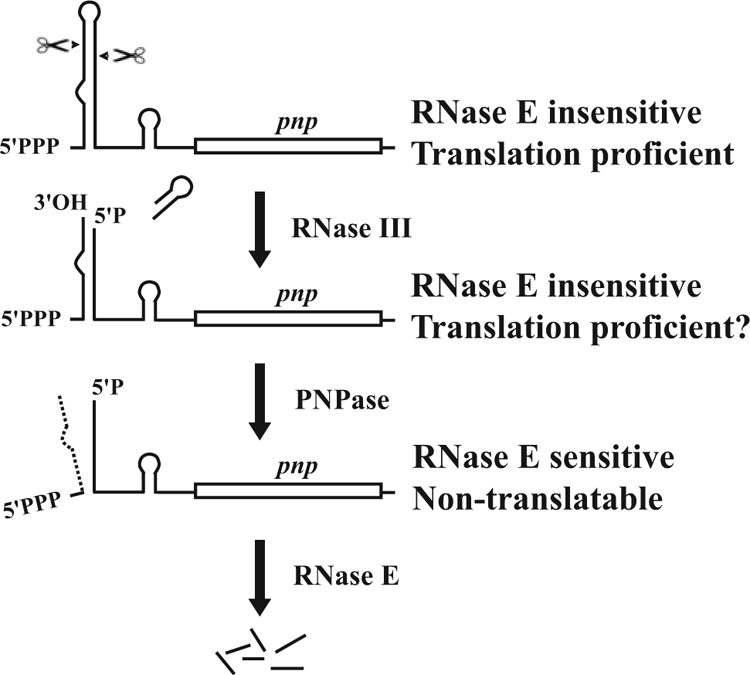
A model for PNPase autogenous regulation. Control of PNPase expression occurs at three sequential steps. Both native (upper drawing) and RNase III-processed (second drawing) pnp mRNAs are stable and translatable. RNase III creates the substrate for PNPase that degrades the small RNA37, thus destroying the double-stranded 5′ stem (third drawing). The processed pnp mRNA with a single-stranded 5′ end (bottom drawing) is not translated and is targeted for RNase E-dependent decay.
DISCUSSION
Early work on PNPase expression control had shown that PNPase posttranscriptionally regulates its own expression by controlling the stability of pnp mRNA. RNase III makes a staggered double-strand cleavage about in the middle of a long hairpin in the 5′-UTR and generates a double-stranded stem in which a small RNA with a protruding 3′ end (RNA37) is paired to the new 5′-monophosphate end of the processed pnp mRNA (48, 58) (Fig. 8). It has been proposed that PNPase would act as a translational repressor by binding the RNase III-matured pnp mRNA (48) and that translation inhibition by PNPase would target the pnp transcript to RNA decay pathways (49). This model was based mainly on analysis of reporter gene expression in wild-type and mutant pnp 5′-UTR-lacZ translational fusions and the mRNA decay rate of such constructs by hybridization of pulse-labeled RNA to DNA-specific probes.
More recently, the same group proposed an elegant new model that replaced the former one. This latest model maintains that the primary pnp transcript and the RNase III-processed mRNA are very stable by virtue of the double-stranded region at the 5′-UTR, whereas the transcript with a single-stranded 5′ end would be unstable. The stabilizing hairpin would be removed by the concerted action of RNase III and PNPase itself. RNase III, by cleaving the hairpin and generating the small RNA37 with a 3′ overhang, would create the substrate for PNPase exonucleolytic activity that would degrade the small RNA37, thus destroying the double-stranded stabilizing structure at the 5′ end (23) (Fig. 8).
However, a possible role of translational repression by PNPase had not been directly addressed in the context of the second model, and in our view, it was not completely ruled out. It thus remained possible that these two non-mutually exclusive mechanisms, removal of a stabilizing structure and translational repression, could operate in concert to autoregulate PNPase expression. For example, it could be assumed that PNPase binds to the pnp translation initiation region of the RNase III-processed pnp mRNA and both acts as a translational repressor and degrades the stabilizing small RNA37. Moreover, PNPase binding to the pnp translation initiation region could be required for RNA37 degradation. The latter hypothesis would be consistent with the observation that both phosphorolytic and RNA binding activities are necessary for autoregulation (4, 16, 22, 34). It thus remained unclear whether PNPase-dependent translation inhibition was implicated in destabilization of this transcript. Moreover, which decay pathway would degrade the processed pnp RNA had not been established.
