

Abstract
Gene delivery has numerous potential applications both clinically and for basic science research. Non-viral vectors represent the long-term future of gene therapy and biomaterials are a critical component for the development of efficient delivery systems. Biomaterial development combined with fundamental studies of virus function and cellular processes will enable the molecular level design of modular vectors. Vectors are being developed based on cationic polymers or lipids that contain functional groups to mediate appropriate interactions with the extracellular environment or to interface with specific cellular processes. This review describes recent progress on the development of biomaterials for non-viral vectors and highlights opportunities for future development. Ultimately, efficient vectors will expand the traditional applications of gene therapy within the clinic and may enable numerous other opportunities within diagnostics, biotechnology, and basic science research.
Keywords: Gene therapy, Vector, Lipoplex, Polyplex, Cell trafficking
1. Introduction
Gene delivery has numerous potential applications both clinically and for basic science research. Typically, gene delivery refers to the use of nucleic acids, such as plasmids, to increase expression of a target protein; however, delivery of RNA (siRNA) and oligonucleotides can be used to decrease target protein production. Clinical therapies are perhaps the most ambitious applications of gene delivery, with ongoing clinical trials for cancer, monogeneic diseases, vascular disease, and infectious diseases [1]. As our understanding of biological processes and disease progression improves, clinical applications will potentially expand into areas such as neurological disorders and tissue regeneration. In addition to these therapeutic applications, gene delivery is used in vitro as a research tool to investigate gene function or regulation within a cellular and physiological context. Initial systems for highly parallel gene delivery have been developed, which could significantly impact biotechnology, diagnostic applications, and basic research [2]. These applications employ both viral and non-viral vectors; however, an increasing concern about immune responses to viral vectors in vivo combined with the ease of production and versatility of plasmids in vitro opens the door for non-viral vector development.
Biomaterial development combined with fundamental studies of virus function and cellular processes will enable the molecular level design of modular non-viral vectors. The term modular non-viral vector indicates that plasmid is complexed with cationic polymers or lipid formulations, which consists of a vector backbone modified with functional groups that mediate environmental interactions and intracellular trafficking to overcome the various barriers to gene transfer (Fig. 1). The vector backbone should enable the formation of stable complexes with nucleic acids, provide low cytotoxicity, and disassemble intracellularly to release the nucleic acid. Environmental interactions are manipulated through the incorporation of functional groups that may stabilize the vector in the extracellular milieu, target a specific tissue or cell type, or maintain the vector within the delivery location. Intracellular trafficking addresses the need to deliver the plasmid to the nucleus for expression, thus bioactive groups can be incorporated to facilitate endosomal escape or nuclear localization. Much of the information regarding these bioactive groups is derived from the study of viruses or the underlying cellular processes. The utility of non-viral vectors would increase with the ability to obtain transgene expression in difficult-to-transfect cell types (e.g. neurons), which currently limits their in vitro and in vivo application. Modifications to the vector or nucleic acids may increase the number of cells expressing the transgene or the duration of expression. The following paragraphs present recent progress in vector design for plasmid delivery and highlight opportunities for future development.
Fig. 1.

Modular design of non-viral vectors schematic. Modules associated with vector design are: vector backbone (grey), functional groups for regulating environmental interactions (purple), and intracellular trafficking (red). The vector backbone, typically containing polymers, lipids, or polysaccharides, is designed for DNA binding and complexation, which can protect against nuclease degradation, create a small, less negatively charged particle that can be internalized by cells, and facilitate some intracellular trafficking. The function of the vector backbone is being augmented by the attachment of groups that address the extracellular and intracellular barriers. The environmental functional groups can serve to limit interactions with serum components, promote specific cell binding or tissue targeting, or facilitate interactions with the extracellular matrix or biomaterials. The intracellular functional groups aim to enhance nuclear accumulation of the DNA either by facilitating endosomal escape, movement along the cytoskeleton, or nuclear pore trafficking. The individual modules can be assembled in different ways (a–c) for complexation with DNA (green), which may affect the structure and function of the resulting non-viral vector. (d) Schematic illustrating the distribution of the modules and DNA throughout the vector cross section, with the desired organization of functional groups regulating the environmental interactions presented primarily on the exterior and the groups for intracellular trafficking protected within the vector interior for activity following internalization. (e) Vectors are internalized by endocytosis and must subsequently escape the endosome for transport to the nucleus. Additionally, the modular components must dissociate from the DNA to allow for transcription. Adapted from Martin-Herranz [3].
