

Abstract
Escherichia coli AcrB is a proton motive force-dependent multidrug efflux transporter that recognizes multiple toxic chemicals having diverse structures. Recent crystallographic studies of the asymmetric trimer of AcrB suggest that each protomer in the trimeric assembly goes through a cycle of conformational changes during drug export (functional rotation hypothesis). In this study, we devised a way to test this hypothesis by creating a giant gene in which three acrB sequences were connected together through short linker sequences. The “linked-trimer” AcrB was expressed well in the inner membrane fraction of ΔacrB ΔrecA strains, as a large protein of ∼300 kDa which migrated at the same rate as the wild-type AcrB trimer in native polyacrylamide gel electrophoresis. The strain expressing the linked-trimer AcrB showed resistance to some toxic compounds that was sometimes even higher than that of the cells expressing the monomeric AcrB, indicating that the linked trimer functions well in intact cells. When we inactivated only one of the three protomeric units in the linked trimer, either with mutations in the salt bridge/H-bonding network (proton relay network) in the transmembrane domain or by disulfide cross-linking of the external cleft in the periplasmic domain, the entire trimeric complex was inactivated. However, some residual activity was seen, presumably as a result of random recombination of monomeric fragments (produced by protease cleavage or by transcriptional/translational truncation). These observations provide strong biochemical evidence for the functionally rotating mechanism of AcrB pump action. The linked trimer will be useful for further biochemical studies of mechanisms of transport in the future.
RND (resistance-nodulation-cell division) family (23) multidrug efflux transporters, such as AcrB of Escherichia coli, not only are responsible for the intrinsic resistance of gram-negative bacteria to many lipophilic agents but also, when overproduced, generate a multidrug-resistant phenotype (13) that is becoming a major clinical problem in organisms like Pseudomonas aeruginosa (25). AcrB has been studied most extensively as a prototype among these RND multidrug transporters and is especially interesting as it allows the extrusion of an extremely wide range of substrates, including basic dyes; antibiotics such as chloramphenicol, tetracyclines, novobiocin, macrolides, and β-lactams; detergents such as sodium dodecyl sulfate (SDS) and Triton X-100; and even simple solvents (8, 13). AcrB exists as a homotrimer in which each subunit contains 12 transmembrane helices (TM1 to TM12) and two large periplasmic domains between TM1 and TM2 and between TM7 and TM8 (12). Recent elucidation of the asymmetric trimer structure through X-ray crystallography (11, 17, 19) led to the notion that the transporter works by a functionally rotating mechanism in which each protomer goes through a cycle of conformational alterations which are facilitated in turn by the complementary alterations in neighboring protomers.
Although this hypothesis can explain the mechanism of drug uptake, binding, and extrusion, it requires direct confirmation because it is based on the static crystal structure of the transporter. We especially wanted to test an essential feature of the rotating mechanism hypothesis, that the inactivation of one single protomer within the trimeric structure should inactivate the pumping function of the entire trimer. For this purpose, genetically or biochemically inactivating the unique copy of the acrB gene is obviously inadequate, as all protomers will be inactive copies. Thus, in this study, we developed a giant gene coding for a covalently linked trimer of AcrB, so that only one of the three linked protomers could be inactivated.
We previously used two approaches to inactivate AcrB. (i) Because AcrB is a proton-drug antiporter (24), protonation/deprotonation of charged amino acid residue(s) within the transmembrane domain is expected to drive the conformational changes needed to produce drug export. Asp407, Asp408, and Lys940 (6) (and a more recently identified Thr978 [22]) appear to form a tight salt bridge/H-bonding network in the transmembrane domain, and each of these residues is essential for function (6, 22). Conversion of any of these “proton relay network” residues to alanine produced inactive proteins. (ii) There is a large external cleft in the periplasmic domain of AcrB. In the structure of the asymmetric AcrB trimer, this cleft is open in two protomers but becomes closed in the “extrusion” protomer (11, 17, 19). We and others found that the pump is inactivated by fixing its conformation through site-directed disulfide cross-linking of residues (18, 21).
By applying these two approaches, this time for only one of the three protomers within the giant linked protein, we could show that the inactivation of one protomer inactivates the entire trimer, thus producing biochemical evidence for the functionally rotating mechanism of AcrB action. The linked trimer will also be useful in future studies of the biochemical mechanism of AcrB functions.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
Bacterial strains and plasmids used in this work are listed in Table 1. E. coli DH5α and DH10B were used for the construction and propagation of various plasmid constructs, the latter strain mostly for large plasmids. Cells were grown in Luria-Bertani (LB) broth supplemented with ampicillin (100 μg/ml), spectinomycin (50 μg/ml), kanamycin (35 μg/ml), tetracycline (10 μg/ml), and/or glucose (0.05 to 0.1%) when needed.
TABLE 1.
E. coli strains and plasmids
| Strain or plasmid | Genotype or description | Reference or source |
|---|---|---|
| Strains | ||
| DH5α | F−endA1 recA1 Φ80lacZΔM15 Δ(lacZYA-argF)U169 hsdR17(rK−mK+) supE44 thi-1 gyrA96 relA1 phoA deoR λ− | 15 |
| DH10B | F−endA1 recA1 galE15 galK16 nupG rpsL ΔlacX74 Φ80lacZΔM15 araD139 Δ(ara leu)7697 mcrA Δ(mrr-hsdRMS-mcrBC) λ− | Invitrogen |
| BLR | F−ompT hsdSB(rB−mB−) gal dcm lon Δ(srl-recA)306::Tn10(Tcr) | Novagen |
| RI90 | MC1000 phoR Δara714 leu+dsbA1::kan | 14 |
| AG100YB | AG100 ΔacrB::Spcr | 21 |
| AG100YBR | AG100YB Δ(srl-recA)306::Tn10(Tcr) | This study |
| BL21 | F−ompT hsdSB(rB−mB−) gal dcm lon | Novagen |
| BL21YB | BL21 ΔacrB::Spcr | This study |
| BL21YBR | BL21YB Δ(srl-recA)306::Tn10(Tcr) | This study |
| BL21YBDR | BL21YB dsbA1::kan Δ(srl-recA)306::Tn10(Tcr) | This study |
| Plasmids | ||
| pUC19 | High-copy-no. cloning vector; amp | |
| pGB2 | Low-copy-no. cloning vector; Spcr | 4 |
| pSPORT1 | Medium-copy-no. cloning and expression vector; amp; lac-inducible expression | Gibco BRL |
| pUCK151A | 6.5-kb BglII fragment containing entire acrAB operon cloned into pUC19 vector in the direction opposite to that of the lac promoter | 9 |
| pSAcrBHis | acrB gene with His4 tag sequence at 3′ end cloned into pSPORT1, producing AcrB protein containing a hexahistidine C-terminal sequence (two intrinsic and four additional histidine residues) under control of the lac promoter | 22 |
| pSCLBH | Derived from pSAcrBHis; codons for two intrinsic cysteines (Cys493 and Cys887) of acrB converted to codons for serines, producing cysteineless and His6-tagged AcrB | 21 |
| pSCLB10His | pSPORT1 containing cysteineless acrB gene with His8 tag sequence at 3′ end, producing cysteineless and His10-tagged AcrB | This study |
| pSCL-F666C/Q830C | Derived from pSCLBH; producing His6-tagged AcrB with double-Cys mutations F666C and Q830C | 21 |
| pS(CLB)2 | pSPORT1 containing two cysteineless acrB units without His tag sequence connected with short linker sequence | This study |
| pS(CLB)3 | pSPORT1 containing three cysteineless acrB units connected with short linker sequences, producing cysteineless linked AcrB trimer in which only the third protomer has His10 tag at C terminus | This study |
| pS(D407A-B2) | First acrB unit in pS(CLB)3 contains nonfunctional proton relay network mutation D407A | This study |
| pS(B2-D407A) | Third acrB unit in pS(CLB)3 contains D407A mutation | This study |
| pS(FQ-B2) | First acrB unit in pS(CLB)3 contains double-Cys mutations F666C and Q830C | This study |
| pS(B-FQ-B) | Second acrB unit in pS(CLB)3 contains F666C and Q830C | This study |
| pS(B2-FQ) | Third acrB unit in pS(CLB)3 contains F666C and Q830C | This study |
Construction of plasmids.