The data we have presented support the most recent model as the only mechanism for PNPase autogenous regulation. In addition, we show that the RNase III-processed pnp transcript devoid of the double-stranded stem is both very unstable and does not seem to be translated and suggest that the high instability of this RNA is not a direct consequence of its poor translatability. Finally, we identify RNase E-dependent decay as the major pathway for degradation of mature pnp mRNA (see model in Fig. 8).
Our search for cis-acting determinants of pnp mRNA posttranscriptional regulation has been performed by Northern blotting of pnp mRNA of an internal pnp deletion mutant in its natural chromosomal context associated with cis mutations, in the presence and absence of ectopically expressed PNPase. This approach, unlike the use of protein-encoding reporter genes, avoids interference by variations of translation efficiency that may be associated with some mutations. Moreover, Northern blotting allows direct visualization of full-length (both native and processed) transcripts, thus avoiding to some extent possible interference by intermediate decay products. Some discrepancies between our results and previously published work (23, 48, 49) may be mostly imputed to the different experimental approaches.
PNPase regulates its own expression by transforming a stable mRNA precursor in an unstable transcript simply by degrading the small RNA37.
The data we have presented are in full agreement with the Jarrige et al. (23) model only. All conditions that prevent formation of a single-stranded 5′ end (mutation in the 5′ leader and/or mutations in RNase III and PNPase) abolish autogenous regulation, i.e., increase the abundance (and stability, when tested) of pnp mRNA in the presence of PNPase. Conversely, deletions that remove the double-stranded 5′ end destabilize the pnp mRNA independently of PNPase. Mutations affecting translation by destroying the Shine-Dalgarno sequence or the start codon also have an adverse effect on mRNA stability in the absence of PNPase. However, the pnp mRNA abundance and half-life are still higher than in the presence of PNPase, indicating that translational repression cannot be the only mechanism by which PNPase targets its own mRNA to decay and that removal of the double-strand structure at the 5′ end is required to fully destabilize the transcript. Thus, our data do not support the hypothesis of translational repression by PNPase previously proposed by Robert Le Meur and Portier (48, 49). Likewise, a 5′ region of the coding sequence previously implicated in autogenous control (49) and the Rho-independent transcription terminator do not appear to be implicated in this process, as deletions ΔpnpL1005 and ΔpnpL1010, which remove the 5′ region and Rho-independent transcription terminator, respectively, did not affect PNPase autoregulation.
Stability and translatability of processed pnp mRNA.
RNase III processing of pnp mRNA facilitates endonucleolytic degradation at several specific cleavage sites (56). Hajnsdorf et al. (20) implicated RNase E in this process and identified in vitro a high-affinity primary site and additional secondary sites for this endonuclease. We have shown in vivo that deletion of this high-affinity site did not affect autogenous regulation. However, inactivation of a thermosensitive RNase E stabilizes pnp mRNA in the presence of PNPase (20; this work). Therefore, it appears that RNase E is the major endoribonuclease responsible for the fast decay of processed pnp mRNA and that the secondary RNase E cleavage sites are sufficient to promote endonucleolytic degradation. Interestingly, the abundance of the small RNA37 did not increase at 44°C, whereas when autoregulation was abolished by lack of PNPase or transiently suppressed during cold acclimation (2, 62), both pnp mRNA abundance and RNA37 abundance increased concomitantly (unpublished data). This suggests that the RNA37 plays a major role in protecting the RNase III-processed pnp mRNA from RNase E.
Current models claim that RNase E-mediated mRNA decay in E. coli is triggered by translation inhibition, whereas translating ribosomes protect mRNA from RNase E endonucleolytic attack (10). We have shown both in vivo and in vitro a tight correlation between translatability and stability of pnp mRNA in PNPase autogenous regulation. However, rapid decay and inefficient translation appear to be two at least partially independent features of the RNase III-processed pnp mRNA lacking the 5′-end duplex. In fact, on the one hand pnp mRNAs harboring mutations that alter its translatability remain more stable in the absence of PNPase, thus suggesting that disruption of the 5′-end RNA duplex is sufficient to further destabilize the transcript; on the other hand, such an mRNA seems to be very poorly translated both in vivo when RNase E is inactivated and in vitro.