2. Synthetic biomaterials for vector backbones
Numerous cationic lipids and polymers have been developed to package DNA for cellular internalization and protect it against degradation, leading to the identification of some structure–function design relationships (Table 1). Cationic lipids have three basic constituents: the polar headgroup, a linker, and a hydrophobic moiety. The cationic headgroup promotes interaction with DNA, whereas the hydrophobic moiety provides self-association to form either micelles or liposomes in the presence of a helper lipid, such as dioleylphosphatidylethanolamine (DOPE). Lipoplexes form a multilayered structure consisting of plasmid sandwiched between the cationic lipids, with hexagonal structures observed for some lipid compositions [7]. Several recent reviews have described the design of the cationic headgroup (e.g. type of amine, linear or T-shape), hydrophobic moiety (e.g. two linear C8–C18, cholesterol), and linker (e.g. degradable, tunable) for optimized transfection [8].
Table 1.
Representative examples of vector backbones for non-viral vectors






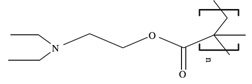

Cationic polymers can be classified in two groups: natural polymers, such as proteins and peptides, and synthetic polymers, such as polyethylenimine (PEI), dendrimers, and polyphosphoesters [9–11]. They spontaneously associate with plasmids by electrostatic interactions due to protonatable amine residues. The ratio of amines in the cationic polymer to phosphates on the plasmid, often referred to as the N/P ratio, affects the effective diameter and zeta potential [12]. High molecular weight polymers tend to form more stable, small complexes relative to low molecular weight polymers, yet low molecular weight polymers can enhance transfection efficiencies, likely due to a decreased cytotoxicity and the increased ability of the plasmid to dissociate from the cationic polymer [13,14]. Block copolymers can potentially regulate assembly and structure of the complex, while providing for multiple functions due to each component. Poly(2-diethylaminoethyl methacrylate) (PDEAEMA) or poly(2-dimethylaminoethyl methacrylate) (PDMAEMA), which have primary and secondary amines to facilitate complexation and intracellular trafficking, can be combined with poly(ethylene oxide) (PEO) or poly(propylene oxide) (PPO) to prevent aggregation and reduce toxicity [15].
The incorporation of polysaccharides into non-viral vectors has demonstrated the ability to reduce toxicity and provide greater stability in physiological fluids (e.g. serum). Polysaccharides (e.g. β-cyclodextrin, polyglucar-amidoamine) can be conjugated with linear cationic polymers allowing for self-assembly with plasmids [16–18]. Smaller carbohydrate moieties and larger distances of the carbohydrates from the charged amino centers produce more cell toxicity, with the latter also enhancing the delivery efficiency [17]. Interestingly, β-cyclodextrin may significantly reduce or avoid the inflammatory response induced by plasmid delivery, though the mechanism is not well understood. Alternatively, polysaccharides, such as chitosan and pullulan, can incorporate the plasmid into nanoparticles that can be internalized by cells [19,20]. In vitro gene delivery with these nanoparticles is similar to or less than traditional lipoplexes; however, in vivo delivery can be enhanced relative to naked DNA or lipoplexes [19].