pSCLB10His, which was used as a source for a cysteineless acrB gene with a His8 tag sequence following the two intrinsic histidines at the 3′ end (10His), was constructed from pSCLBH (21) and pG(T7)AcrB10His (Y. Takatsuka and H. Nikaido, unpublished data). The latter, a plasmid based on pGB2 (4), was generated through several steps and contains, under the control of the T7 promoter derived from the pET21a(+) vector (Novagen), the acrB(10His) gene with two intrinsic cysteines (Cys493 and Cys887), which was created by the overlap extension method with pSAcrBHis (22) and pUCK151A (9) as template plasmids, using the primers NSFw (22), 10HisRv (5′-CGTTGTATCAGTGGTGGTGGTGATGGTGGTGGTGATGATGATCGA-3′), HisFw (22), and M13Rv(−48) (5′-AGCGGATAACAATTTCACACAGGA-3′) for the first PCR amplification and the primers NSFw and CB104Rv (22) for the second PCR. The codon for Cys887 of acrB(10His) in pG(T7)AcrB10His was converted to a codon for serine by site-directed mutagenesis using primers described previously (21). The resulting plasmid, pG(T7)AcrB10His(C887S), was digested with SacII and BamHI, and a 1-kb SacII-BamHI fragment containing about one-third of the 3′ end of acrB with C887S and 10His mutations was inserted into the SacII-BamHI-restricted pSCLBH.
pS(CLB)3 was constructed through three major steps (Fig. 1). This plasmid contains a gene composed of three cysteineless acrB sequences in which the first and the second acrB sequences have the deletion of codons for two intrinsic histidines at the 3′ end (ΔHis) and the third acrB sequence has a C-terminal His8 tag sequence (10His), all connected together through short linker sequences. As the linker sequence, we used a 135-bp internal sequence from acrB, which encodes 45 amino acid residues from Met496 to Arg540 (∼5 kDa), corresponding to the cytosolic α-helix (Iα) and its flanking regions. Because of the failure of the initial attempts to create a giant gene with three acrB sequences linked in tandem in a pUC19-based plasmid (see Results), we redesigned the strategy to use the medium-copy-number plasmid pSPORT1. In the first step (step I in Fig. 1), the three plasmids each containing a cysteineless acrB sequence were constructed based on pSPORT1 or pUC19 (only for pUBΔH-linkXSac). The acrB and linker sequences were amplified by PCR using Pfu Ultra high-fidelity DNA polymerase (Stratagene) with appropriate plasmids containing acrB as a template and were cloned into corresponding restriction sites of pSPORT1, pUC19, or their derivatives. The primers used were as follows (underlining shows the restriction site given after the sequence, and italics show the N-terminal fMet residue of the acrB gene): for acrB amplifications, NSFw (22), MetSacFw (5′-ATGCATGAGCTCATGCCTAATTTCTTTATCGATC-3′; SacI), MetBFw (5′-CGCGGATCCATGCCTAATTTCTTTATCGATC-3′; BamHI), CdHisXRv (5′-CGCTCTAGAATCGACAGTATGGCTGTGCT-3′; XbaI), and CH104Rv (5′-ATGCATAAGCTTTTATCCGTGGTTAATACTG-3′; HindIII); and for linkers, linkXFw (5′-GGCTCTAGAATGCTGAAACCGATTGCCA-3′; XbaI), linkSacRv (5′-ATGCATGAGCTCACGCCCCGTACTGCGCAG-3′; SacI), and linkBRv (5′-CGCGGATCCACGCCCCGTACTGCGCAG-3′; BamHI). The primer NSFw contains an SalI restriction site. In the primer CdHisXRv, the codons for two intrinsic histidines at the 3′ end of acrB are deleted and followed directly by an XbaI site. The entire cloned regions of PCR products in each plasmid were sequenced to confirm that there were no unintended sequence alterations. pUBΔH-linkXSac was generated from pUC19 and contains a 3.3-kb SalI-SacI fragment of the cysteineless acrB(ΔHis) gene, preceded by the upstream region, including the Shine-Dalgarno sequence, and followed by the linker sequence in frame through the XbaI site. pSMBΔH-linkXB contains a 3.3-kb cysteineless acrB(ΔHis) sequence, followed by the linker sequence and cloned into the SacI-BamHI site of pSPORT1. In the plasmid pSMB10, cysteineless acrB(10His) from the initiation methionine codon, followed by the extended 3′ region, including the transcription termination loop, was cloned into the BamHI-HindIII site of pSPORT1. The details of the methods used for the construction of these plasmids are available from the authors upon request. In the second step (Fig. 1), the 3.3-kb SalI-SacI fragment from pUBΔH-linkXSac was inserted in front of the acrB(ΔHis) sequence in pSMBΔH-linkXB in frame. The resulting plasmid, pS(CLB)2, expresses the cysteineless linked dimer under the control of the lac promoter of pSPORT1. Finally, in the last step (Fig. 1), the 3.3-kb BamHI-HindIII fragment from pSMB10 was inserted into the corresponding site of pS(CLB)2 behind the second linker sequence in frame, and the pS(CLB)3 (pSPORT1 based; ∼14 kb) was obtained.
FIG. 1.

Construction of the plasmid pS(CLB)3 for the expression of the cysteineless linked trimer of AcrB. Construction of the giant gene containing three acrB sequences connected by linkers was achieved through three major steps. A 135-bp internal sequence of acrB was used as the linker. See text for details. S.D., Shine-Dalgarno sequence; term. loop, termination loop.