The protective role of the 5′-end hairpin can be easily reconciled with current models on mRNA decay mechanisms. It is known that a 5′-end triphosphorylated RNA is not a good substrate for RNase E (14, 24, 33, 61). It may be that in the RNase III-processed mRNA, the dangling RNA37 in the duplex region hinders the new monophosphate 5′ end and thus remains, like the native mRNA, inaccessible to RNase E. Recently, however, an RNA pyrophosphohydrolase (RppH) that removes a pyrophosphate from native RNAs has been found in E. coli (11). Since disruption of the double-strand stem via degradation of RNA37 seems to be required to trigger RNase E-mediated decay, it may be assumed that the 5′-end hairpin protects not only the recessed monophosphate 5′ end of the RNase III-processed transcript, but also, to some extent, the triphosphate 5′ end of native pnp mRNA from RppH activity, thus preventing RNase E-mediated decay until the RNase III-processed 5′-triphosphate stem is destroyed by PNPase degradation. Alternatively, the 5′ double-strand structure protects the pnp mRNA from RNase E irrespective of the 5′-end phosphate status.
Another puzzling issue is how a double-stranded 5′ stem makes the pnp mRNA translatable, whereas the processed single-stranded 5′ end is not. Comparison of secondary structures of native and RNase III-processed pnp mRNA predicted by MFOLD (63) did not show differences that could obviously suggest differential translation inhibition between the two forms. Structural studies of the alternative configurations that could be adopted by the pnp 5′-UTR will be essential to hint at reliable models. In addition, to our knowledge, nothing is known about the influence of 5′-end phosphorylation status on translation in E. coli. We cannot exclude that this feature could independently influence both stability and translatability of pnp mRNA.
Supplementary Material
Acknowledgments
We thank Elisa Gaetani, Anna Brandi, and Mara Giangrossi for technical help.
This research was supported by joint grants from the Ministero dell'Istruzione, dell'Università e della Ricerca, and Università degli Studi di Milano (PRIN 2005).
Footnotes
Published ahead of print on 9 January 2009.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Babitzke, P., L. Granger, J. Olszewski, and S. R. Kushner. 1993. Analysis of mRNA decay and rRNA processing in Escherichia coli multiple mutants carrying a deletion in RNase III. J. Bacteriol. 175229-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beran, R. K., and R. W. Simons. 2001. Cold-temperature induction of Escherichia coli polynucleotide phosphorylase occurs by reversal of its autoregulation. Mol. Microbiol. 39112-125. [DOI] [PubMed] [Google Scholar]
- 3.Bermudez-Cruz, R. M., F. Fernandez-Ramirez, L. Kameyama-Kawabe, and C. Montanez. 2005. Conserved domains in polynucleotide phosphorylase among eubacteria. Biochimie 87737-745. [DOI] [PubMed] [Google Scholar]
- 4.Briani, F., M. Del Favero, R. Capizzuto, C. Consonni, S. Zangrossi, C. Greco, L. De Gioia, P. Tortora, and G. Dehò. 2007. Genetic analysis of polynucleotide phosphorylase structure and functions. Biochimie 89145-157. [DOI] [PubMed] [Google Scholar]
- 5.Bycroft, M., T. J. Hubbard, M. Proctor, S. M. Freund, and A. G. Murzin. 1997. The solution structure of the S1 RNA binding domain: a member of an ancient nucleic acid-binding fold. Cell 88235-242. [DOI] [PubMed] [Google Scholar]
- 6.Carpousis, A. J., G. Van Houwe, C. Ehretsmann, and H. M. Krisch. 1994. Copurification of E. coli RNase E and PNPase: evidence for a specific association between two enzymes important in RNA processing and degradation. Cell 76889-900. [DOI] [PubMed] [Google Scholar]
- 7.Chou, J. Y., and M. F. Singer. 1971. Deoxyadenosine diphosphate as a substrate and inhibitor of polynucleotide phosphorylase of Micrococcus luteus. 3. Copolymerization of adenosine diphosphate and deoxyadenosine diphosphate. J. Biol. Chem. 2467505-7513. [PubMed] [Google Scholar]
- 8.Condon, C. 2007. Maturation and degradation of RNA in bacteria. Curr. Opin. Microbiol. 10271-278. [DOI] [PubMed] [Google Scholar]