Identifying the structure–function relationships for the cationic lipids and polymers can be facilitated by combinatorial chemistry techniques combined with high through-put screening procedures [12,21]. For the previously described polymers and lipids, structure–function relationships have been investigated through an iterative process, whereas combinatorial approaches can facilitate the generation and investigation of diverse libraries. Poly(β-amino esters) have been selected for their in vitro efficiency and their ability to be synthesized by the conjugate addition of primary or secondary amines to a diacrylate. The parallel syntheses of 20 different secondary amine monomers with seven different diacrylate monomers were initially screened for cellular internalization and transgene expression, leading to the identification of 10 candidate polymers [12]. These 10 polymers were subsequently examined for size, zeta potential, and the lysosomal pH environment. Polymers producing particles with diameters less than 250nm, positive zeta potentials, and a near neutral pH environment that suggests lysosome escape, produced the highest transfection efficiencies. A second-generation library was developed consisting of 70 different primary structures and 6–12 different molecular weights [21]. The most effective polymers had molecular weights greater than 10,000 Da, polymer/DNA ratios of 40:1 or higher, and condensed DNA into complexes with diameters less than 150 nm, with the smallest particles being the most effective. The top nine polymers were all formed from amino alcohols. For polymers that can be synthesized in this manner, this screening approach can significantly increase the rate at which chemical structures with maximal gene transfer can be identified.
3. Environmental interactions
For both in vitro and in vivo applications, non-viral vectors must be designed with consideration of their interactions with serum components, the extracellular milieu within the tissue or cell culture media, and binding to the cell surface. Many of the vectors described in the previous section interact with serum proteins that can inactivate the complex or promote clearance from the desired tissue, which limits the opportunity for cellular internalization. Cellular internalization can be enhanced by the addition of receptor ligands to the vector, which can increase binding to cell surface receptors and may allow for the targeting of specific cell types [22–26]. Interactions with serum components can be minimized by modifying the vector with PEG groups [27]. Alternatively, biomaterials can potentially shield vectors from the extracellular environment or prevent clearance from the tissue by encapsulation within a polymer or immobilization to a surface [28]. Biomaterials can be fabricated as nanospheres or microspheres for minimally invasive delivery or can be formed into three-dimensional structures for use in medical devices or tissue engineering. The latter structures, also termed scaffolds, function to create and maintain a space and provide mechanical support. They define a micro-environment designed to regulate cell migration, proliferation, and differentiation; however, applications to gene delivery will require that cellular processes associated with gene transfer are considered. For example, the mechanical properties of the material influence cell proliferation and differentiation and can also affect vector internalization and unpacking, and transgene expression [29].
Encapsulation within biomaterials can shield the vector from the extracellular environment and provide localized release. Naked plasmids have been encapsulated within biomaterials for sustained release, with the capacity to induce long-term gene expression and also promote tissue formation [30]. The surface area of the scaffold and DNA dose regulate the extent and duration of transgene expression. The release of DNA complexes can achieve similar or greater levels of transgene expression than naked DNA, while significantly reducing the quantities of DNA required [31]. The release profile for DNA complexes does not match that for naked DNA and modifications to the cationic polymers, such as PEGylation, have been incorporated to facilitate delivery and enhance transgene expression [32]. An approach similar to encapsulation involves multilayered polyelectrolyte assemblies consisting of alternate layers of DNA and a synthetic degradable polyamine [33]. Film growth up to 100 nm was observed, with DNA released from the surface and dissociated from the polyamine. Although this system only released naked DNA, it may be developed to release DNA complexes.