To construct the plasmid containing the giant gene with one of the three protomers containing point mutation(s), in the cases of the D407A mutation in the first or the third protomer [pS(D407A-B2) or pS(B2-D407A)] and in the case of the double-Cys mutation (F666C and Q830C) in the third protomer [pS(B2-FQ)], mutation(s) were introduced by site-directed mutagenesis into the plasmids at step I (Fig. 1). For double-Cys mutation in the first or the second protomer [pS(FQ-B2) or pS(B-FQ-B)], acrB(ΔHis) sequences with mutations and without linkers were amplified by PCR with pSCL-F666C/Q830C (21), using the primers NSFw (or MetSacFw) and CdHisXRv, and put into the SalI (or SacI)-XbaI sites of the plasmids at step I. After the sequences were confirmed, the 3.3-kb SalI-SacI, SacI-BamHI, or BamHI-HindIII fragments were replaced into corresponding sites of pS(CLB)3 or inserted into pS(CLB)2. The region(s) containing mutation(s) in the resulting linked-trimer plasmids were sequenced, and it was confirmed that they contained, as expected, a mixture of both the wild-type and the mutant sequences.
Site-directed mutagenesis.
Point mutations were introduced by PCR as described previously (22). The sequences of all primers used are available upon request.
Construction of strains.
All strains were constructed by P1cml,clr100-mediated transduction (10), and the presence of ΔacrB::Spcr and ΔrecA::Tn10 genes was confirmed by PCR. For the stable maintenance of the plasmids carrying tandem-repeat acrB sequences, all strains used for assay in this study were transduced with the ΔrecA::Tn10 allele of strain BLR (Novagen) at the last step of their construction.
BL21YB, an acrB deletion mutant (90% of the acrB gene was replaced by the spectinomycin resistance [Spcr] gene aadA) with a protease-deficient E. coli B background (lon and ompT), was constructed by transduction of ΔacrB::Spcr from AG100YB (21) into BL21 (Novagen). BL21YB was then transduced with dsbA1::kan of strain RI90 (14), resulting in strain BL21YBD. Finally, the Δ(srl-recA)::Tn10 allele was transduced into BL21YB and BL21YBD, resulting in BL21YBR and BL21YBDR, respectively.
Analysis of AcrB expression and localization in the cells.
Fresh transformant colonies of AG100YBR or BL21YBR with pSPORT1-derived plasmids were inoculated in LB medium with ampicillin and grown overnight without shaking at 30°C without isopropyl-β-d-thiogalactopyranoside (IPTG) induction. The optical density at 660 nm (OD660) reached 0.5 to 0.8. Cells were harvested, resuspended in 10 mM HEPES-KOH (pH 7.5) buffer at an OD660 of 12 (for whole-cell analysis) or 30 (for fractionation), and broken by sonication in the presence of complete, EDTA-free protease inhibitor cocktail (Roche). For the analysis of whole cells, the sonicated suspensions were treated with the sample buffer, and proteins were resolved by SDS-7.5% polyacrylamide gel electrophoresis (PAGE). For the analysis of the localization of AcrB in the cells, unbroken cells of the sonicated suspensions were removed by low-speed centrifugation and, after ultracentrifugation at 150,000 × g for 30 min at 4°C, the supernatant was collected as the soluble fraction. The pellet was resuspended in 10 mM HEPES-KOH (pH 7.5) buffer containing 1.5% (wt/vol) N-lauroylsarcosine for solubilization of inner membrane proteins, and after repetition of the ultracentrifugation step, the supernatant and the pellet were used as the inner membrane and the (insoluble) residue fraction, respectively. Proteins resolved by SDS-PAGE were transferred to a nitrocellulose membrane (GE Water & Process Technologies) for Western blot analysis using polyclonal rabbit anti-AcrB (24) or tetra-His (Qiagen) primary antibody and an alkaline phosphatase-conjugated anti-rabbit (Sigma) or anti-mouse (Bio-Rad) secondary antibody, and then protein-antibody conjugates were visualized with nitroblue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate.
Blue native PAGE.
The inner membrane fraction was subjected to Blue native PAGE, and AcrB band(s) were detected by Western blot analysis. BL21YBR cells harboring pSPORT1-derived plasmids were grown overnight in LB broth supplemented with ampicillin at 30°C without shaking (the OD660 reached ∼0.8). Cells from a 10-ml culture were collected, resuspended in 10 mM HEPES-KOH (pH 7.5) buffer at an OD660 of 30, and broken by sonication in the presence of complete, EDTA-free protease inhibitor cocktail. The inner membrane fraction was prepared as described above and treated with BN-sample buffer (5× solution; 0.5 M 6-aminocaproic acid-50 mM BisTris buffer [pH was adjusted to 7.0 with HCl], 30% [wt/vol] sucrose, 5% [wt/vol] Coomassie brilliant blue R-250). Blue native PAGE was performed with a modification of the method described by Schägger and von Jagow (16). Proteins were resolved on Tris-HCl precast gel (4 to 15% linear gradient; Bio-Rad) using the electrode buffer of the Laemmli system with SDS omitted (25 mM Tris-192 mM glycine buffer [pH was adjusted to 8.3 with HCl]). Electrophoresis was performed at 4°C and started at 60 V with cathode buffer containing 0.01% (wt/vol) Coomassie brilliant blue. Once the front ran two-thirds of the gel, the cathode buffer was changed to the buffer without Coomassie (to reduce the background for Western blotting), and voltage was then set to 100 to 120 V. The total running times were 3 to 4 h. After the gel was rinsed, the proteins were transferred to a nitrocellulose membrane for Western blot analysis as described above, using a horseradish peroxidase-conjugated anti-rabbit immunoglobulin G (Pierce) as a secondary antibody and Western lightning chemiluminescence reagent plus (Perkin-Elmer Life Sciences, Inc.) for the detection of protein-antibody conjugates.
Drug susceptibility assays.
E. coli cells harboring pSPORT1-derived plasmids were tested for drug susceptibilities, at 37°C and without IPTG induction, by two different methods. The MICs of cholic acid, erythromycin, ethidium bromide, and novobiocin were determined by using a twofold-dilution method with LB broth containing ampicillin (100 μg/ml). The volume in each tube was 300 μl. Mid-exponential-phase cultures from a single colony of the fresh transformant were diluted to 0.1 OD660 unit with LB broth containing ampicillin, and 10 μl was inoculated into each test tube. Each experiment was repeated at least three times.
The drug susceptibilities of these same cultures were measured with a second method using a gradient plate, as described previously (3, 21, 22). A linear concentration gradient of cholic acid (Sigma) was prepared in square LB agar plates containing 10,000, 3,000 to 5,000, and 3,000 or 4,000 μg/ml of cholic acid in the lower layer for AG100YBR, BL21YBR, and BL21YBDR, respectively. All mutants were assayed at least three times, using strains harboring wild-type cysteineless pSCLBH [or pS(CLB)3 in the case of the disulfide cross-linking experiment] or the vector pSPORT1 as positive and negative controls, respectively. Under these conditions, the differences in the lengths of growth of the two control strains were between 10 and 40 mm. The relative activity of each linked or mutated AcrB protein was calculated as described previously (21, 22), and the activities of controls with full efflux activity [pSCLBH or pS(CLB)3] and no efflux activity (pSPORT1) were defined as 100 and 0%, respectively.
Ethidium accumulation assay and effect of methanethiosulfonate (MTS) cross-linking reagents.