- 9.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 976640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deana, A., and J. G. Belasco. 2005. Lost in translation: the influence of ribosomes on bacterial mRNA decay. Genes Dev. 192526-2533. [DOI] [PubMed] [Google Scholar]
- 11.Deana, A., H. Celesnik, and J. G. Belasco. 2008. The bacterial enzyme RppH triggers messenger RNA degradation by 5′ pyrophosphate removal. Nature 451355-358. [DOI] [PubMed] [Google Scholar]
- 12.Dehò, G., S. Zangrossi, P. Sabbattini, G. Sironi, and D. Ghisotti. 1992. Bacteriophage P4 immunity controlled by small RNAs via transcription termination. Mol. Microbiol. 63415-3425. [DOI] [PubMed] [Google Scholar]
- 13.Donovan, W. P., and S. R. Kushner. 1986. Polynucleotide phosphorylase and ribonuclease II are required for cell viability and mRNA turnover in Escherichia coli K-12. Proc. Natl. Acad. Sci. USA 83120-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng, Y., T. A. Vickers, and S. N. Cohen. 2002. The catalytic domain of RNase E shows inherent 3′ to 5′ directionality in cleavage site selection. Proc. Natl. Acad. Sci. USA 9914746-14751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fontanella, L., S. Pozzuolo, A. Costanzo, R. Favaro, G. Dehò, and P. Tortora. 1999. Photometric assay for polynucleotide phosphorylase. Anal. Biochem. 269353-358. [DOI] [PubMed] [Google Scholar]
- 16.García-Mena, J., A. Das, A. Sanchez-Trujillo, C. Portier, and C. Montanez. 1999. A novel mutation in the KH domain of polynucleotide phosphorylase affects autoregulation and mRNA decay in Escherichia coli. Mol. Microbiol. 33235-248. [DOI] [PubMed] [Google Scholar]
- 17.Ghisotti, D., R. Chiaramonte, F. Forti, S. Zangrossi, G. Sironi, and G. Dehò. 1992. Genetic analysis of the immunity region of phage-plasmid P4. Mol. Microbiol. 63405-3413. [DOI] [PubMed] [Google Scholar]
- 18.Ghora, B. K., and D. Apirion. 1978. Structural analysis and in vitro processing to p5 rRNA of a 9S RNA molecule isolated from an rne mutant of E. coli. Cell 151055-1066. [DOI] [PubMed] [Google Scholar]
- 19.Godefroy, T., M. Cohn, and M. Grunberg-Manago. 1970. Kinetics of polymerization and phosphorolysis reactions of E. coli polynucleotide phosphorylase. Role of oligonucleotides in polymerization. Eur. J. Biochem. 12236-249. [DOI] [PubMed] [Google Scholar]
- 20.Hajnsdorf, E., A. J. Carpousis, and P. Régnier. 1994. Nucleolytic inactivation and degradation of the RNase III processed pnp message encoding polynucleotide phosphorylase of Escherichia coli. J. Mol. Biol. 239439-454. [DOI] [PubMed] [Google Scholar]
- 21.Hajnsdorf, E., and P. Régnier. 1999. E. coli RpsO mRNA decay: RNase E processing at the beginning of the coding sequence stimulates poly(A)-dependent degradation of the mRNA. J. Mol. Biol. 2861033-1043. [DOI] [PubMed] [Google Scholar]
- 22.Jarrige, A., D. Bréchemier-Baey, N. Mathy, O. Duche, and C. Portier. 2002. Mutational analysis of polynucleotide phosphorylase from Escherichia coli. J. Mol. Biol. 321397-409. [DOI] [PubMed] [Google Scholar]
- 23.Jarrige, A. C., N. Mathy, and C. Portier. 2001. PNPase autocontrols its expression by degrading a double-stranded structure in the pnp mRNA leader. EMBO J. 206845-6855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang, X., and J. G. Belasco. 2004. Catalytic activation of multimeric RNase E and RNase G by 5′-monophosphorylated RNA. Proc. Natl. Acad. Sci. USA 1019211-9216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaberdin, V. R., and U. Bläsi. 2006. Translation initiation and the fate of bacterial mRNAs. FEMS Microbiol. Rev. [DOI] [PubMed]
- 26.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227680-685. [DOI] [PubMed] [Google Scholar]