Alternatively, non-viral vectors can be designed to interact with biomaterial surfaces through either non-specific interactions or specific functional groups, resulting in their immobilization. This interaction between the vector and the material is similar to virus binding to extracellular matrix proteins [34]. Proteins are known to non-specifically adsorb to biomaterials through electrostatic, van der Waals, and hydrophobic interactions, and these interactions likely support the binding of DNA complexes. DNA complexes can adsorb directly to natural or synthetic biomaterials, or to pre-adsorbed proteins on these materials. The properties of the complex (e.g. size, N/P ratio) and the substrate (e.g. hydrophobicity) affect transfection. Hydrophilic, ionic surfaces support greater transgene expression than hydrophilic, non-ionic or hydrophobic surfaces [35]. Alternatively, functional groups can be attached to the complex that enables specific binding to complementary groups on the biomaterial. The biotin–avidin linkage has been utilized to immobilize poly(L-lysine) (PLL) and PEI complexes to a Neutravidin-modified surface [36]. The number of biotin groups in the complex and the distribution of biotin groups among the cationic polymers determined the transfection efficiency, likely due to the need to balance substrate binding with cellular internalization. The substrate-mediated delivery approach may be adaptable to numerous materials ranging from natural polysaccharides to synthetic polymers [37]. Taken together, these observations indicate that vectors can be designed for interaction with the extracellular milieu or with biomaterials designed for various applications (Table 2).
Table 2.
Functional groups incorporated into the vector for regulating environmental interactions
| Targeting ligands | Target | Cell types | Refs. |
|---|---|---|---|
| Folate | Folate receptor | Malignant cells e.g. ovarian carcinomas | [22] |
| Transferrin | Transferrin receptor | Malignant cells e.g. leukemia cells | [23] |
| RGD | Integrin | Broad range of cells e.g. endothelial cells | [24] |
| EGF (epidermal growth factor) | EGF receptor | Malignant cells e.g. epidermal carcinoma cells | [25] |
| Anti-CD3 | CD3 | T-cells | [26] |
| Vector stabilization | Mechanism | Refs. | |
| PEG (poly(ethylene glycol)) | Provides stability against serum components | [22] | |
| Polysaccharides (e.g. β-cyclodextrin, chitosan, polyglucaramidoamine) | Reduces toxicity and inflammation, provides stability against serum components | [17,19,52] | |
| Biomaterial immobilization | Material | Refs. | |
| Biotin | Neutravidin modified hyaluronic acid | [36] | |
| Non-specific interactions (e.g. charge) | Poly(lactide-co-glycolide) (PLG) | [30] | |
4. Intracellular trafficking
Following extracellular transport to the target cell, the vector is internalized and must be designed to mediate trafficking through the cell. Cellular internalization occurs primarily by endocytosis and the vectors must escape the endosome to avoid lysosomal degradation. The vector must then be transported to the nucleus and cross the nuclear membrane for subsequent transcription. Nuclear localization occurs either following cell division or through transport through the nuclear pore complex. The rate-limiting step for this system is dependent upon the vector. Lipoplexes were limited by cellular uptake, whereas polyplexes were limited by nuclear import [38]. Therefore, the incorporation of bioactive groups into the vector that interface with one or more of these cellular processes can potentially increase gene transfer efficiency (Table 3). Viruses, which are able to interface with the intracellular processes, require 2–3 orders of magnitude less DNA for transgene expression relative to non-viral vectors [38].
Table 3.
Intracellular trafficking can be enhanced by the incorporation of specific functional groups onto the vector backbone
| Endosomal escape | Mechanism | Sequence | Refs. |
|---|---|---|---|
| INF (influenza virus-derived sequence) | Endosomolytic at pH 5.0 by adopting an amphipathic α-helical conformation | GLFEAIEGFIENGWEGMIDGWYG (or GGC) | [41] |
| KALA | PH-dependent α-helical conformational change disrupts endosome | WEAKLAKALAKALAKHLAKALAKALKACEA | [43] |
| Nuclear localization | Nuclear binding protein | Sequence | Refs. |
| SV40 | Importin-α | PKKKRKV | [53] |
| M9 | Transportin | GNQSSNFGPMKGGNFGGRSSGPYGGGGQYFAKPRNQGGY | [54] |
| Pharmacological agents | Effect | Refs. | |
| Dexamethasone spermine | Reduces cytokine production, reduces inflammation | [47] | |
| Chloroquine | Enhances endosomal escape, but limited by systemic toxicity | [48] | |
Small molecule peptides and proteins have been attached to either the plasmid or to the cationic lipid or polymer to promote endosomal escape and nuclear localization [39,40]. Importantly, these bioactive factors must be incorporated without inhibiting their function or disrupting vector assembly. Fusogenic peptides have been incorporated to disrupt membranes, which may facilitate either cell entry without endocytosis or endosomal escape following endocytosis [41–43]. Nuclear localization can be enhanced by the attachment of nuclear localization signals (NLS), which are oligopeptides that bind to the cytoplasmic receptors (importins) responsible for binding and transport through the nuclear pore complex [44]. Nuclear localization is perhaps the most difficult hurdle in successful gene transfer and overcoming this step in transfection would enhance the application to non-dividing cells, which is common in vivo and in many difficult to transfect cell types in vitro. In dividing cells, plasmids can enter the nucleus when the nuclear membrane breaks down and then reforms. In non-dividing cells, the plasmid must cross the nuclear pore complex (NPC), which allows entry by free diffusion for only small molecules (≤60 kDa). However, success with NLS has been varied: some papers have reported that inclusion of NLS-containing proteins or peptides increases gene transfer and expression, while others have found no such enhancement [44].