A “real-time” disulfide cross-linking experiment using a fast-acting MTS cross-linker was performed as described previously (21). A single colony of the freshly transformed BL21YBDR with pSPORT1-derived plasmids was inoculated into LB broth with ampicillin and grown overnight without shaking at 30°C. The cultures (70 to 100 μl, with an adjusted initial OD660 of ∼0.08 for all strains assayed at the same time) were diluted into 10 ml of LB medium with ampicillin and grown with shaking at 37°C to an OD660 of 0.8 to 0.9 without IPTG induction. Cells were then harvested at room temperature, washed once, and resuspended in 50 mM sodium phosphate buffer (pH 7.0) containing 0.1 M NaCl and 0.1% (vol/vol) glycerol. The accumulation of ethidium by cells was monitored by using a Shimadzu RF-5301PC spectrofluorometer as described previously (21). The final concentration of ethidium bromide used was 5 μM, and the concentration of bacterial cells corresponded to an OD660 of 0.2 in the final volume of 2 ml. The excitation and emission wavelengths were 520 and 590 nm, respectively. The widths of the slits were 5 and 10 nm, respectively. After 2 min of preincubation with ethidium bromide, 2.5 mM 1,2-ethanediyl bismethanethiosulfonate (MTS-2-MTS) and 2.5 mM pentyl MTS (5-MTS) (Toronto Research Chemicals, Toronto, Ontario, Canada), freshly dissolved in dimethyl sulfoxide-ethyl acetate (3:1, vol/vol), were added to the cells to a final concentration of 4 μM, and the accumulation of ethidium was followed.
RESULTS
Construction of a giant gene with three acrB sequences connected by linker sequences.
To create a giant gene in which three acrB sequences are linked in tandem, we chose as the linker a 135-bp internal sequence of acrB which encodes an α-helix (Iα) and its flanking regions, corresponding to Met496 to Arg540 (∼5 kDa). In the crystal structure (12), Iα is located between TM6 and TM7 and connects the N-terminal and C-terminal halves of the AcrB protomer on the cytosolic surface of the transmembrane domain.
Initially we tried to make all plasmids with the pUC19 (or pUC18) vector, expecting that its small size (2,686 bp) would allow it to accommodate large inserts (∼10 kb). However, the plasmid containing two acrB genes, corresponding to step II of the construction strategy (Fig. 1), could not be obtained after many trials with different approaches. Even in the step corresponding to step I, the colonies of E. coli harboring pUC-based acrB plasmid were small on LB plates, and the addition of glucose to the medium was needed to avoid growth inhibition and to maintain the plasmid.
The construction of the giant gene was finally achieved by a redesigned strategy (Fig. 1) using the medium-copy-number plasmid pSPORT1. Despite the larger size of the vector (4,110 bp), this strategy allowed us to obtain pS(CLB)2 and pS(CLB)3 without any problems even without the addition of glucose to the medium. Plasmid pS(CLB)3 (∼14 kb) contains three cysteineless acrB sequences; among them the first and the second ones have deletion of codons for the two endogenous histidines at the 3′ end (ΔHis) and the third sequence has the additional His8 tag sequence next to the two intrinsic histidine codons at the 3′ end (10His). The three acrB sequences are connected together through the linker sequences, and the giant gene is under the control of the lac promoter. Since the restriction sites used to connect acrB genes and linkers in frame, XbaI, SacI, and BamHI, are expected to be translated, pS(CLB)3 should produce a single giant protein of 350 kDa, with no Cys residue in the entire sequence and with the 10His tag sequence only at the C terminus.
Expression and localization analysis of the products from pS(CLB)3.
To examine the protein(s) expressed from pS(CLB)3 in E. coli, we first constructed strain AG100YBR, a ΔacrB AG100YB strain (21) also containing the ΔrecA mutation, in order to prevent homologous recombination between tandemly repeated acrB sequences. Since we noticed that the products were more stable in the protease-deficient host strain (see below), the ΔacrB ΔrecA strain BL21YBR generated from BL21 was also used.
Western blotting analysis of the whole-cell proteins from the exponential-phase cells harboring pS(CLB)3 without IPTG induction showed the presence of a large protein of ∼300 kDa which reacted with anti-AcrB antibody (not shown). The ∼300-kDa protein was also detected with anti-His4 antibody (data not shown), indicating that the giant gene was translated completely up to the C-terminal end containing the histidine tag sequence.
The total cell proteins were further fractionated into three fractions, the soluble fraction, inner membrane, and insoluble residue (containing the outer membrane), as described in Materials and Methods, and analyzed by Western blotting (Fig. 2A). The 300-kDa protein was mainly localized in the inner membrane fraction in both host strains, AG100YBR and BL21YBR, similar to the monomeric cysteineless and hexahistidine-tagged AcrB (CL-AcrBHis) expressed from pSCLBH. In addition to the ∼300-kDa protein, much smaller amounts of an approximately 220-kDa protein, presumably corresponding to a linked dimer of AcrB, were also detected in the inner membrane fractions from the cells harboring pS(CLB)3. In the host strain AG100YBR, the monomeric form of AcrB (110 kDa) was also detected faintly. The intensities of the 220-kDa and 110-kDa bands in the inner membrane fraction from pS(CLB)3-harboring AG100YBR were 20% and 11%, respectively, of the intensity of the 300-kDa band, as estimated by the use of the NIH ImageJ program, while they were 10% and 2% in BL21YBR (Fig. 2A), suggesting that the giant 300-kDa protein is more stable in the BL21YBR host. We also noted that the total amounts of AcrB sequences expressed in BL21YBR, determined by Western blotting with anti-AcrB antibody, was almost always higher from pS(CLB)3 than from CL-AcrBHis (Fig. 2A). These features regarding stability and expression levels will be discussed below in connection with the function of these proteins.
FIG. 2.

Expression and localization of the linked-trimer AcrB. AcrB proteins were analyzed by Western blotting using a polyclonal anti-AcrB antibody. (A) Total cell proteins from noninduced ΔacrB ΔrecA host strains AG100YBR and BL21YBR containing each plasmid were fractionated into three fractions and separated by SDS-PAGE, as described in Materials and Methods. The amount of protein in each lane corresponds to that from ∼3.6 × 108 cells. Fractions: S, soluble fraction; IM, inner membrane proteins; R, insoluble residue (containing the outer membrane). (B) Blue native PAGE. Inner membrane proteins were prepared from BL21YBR (ΔacrB ΔrecA) cells containing pSPORT1-derived plasmids and separated by Blue native PAGE, as described in Materials and Methods. The amount of protein in each lane corresponds to that from ∼7 × 109 cells for pSPORT1 and pSCLBH and ∼1 × 109 cells for pS(CLB)3. AcrB was detected with anti-AcrB polyclonal antibody, as described in Materials and Methods. The product from pS(CLB)3 migrates at the same rate as wild-type AcrB trimer.
The proteins in the inner membrane fractions from BL21YBR cells harboring pSPORT1, pSCLBH, or pS(CLB)3 were separated on Blue native gel and analyzed by Western blotting using anti-AcrB antibody (Fig. 2B) as described in Materials and Methods. Because of the different expression levels of AcrB between pSCLBH and pS(CLB)3 in BL21YBR mentioned above, about seven-times-larger amounts of proteins were applied for pSPORT1 and pSCLBH lanes than for pS(CLB)3. The product from pS(CLB)3 migrated at the same rate as the wild-type AcrB trimer (∼360 kDa), suggesting that the quaternary structure of the ∼300-kDa protein is similar to that of the wild-type AcrB trimer.