- 27.La Teana, A., A. Brandi, M. Falconi, R. Spurio, C. L. Pon, and C. O. Gualerzi. 1991. Identification of a cold shock transcriptional enhancer of the Escherichia coli gene encoding nucleoid protein H-NS. Proc. Natl. Acad. Sci. USA 8810907-10911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lessl, M., D. Balzer, R. Lurz, V. L. Waters, D. G. Guiney, and E. Lanka. 1992. Dissection of IncP conjugative plasmid transfer: definition of the transfer region Tra2 by mobilization of the Tra1 region in trans. J. Bacteriol. 1742493-2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leszczyniecka, M., R. DeSalle, D. C. Kang, and P. B. Fisher. 2004. The origin of polynucleotide phosphorylase domains. Mol. Phylogenet. Evol. 31123-130. [DOI] [PubMed] [Google Scholar]
- 30.Li, X. M., and L. J. Shapiro. 1993. Three-step PCR mutagenesis for ‘linker scanning.’ Nucleic Acids Res. 213745-3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lopez, P. J., I. Marchand, S. A. Joyce, and M. Dreyfus. 1999. The C-terminal half of RNase E, which organizes the Escherichia coli degradosome, participates in mRNA degradation but not rRNA processing in vivo. Mol. Microbiol. 33188-199. [DOI] [PubMed] [Google Scholar]
- 32.Lorow, D., and J. Jessee. 1990. Max efficiency DH10B™: a host for cloning methylated DNA. Focus 1219-20. [Google Scholar]
- 33.Mackie, G. A. 1998. Ribonuclease E is a 5′-end-dependent endonuclease. Nature 395720-723. [DOI] [PubMed] [Google Scholar]
- 34.Matus-Ortega, M. E., M. E. Regonesi, A. Pina-Escobedo, P. Tortora, G. Dehò, and J. García-Mena. 2007. The KH and S1 domains of Escherichia coli polynucleotide phosphorylase are necessary for autoregulation and growth at low temperature. Biochim. Biophys. Acta 1769194-203. [DOI] [PubMed] [Google Scholar]
- 35.Miczak, A., V. R. Kaberdin, C. L. Wei, and S. Lin-Chao. 1996. Proteins associated with RNase E in a multicomponent ribonucleolytic complex. Proc. Natl. Acad. Sci. USA 933865-3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mohanty, B. K., and S. R. Kushner. 2000. Polynucleotide phosphorylase functions both as a 3′→5′ exonuclease and a poly(A) polymerase in Escherichia coli. Proc. Natl. Acad. Sci. USA 9711966-11971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mohanty, B. K., and S. R. Kushner. 2006. The majority of Escherichia coli mRNAs undergo post-transcriptional modification in exponentially growing cells. Nucleic Acids Res. 345695-5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Musco, G., G. Stier, C. Joseph, M. Castiglione Morelli, M. Nilges, T. J. Gibson, and A. Pastore. 1996. Three-dimensional structure and stability of the KH domain: molecular insights into the fragile X syndrome. Cell 85237-245. [DOI] [PubMed] [Google Scholar]
- 39.Portier, C., L. Dondon, M. Grunberg-Manago, and P. Régnier. 1987. The first step in the functional inactivation of the Escherichia coli polynucleotide phosphorylase messenger is a ribonuclease III processing at the 5′ end. EMBO J. 62165-2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Portier, C., C. Migot, and M. Grunberg-Manago. 1981. Cloning of E. coli pnp gene from an episome. Mol. Gen. Genet. 183298-305. [DOI] [PubMed] [Google Scholar]
- 41.Portier, C., and P. Régnier. 1984. Expression of the rpsO and pnp genes: structural analysis of a DNA fragment carrying their control regions. Nucleic Acids Res. 126091-6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Py, B., C. F. Higgins, H. M. Krisch, and A. J. Carpousis. 1996. A DEAD-box RNA helicase in the Escherichia coli RNA degradosome. Nature 381169-172. [DOI] [PubMed] [Google Scholar]
- 43.Régnier, P., and M. Grunberg-Manago. 1990. RNase III cleavages in non-coding leaders of Escherichia coli transcripts control mRNA stability and genetic expression. Biochimie 72825-834. [DOI] [PubMed] [Google Scholar]
- 44.Régnier, P., M. Grunberg-Manago, and C. Portier. 1987. Nucleotide sequence of the pnp gene of Escherichia coli encoding polynucleotide phosphorylase. Homology of the primary structure of the protein with the RNA-binding domain of ribosomal protein S1. J. Biol. Chem. 26263-68. [PubMed] [Google Scholar]
- 45.Régnier, P., and E. Hajnsdorf. 1991. Decay of mRNA encoding ribosomal protein S15 of Escherichia coli is initiated by an RNase E-dependent endonucleolytic cleavage that removes the 3′ stabilizing stem and loop structure. J. Mol. Biol. 217283-292. [DOI] [PubMed] [Google Scholar]