The linked trimer of AcrB is fully functional.
The transport activity of the linked trimer was evaluated by examining the drug susceptibilities of plasmid-containing ΔacrB ΔrecA strains AG100YBR and BL21YBR. Two different methods, MIC determination by the broth dilution method and gradient plate assay, were employed as described in Materials and Methods. To avoid the nonreproducible drug susceptibility patterns caused by the strong overexpression of AcrB alone (22), acrB sequences were expressed without IPTG induction. We previously observed variation in resistance levels following storage of the transformed cells of another host strain, HNCE1a (5, 21). However, this was not much of a problem with the recA-null host strains AG100YBR and BL21YBR, and reproducible results were obtained with the transformed colonies stored at 4°C for up to 5 days.
The measurement of MICs against cholic acid, erythromycin, ethidium bromide, and novobiocin showed that the AG100YBR cells harboring pS(CLB)3 were resistant to these compounds at about the same level as pSCLBH-harboring cells (Table 2), suggesting that the linked trimer functions fully in intact cells. With BL21YBR as the host, the drug resistance produced by the linked trimer was often higher than that produced by the monomeric acrB gene in pSCLBH (Table 2). For example, the MIC for cholic acid was reproducibly twice higher with pS(CLB)3 than with pSCLBH. This tendency could be observed even more clearly in the results of the gradient plate assay of cholic acid resistance (Fig. 3). In the host strain BL21YBR, the relative activity of the linked trimer was 2.6 times higher than that of the monomeric CL-AcrBHis (product from pSCLBH), while that in AG100YBR was about the same as that of CL-AcrBHis. The reason for this influence of the host strains is most likely the greater stability of the linked trimer in the protease-deficient (lon and ompT) strain BL21YBR, as well as the enhanced expression level of the linked trimer in comparison with the expression of the monomer in BL21YBR. As shown in Fig. 4, Western blot analysis of the whole-cell proteins revealed that several proteins smaller than 300 kDa, especially the apparently monomeric form of 110 kDa, were seen in AG100YBR expressing the linked trimer. In contrast, in BL21YBR containing pS(CLB)3, only one 220-kDa band was detected other than the main 300-kDa band, similar to the result shown in Fig. 2, mentioned above, suggesting again that the linked trimer is more stable in BL21YBR. Figure 4 also shows that the expression level of the linked trimer was much higher (almost four times) than that of wild-type CL-AcrBHis in BL21YBR. The cause for this difference in expression levels is not understood so far.
TABLE 2.
Drug susceptibilities of the ΔacrB ΔrecA strains expressing wild-type or linked AcrB proteins
| Plasmid | MICa (μg/ml) for host strain:
|
|||||||
|---|---|---|---|---|---|---|---|---|
| AG100YBR
|
BL21YBR
|
|||||||
| Cholic acid | Erythromycin | Ethidium bromide | Novobiocin | Cholic acid | Erythromycin | Ethidium bromide | Novobiocin | |
| pSPORT1 | 5,000 | 4 | 25 | 4-8 | 2,500 | 2-4 | 6.25 | 8-16 |
| pSCLBH | 10,000-20,000 | 64 | 50-100 | 64 | 5,000 | 32 | 25 | 64 |
| pS(CLB)3 | 10,000-20,000 | 64 | 50-100 | 64 | 10,000 | 32 | 25-50 | 64-128 |
| pS(CLB)2 | 10,000 | 16 | 25-50 | 16 | 2,500-5,000 | 8 | 12.5 | 16-32 |
| pS(B2-D407A) | 10,000 | 16-32 | 50 | 32 | 2,500-5,000 | 16 | 12.5-25 | 32 |
| pS(D407A-B2) | 5,000-10,000 | 8 | 25-50 | 8-16 | 2,500-5,000 | 8 | 12.5 | 16-32 |
MICs that are fourfold or more lower than those of the pS(CLB)3-containing strain are shown in italic boldface.
FIG. 3.
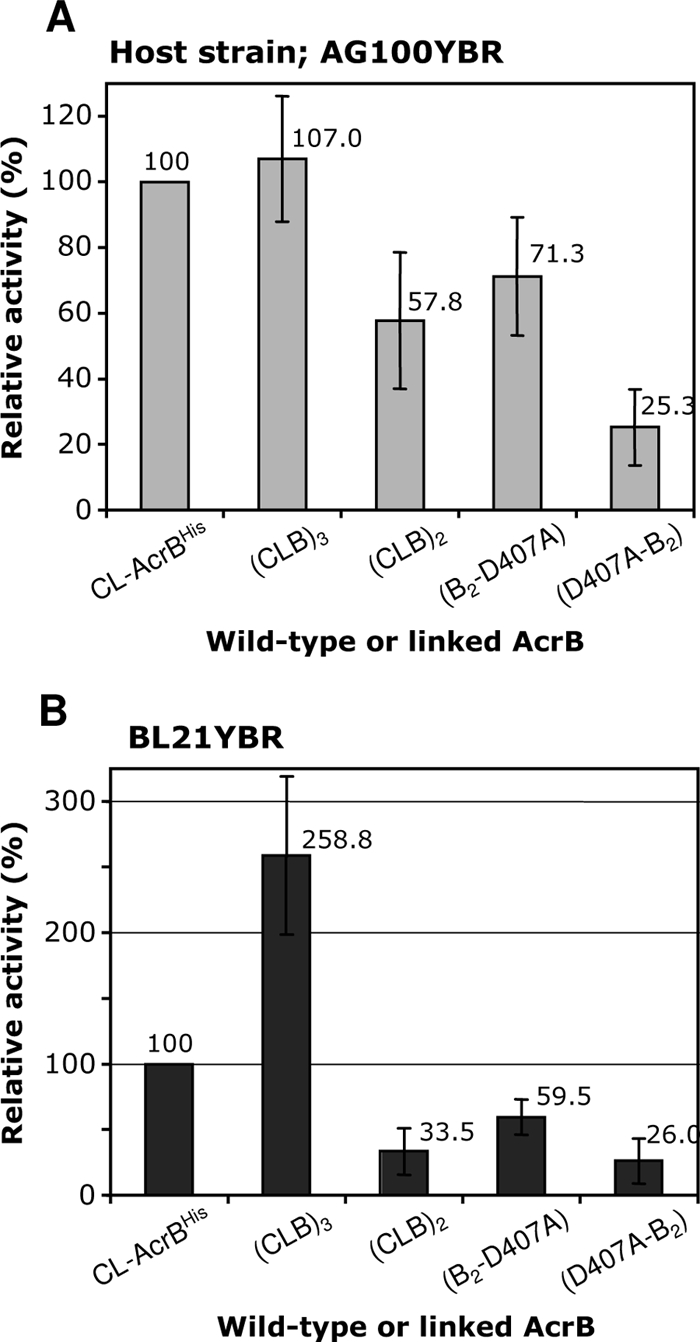
Activities of linked AcrB proteins. Wild-type cysteineless AcrB (CL-AcrBHis) and linked AcrB proteins were expressed in the ΔacrB ΔrecA host strains with different backgrounds, AG100YBR and BL21YBR, and their efflux activities were estimated from their levels of resistance to cholate by using the gradient plate method. For details, see Materials and Methods. Error bars show standard deviations.
FIG. 4.

Levels expression of the monomeric and linked AcrB proteins in AG100YBR and BL21YBR. The cells containing pSPORT1-derived plasmids were grown overnight without shaking and without induction at 30°C, and whole-cell proteins were analyzed, as described in Materials and Methods. A fixed amount of sonicated cells (1.2 × 108 cells) was applied to each lane, and proteins were separated by SDS-PAGE. AcrB was detected with a rabbit polyclonal antibody. The linked trimers (∼300 kDa) were more stable in the protease-deficient host strain, BL21YBR. The expression levels of linked trimers were higher than that of the monomeric AcrB (110 kDa) expressed from pSCLBH in BL21YBR. The bands at 220 kDa probably represent a linked dimer of AcrB. The bands are shown by arrows at right, M, molecular size marker.
One concern was that the drug resistance of the cells containing pS(CLB)3 might be due to the contribution of the small amounts of the monomeric AcrB (Fig. 4 and Fig. 2A), presumably generated by the degradation of the linked trimers or transcriptional or translational truncation. To exclude this possibility, we also examined the drug susceptibilities of the cells harboring pS(CLB)2. pS(CLB)2 is expected to produce a linked dimer (an abnormal artifact for the cells) and was found to also produce small amounts of monomeric AcrB at a somewhat higher level than did pS(CLB)3 (Fig. 4).
As shown in Fig. 3B, the resistance level of the cells containing pS(CLB)3 was dramatically higher than that of those harboring pS(CLB)2, although the latter produced somewhat more of the monomeric AcrB, thereby showing that the efflux activity of the former cells cannot be explained by the presence of monomeric fragments. The cells containing pS(CLB)2, however, showed some marginal resistance in comparison with the resistance level of cells containing the vector pSPORT1 alone (Table 2). This low level of activity is indeed likely due to the existence of the small amounts of the monomeric AcrB fragments.
Inactivation of a single protomer in the linked trimer inactivates the entire complex.
The goal of this study was to test the functionally rotating mechanism hypothesis of AcrB action (11, 17, 19) by inactivating only one protomer of the trimeric structure. The establishment of the functional, linked trimer allowed us to determine the functional consequences of genetically inactivating only one of the three acrB sequences by altering one of the residues involved in the tight salt bridge/H-bonding structure, or so-called proton relay machinery, in the transmembrane domain (20, 22). We changed Asp407 into alanine, a mutation that completely abrogates the activity of the pump if present in all the subunits of the AcrB trimer (22); however, this time we introduced this change only in either the first [pS(D407A-B2)] or the last [pS(B2-D407A)] of the three acrB sequences in the giant gene.
The mutant genes were expressed as strongly as the unmodified linked-trimer gene (Fig. 4). However, these mutant proteins were not functional. In terms of MIC, cells containing these mutant plasmids showed only marginal activity, similar to the activity of those containing pS(CLB)2 (Table 2). When the relative activities of B2-D407A and D407A-B2 in BL21YBR in the cholic acid gradient plate assay (Fig. 3B) were calculated with (CLB)3 as 100%, they were only 23% and 10%, respectively. The residual activity of the products of these mutant plasmids is almost certainly due to the small amounts of 110-kDa AcrB fragments present, because the residual activity of the mutant plasmids, as well as of pS(CLB)2, was much more pronounced in AG100YBR than in BL21YBR (compare Fig. 3A with Fig. 3B), where the degradation of the linked trimer is less pronounced (Fig. 2 and Fig. 4). We also note that the higher level of activity of B2-D407A than of D407A-B2 suggests a significant contribution of premature termination in transcription or translation, as the former construct will generate more of the active monomeric AcrB unit than the latter in this mechanism. These results, then, indicate that the inactivation of only one protomer causes the loss of function of the entire trimer and supply strong evidence for the functional rotation mechanism.
Site-directed disulfide cross-linking in only one protomer inactivates the entire trimer.
As stated in the introduction, we demonstrated in a previous report (21) that the forced closing of the large external cleft in the periplasmic domain by site-directed disulfide cross-linking caused the loss of function of AcrB. The functional rotation mechanism of AcrB (11, 17, 19) predicts that if one of the three protomers becomes inactivated, then the pumping action by the entire trimer comes to a halt. To test this hypothesis, we used linked trimers containing double Cys mutations in only one protomer.
We introduced Cys residues to replace Phe666 and Gln830, located on opposite sides of the large external cleft of periplasmic domain. The distance between F666 and Q830 in the extrusion protomer is 4.4 Å as measured between the ends of the side-chains; in contrast, these distances are over 9 Å in the access and binding protomers. Thus, the two Cys residues are expected to produce a disulfide bond only in one protomer in the AcrB trimer. As shown in our previous report (21), AcrB trimers composed entirely of the double-Cys mutant protein CL-F666C/Q830C were inactive. When we estimated the number of free Cys residues left in the double-Cys mutants by using sulfhydryl-specific labeling with biotin-maleimide, we noted that many of the Cys sulfhydryl groups were still free for modification after the nearly complete inactivation of the pump by disulfide cross-linking (21). This is consistent with the functionally rotating model that predicted that only one of three protomers would have a conformation favoring the disulfide bond formation between the two Cys residues introduced in the cleft. However, a more direct piece of evidence was desirable.
Double-Cys mutations, F666C/Q830C, were introduced into only one of the three acrB sequences in the linked acrB giant gene, and the plasmids pS(FQ-B2), pS(B-FQ-B), and pS(B2-FQ) were obtained. (Here the notation FQ-B2, for example, indicates the giant gene in which only the first acrB sequence was modified by the introduction of the F666C and Q830C mutations [FQ]). They were thus expected to produce the linked heterotrimers containing F666C/Q830C mutations in the first, second, and third acrB sequence, respectively. The plasmid pSCL-F666C/Q830C, containing a single acrB gene producing a monomeric mutant protein, was also used, as a control. We showed earlier (21) that the transport activity, lost in some double-Cys mutants of AcrB, was restored in the dsbA strain, which slows down disulfide-bond formation in the periplasm (2, 14). These plasmids were therefore introduced into a pair of isogenic strains, BL21YBR (ΔacrB ΔrecA) and BL21YBDR (ΔacrB dsbA1 ΔrecA), and their transport activities were evaluated on gradient plates containing cholate (Fig. 5).
FIG. 5.

The activities of linked-heterotrimer AcrB with double-Cys mutation in only one protomer in dsbA+ and dsbA host strains. Efflux activities were estimated from the levels of resistance to cholate by using the gradient plate method. Disulfide cross-linking in any of the protomers of the linked heterotrimer inactivated the entire complex, although residual activities were generated, presumably from the assembly of monomeric fragments. Since the levels of pumping activity in cells expressing the linked trimer were about 2.6 times and 1.8 times higher than that in cells expressing monomers from CL-AcrBHis in BL21YBR (see Fig. 3B) and BL21YBDR (data not shown), respectively, the relative activities shown were normalized to the activities of (CLB)3 and CL-AcrBHis for other linked heterotrimers and CL-F666C/Q830C, respectively. Gray bars, host strain BL21YBR; black bars, BL21YBDR. Error bars show standard deviations.