- 46.Régnier, P., and C. Portier. 1986. Initiation, attenuation and RNase III processing of transcripts from the Escherichia coli operon encoding ribosomal protein S15 and polynucleotide phosphorylase. J. Mol. Biol. 18723-32. [DOI] [PubMed] [Google Scholar]
- 47.Regonesi, M. E., F. Briani, A. Ghetta, S. Zangrossi, D. Ghisotti, P. Tortora, and G. Dehò. 2004. A mutation in polynucleotide phosphorylase from Escherichia coli impairing RNA binding and degradosome stability. Nucleic Acids Res. 321006-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Robert-Le Meur, M., and C. Portier. 1992. E. coli polynucleotide phosphorylase expression is autoregulated through an RNase III-dependent mechanism. EMBO J. 112633-2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Robert-Le Meur, M., and C. Portier. 1994. Polynucleotide phosphorylase of Escherichia coli induces the degradation of its RNase III processed messenger by preventing its translation. Nucleic Acids Res. 22397-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning. A laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 51.Sarkar, D., and P. B. Fisher. 2006. Polynucleotide phosphorylase: an evolutionary conserved gene with an expanding repertoire of functions. Pharmacol. Ther. 112243-263. [DOI] [PubMed] [Google Scholar]
- 52.Sasaki, I., and G. Bertani. 1965. Growth abnormalities in Hfr derivatives of Escherichia coli strain C. J. Gen. Microbiol. 40365-376. [DOI] [PubMed] [Google Scholar]
- 53.Smith, D. B., and L. M. Corcoran. 2001. Expression and purification of glutathione-S-transferase fusion proteins, chapter 16, unit 16.7. In F. M. Ausubel (ed.), Current protocols in molecular biology. John Wiley & Sons, Hoboken, NJ. doi: 10.1002/047114727.mb1607s28. [DOI] [PubMed]
- 54.Stickney, L. M., J. S. Hankins, X. Miao, and G. A. Mackie. 2005. Function of the conserved S1 and KH domains in polynucleotide phosphorylase. J. Bacteriol. 1877214-7221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Symmons, M. F., G. H. Jones, and B. F. Luisi. 2000. A duplicated fold is the structural basis for polynucleotide phosphorylase catalytic activity, processivity, and regulation. Struct. Fold. Des. 81215-1226. [DOI] [PubMed] [Google Scholar]
- 56.Takata, R., M. Izuhara, and K. Akiyama. 1992. Processing in the 5′ region of the pnp transcript facilitates the site-specific endonucleolytic cleavages of mRNA. Nucleic Acids Res. 20847-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takata, R., M. Izuhara, and K. Hori. 1989. Differential degradation of the Escherichia coli polynucleotide phosphorylase mRNA. Nucleic Acids Res. 177441-7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Takata, R., T. Mukai, and K. Hori. 1985. Attenuation and processing of RNA from the rpsO-pnp transcription unit of Escherichia coli. Nucleic Acids Res. 137289-7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thang, M. N., D. C. Thang, and M. Grunberg-Manago. 1967. An altered polynucleotide phosphorylase in E. coli mutant Q13. Biochem. Biophys. Res. Commun. 28374-379. [DOI] [PubMed] [Google Scholar]
- 60.Wade, H. E., and H. K. Robinson. 1966. Magnesium ion-independent ribonucleic acid depolymerases in bacteria. Biochem. J. 101467-479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Walsh, A. P., M. R. Tock, M. H. Mallen, V. R. Kaberdin, A. von Gabain, and K. J. McDowall. 2001. Cleavage of poly(A) tails on the 3′-end of RNA by ribonuclease E of Escherichia coli. Nucleic Acids Res. 291864-1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zangrossi, S., F. Briani, D. Ghisotti, M. E. Regonesi, P. Tortora, and G. Dehò. 2000. Transcriptional and post-transcriptional control of polynucleotide phosphorylase during cold acclimation in Escherichia coli. Mol. Microbiol. 361470-1480. [DOI] [PubMed] [Google Scholar]
- 63.Zuker, M., D. Mathews, and D. Turner. 1999. Algorithms and thermodynamics for RNA secondary structure prediction: a practical guide, p. 11-43. In J. Barciszewski and B. Clark (ed.), RNA biochemistry and biotechnology. Kluwer Academic Publishers, Dordrecht, The Netherlands.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.