The activity of the monomeric double-Cys mutant CL-F666C/Q830C increased from 12% in the dsbA+ host strain BL21YBR to 47% in the dsbA strain BL21YBDR (Fig. 5), as expected. Interestingly, all three linked heterotrimers containing the F666C/Q830C double-Cys mutation in only one protomer showed a tendency similar to that of CL-F666C/Q830C; the activities were low in BL21YBR, in which disulfide-bond formation was allowed, but were restored in the dsbA host (from 19% in BL21YBR to 69% for FQ-B2, from 28% to 80% for B-FQ-B, and from 40% to 85% for B2-FQ, respectively). As was noted with the D407A mutation, apparently premature termination of transcription/translation allowed the generation of active monomers if the mutant unit was located closer to the 3′ terminus of the giant gene, explaining the higher activities of B-FQ-B and, especially, B2-FQ in BL21YBR. These results thus suggest strongly that the inactivation of any of the linked protomers through disulfide-bond formation in the cleft inactivated the entire AcrB heterotrimer, supporting the functionally rotating mechanism of AcrB. Western blot analysis of the whole-cell proteins showed that the expression levels of all three linked heterotrimers were similar to that of (CLB)3 in both host strains and significantly higher than that of the monomeric AcrB (data not shown).
Finally, in order to confirm further that disulfide-bond formation is responsible for the inactivation of linked AcrB heterotrimers, we performed a “real-time” cross-linking experiment using a fast-acting disulfide cross-linking agent, an MTS cross-linker, as described previously (21). MTS-2-MTS (∼5.2-Å spacer) was added to BL21YBDR cells expressing each linked heterotrimer with F666C/Q830C mutations in only one protomer in the presence of ethidium bromide. The linked heterotrimers were active in the efflux of ethidium, so that only very slow entry of ethidium, which causes fluorescence following its binding to nucleic acids, was seen initially (Fig. 6B, C, and D). However, upon the addition of MTS-2-MTS, cross-linking apparently occurred in one protomer, so that the entire linked AcrB trimer became inactivated regardless of the position of the altered protomer, inducing rapid ethidium accumulation and increased fluorescence (curve 3 in Fig. 6B, C, and D). These results again strongly support the notion that the trimer acts by a functionally rotating mechanism. In contrast, MTS-2-MTS had only a very small effect on the ethidium entry rate in cells expressing the cysteineless linked-trimer AcrB (CLB)3 (Fig. 6A). As a control reagent, a non-cross-linker, 5-MTS (whose length is similar to that of MTS-2-MTS) was used; this produced no AcrB inactivation in any of the mutants (Fig. 6). As another positive control, 40 μM (final concentration) of carbonyl cyanide m-chlorophenylhydrazone, a proton conductor, was added to the cells expressing (CLB)3, resulting in a rapid influx of ethidium because of the inactivation of AcrB due to the loss of the proton motive force (Fig. 6A).
FIG. 6.

Effect of cross-linker addition on ethidium accumulation in BL21YBDR cells expressing linked-heterotrimer AcrB with double-Cys mutation in only one protomer. Cellular accumulation of ethidium was monitored continuously by measuring the fluorescence of the ethidium-nucleic acid complex at excitation and emission wavelengths of 520 and 590 nm, respectively. After 2 min of incubation with 5 μM ethidium bromide (arrows), an MTS reagent or solvent alone (dimethyl sulfoxide-ethyl acetate [3:1, vol/vol]) was added to 2 ml of cell suspension. Additions were as follows: curve 1, 3.2 μl of solvent; curve 2, 4 μM 5-MTS (3.2 μl of a 2.5 mM stock solution); curve 3, 4 μM MTS-2-MTS (3.2 μl of a 2.5 mM stock solution). A.U., arbitrary unit.
DISCUSSION
The crystallographic elucidation of the structure of the asymmetric AcrB trimer (11, 17, 19) showed that each protomer takes a significantly different conformation from its neighbors, and the crystallization of drug-liganded AcrB allowed Murakami et al. (11) to propose that the three conformations represent the successive stages of drug transport, i.e., drug access, drug binding, and drug extrusion, the functionally rotating hypothesis. The conformational changes are especially pronounced in the periplasmic domain, resulting in the closure of the large, external cleft only in the extrusion protomer. Disulfide-induced closure of this cleft inactivated the AcrB transporter, a result consistent with this functional rotation mechanism (21).
We have earlier shown that mutations in the tightly interacting salt bridge/H-bonding (or proton relay) network in the transmembrane domain of AcrB inactivate the transporter and produce an extended alteration of its conformation, especially in the transmembrane domain (20, 22). In fact, the altered conformation of side-chains in the salt bridge/H-bonding network in these mutants almost exactly mimics that found in the extrusion protomer of the asymmetric AcrB trimer. Nevertheless, a large conformational alteration seen in the extrusion protomer of the wild-type AcrB could not be found in the trimers of our mutants, presumably because this conformational change requires the complementary, accommodative alterations in the conformations of the neighboring protomers.
Also, the functional rotation mechanism of AcrB predicts that if one of the three protomers is defective in the proton relay network, then the pumping action by the entire trimer comes to a halt.
According to the functionally rotating mechanism hypothesis, a heterotrimeric AcrB with one mutated, inactivated AcrB and two wild-type protomers should be inactive (and should allow the expected conformational alteration in the periplasmic domain of the mutant protein). We first tried to produce such heterotrimers by coexpression of wild-type and mutant AcrB. However, this approach allows too much random mixing of protomers, and the interpretation of the results became too complex. Thus, in this study, we developed an alternative approach of creating a giant gene in which three acrB sequences were connected together through short linker sequences. This linked trimer was expressed well as a single giant protein and produced levels of drug resistance often higher than that produced by the monomeric acrB gene.
The linked heterotrimer containing only one protomer with a proton relay network mutation lost its function. Furthermore, the experiment using the linked heterotrimer with disulfide cross-linking in only one protomer showed that the inactivation of any of the three protomers caused the loss of function of the entire complex. When only one of the three component AcrB proteins was made to contain the Phe666Cys and Gln830Cys mutations, then cross-linking of these two cysteine residues by a fast-acting MTS cross-linker instantaneously inactivated the entire trimer, regardless of the position of the altered protomer in the giant sequence (Fig. 6). These results strongly support the notion that the trimer acts by a functional rotating mechanism.
The interpretation of the results, however, was often not completely straightforward, because the plasmids expressing the mutant proteins sometimes gave low but significant activity (Table 2 and Fig. 3 and 5). We believe that this is due to the production of monomeric AcrB through degradation of the trimeric protein or through premature truncation in transcription or translation. The importance of the first factor is suggested by the observation that BL21-based host strains, lacking two proteases (lon and ompT), always gave cleaner results than the AG100-based strains (Fig. 3 and 4) and produced monomer and dimer bands of lesser density (Fig. 4). The significance of the latter factor is supported by the observation that the residual activity produced by the mutant genes was always stronger when the first and the second sequences were wild type, thus permitting the production of more active monomers from premature truncation processes.
In the host strain BL21YBR, the steady-state expression level of the linked trimer was much higher than that of the wild-type monomeric protein (Fig. 4), at least in part explaining the higher resistance caused by the linked trimer (Table 2 and Fig. 3B). We do not know what causes this high level of expression, but it seems possible that the covalent connection of protomers through the linker may cause a more efficient folding and assembly of the trimeric units. It is also unclear whether the difference between the BL21-derived hosts and the AG100-based hosts can be explained entirely by the absence of two proteases in the former, because these strains come from two very different backgrounds, the former from E. coli B and the latter from E. coli K12.
The giant gene containing more than one acrB unit could not be obtained with a very-high-copy-number plasmid, the pUC vector. It was observed earlier that the overexpression of AcrB alone is sometimes harmful to cells. A simple explanation for this phenomenon is that membrane integrity is damaged when excessive numbers of AcrB molecules are trying to become inserted. However, an interesting recent report (1) showed that BamA (formally YaeT) and AcrB were involved in contact-dependent growth inhibition in E. coli. In this light, overexpressed AcrB might cause growth inhibition of the cells through the increased regulation by this cell-to-cell-contact growth inhibition system. In this connection, AcrEF, a close homolog of AcrAB, is known to affect cell division (7).
Acknowledgments
This study was supported by a research grant from the U.S. Public Health Service (AI-09644).
We thank Jason A. Hall for constructive criticism.
Footnotes
Published ahead of print on 5 December 2008.
REFERENCES
- 1.Aoki, S. K., J. C. Malinverni, K. Jacoby, B. Thomas, R. Pamma, B. N. Trinh, S. Remers, J. Webb, B. A. Braaten, T. J. Silhavy, and D. A. Low. 2008. Contact-dependent growth inhibition requires the essential outer membrane protein BamA (YaeT) as the receptor and the inner membrane transport protein AcrB. Mol. Microbiol. 70323-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bardwell, J. C., K. McGovern, and J. Beckwith. 1991. Identification of a protein required for disulfide bond formation in vivo. Cell 67581-589. [DOI] [PubMed] [Google Scholar]
- 3.Bryson, V., and W. Szybalski. 1952. Microbial selection. Science 11645-51. [PubMed] [Google Scholar]
- 4.Churchward, G., D. Belin, and Y. Nagamine. 1984. A pSC101-derived plasmid which shows no sequence homology to other commonly used cloning vectors. Gene 31165-171. [DOI] [PubMed] [Google Scholar]
- 5.Elkins, C. A., and H. Nikaido. 2002. Substrate specificity of the RND-type multidrug efflux pumps AcrB and AcrD of Escherichia coli is determined predominantly by two large periplasmic loops. J. Bacteriol. 1846490-6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guan, L., and T. Nakae. 2001. Identification of essential charged residues in transmembrane segments of the multidrug transporter MexB of Pseudomonas aeruginosa. J. Bacteriol. 1831734-1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lau, S. Y., and H. I. Zgurskaya. 2005. Cell division defects in Escherichia coli deficient in the multidrug efflux transporter AcrEF-TolC. J. Bacteriol. 1877815-7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li, X. Z., and H. Nikaido. 2004. Efflux-mediated drug resistance in bacteria. Drugs 64159-204. [DOI] [PubMed] [Google Scholar]
- 9.Ma, D., D. N. Cook, M. Alberti, N. G. Pon, H. Nikaido, and J. E. Hearst. 1993. Molecular cloning and characterization of acrA and acrE genes of Escherichia coli. J. Bacteriol. 1756299-6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 11.Murakami, S., R. Nakashima, E. Yamashita, T. Matsumoto, and A. Yamaguchi. 2006. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature 443173-179. [DOI] [PubMed] [Google Scholar]
- 12.Murakami, S., R. Nakashima, E. Yamashita, and A. Yamaguchi. 2002. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature 419587-593. [DOI] [PubMed] [Google Scholar]
- 13.Nikaido, H. 1996. Multidrug efflux pumps of gram-negative bacteria. J. Bacteriol. 1785853-5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rietsch, A., D. Belin, N. Martin, and J. Beckwith. 1996. An in vivo pathway for disulfide bond isomerization in Escherichia coli. Proc. Natl. Acad. Sci. USA 9313048-13053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 16.Schägger, H., and G. von Jagow. 1991. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 199223-231. [DOI] [PubMed] [Google Scholar]
- 17.Seeger, M. A., A. Schiefner, T. Eicher, F. Verrey, K. Diederichs, and K. M. Pos. 2006. Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science 3131295-1298. [DOI] [PubMed] [Google Scholar]
- 18.Seeger, M. A., C. von Ballmoos, T. Eicher, L. Brandstatter, F. Verrey, K. Diederichs, and K. M. Pos. 2008. Engineered disulfide bonds support the functional rotation mechanism of multidrug efflux pump AcrB. Nat. Struct. Mol. Biol. 15199-205. [DOI] [PubMed] [Google Scholar]
- 19.Sennhauser, G., P. Amstutz, C. Briand, O. Storchenegger, and M. G. Grutter. 2007. Drug export pathway of multidrug exporter AcrB revealed by DARPin inhibitors. PLoS Biol. 5e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su, C. C., M. Li, R. Gu, Y. Takatsuka, G. McDermott, H. Nikaido, and E. W. Yu. 2006. Conformation of the AcrB multidrug efflux pump in mutants of the putative proton relay pathway. J. Bacteriol. 1887290-7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takatsuka, Y., and H. Nikaido. 2007. Site-directed disulfide cross-linking shows that cleft flexibility in the periplasmic domain is needed for the multidrug efflux pump AcrB of Escherichia coli. J. Bacteriol. 1898677-8684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takatsuka, Y., and H. Nikaido. 2006. Threonine-978 in the transmembrane segment of the multidrug efflux pump AcrB of Escherichia coli is crucial for drug transport as a probable component of the proton relay network. J. Bacteriol. 1887284-7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tseng, T. T., K. S. Gratwick, J. Kollman, D. Park, D. H. Nies, A. Goffeau, and M. H. Saier, Jr. 1999. The RND permease superfamily: an ancient, ubiquitous and diverse family that includes human disease and development proteins. J. Mol. Microbiol. Biotechnol. 1107-125. [PubMed] [Google Scholar]
- 24.Zgurskaya, H. I., and H. Nikaido. 1999. Bypassing the periplasm: reconstitution of the AcrAB multidrug efflux pump of Escherichia coli. Proc. Natl. Acad. Sci. USA 967190-7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ziha-Zarifi, I., C. Llanes, T. Köhler, J. C. Péchère, and P. Plésiat. 1999. In vivo emergence of multidrug-resistant mutants of Pseudomonas aeruginosa overexpressing the active efflux system MexA-MexB-OprM. Antimicrob. Agents Chemother. 43287-291. [DOI] [PMC free article] [PubMed] [Google Scholar]