Pharmacological agents have been employed to enhance the efficacy of the vector and promote transgene expression, which presents opportunities to translate them for in vivo application. A significant barrier to in vivo gene delivery is inflammation, as this response to the vector can reduce the biological activity of the secreted protein, eliminate transfected cells, or prevent repeated dosing [45]. Gene delivery can up-regulate inflammatory cytokines, such TNF-α, IL-1, IL-6, IL-12, and IFN-γ, which may inhibit promoter activity and attenuate gene transcription [45,46]. Glucocorticoid hormones, such as dexamethasone, delivered along with lipoplexes, showed reduced cytokine production and increased transfection relative to lipoplex delivery alone. A cationic corticosteroid was developed as a non-viral vector by coupling dexamethasone with spermine to create dexamethasone–spermine (DS) [47]. In vivo injection of an inflammatory agent showed that DS was able to reduce the accumulation of neutrophils, confirming that DS had anti-inflammatory activity. Another example of a pharmacological agent in gene delivery is chloroquine, which has been widely used in vitro to promote endosomal escape for non-viral vectors; however, the toxicity of this agent prohibits its systemic delivery in vivo [48]. These examples indicate the potential to incorporate pharmacologically active agents into non-viral vectors, which can reduce inflammation or alter other cellular processes to enhance transgene expression.
Numerous opportunities remain to increase gene transfer by designing vectors to interface with specific cellular processes, with functional groups derived from understanding viruses or cellular processes. For example, viral vectors use dynein-based active linear translocation along microtubules to travel at velocities that average an order of magnitude greater than for non-viral vectors [49]. Non-viral vectors engineered with functional groups that allow for directed motion along the cytoskeleton could increase accumulation at the nucleus and decrease the amount of DNA required. Some viruses are able to achieve stable transgene expression by insertion into defined locations within the chromosome. Defining a location within the chromosome for insertion is critical to avoid disruption of an essential gene [50]. Peptide nucleic acids (PNAs) have been developed that are able to bind tightly to specific DNA sequences. Incorporation of PNAs could potentially target the transgene to a specific chromosomal location. Alternatively, engineered zinc-finger proteins with nuclease activity were developed to recognize a unique chromosomal site and induce a double-strand break. At this break site, the chromosome can recombine with an extrachromosomal sequence of interest [51]. Finally, the nucleic acid itself could be engineered to avoid silencing by methylation, thereby extending the duration of transgene expression.
5. Conclusions
Non-viral vectors capable of efficient gene delivery would significantly impact in clinical therapies, diagnostics, and basic science research. The extracellular and intracellular barriers that must be overcome create opportunities for the development of non-viral vectors that have appropriate interactions with the extracellular milieu, while interfacing with specific intracellular processes. Although non-viral vectors are advantageous for their safety profile, relative ease of production, and versatility, their potential will not be realized until the development of efficient vectors capable of transfecting a variety of cell types, both in vitro and in vivo. Continued development of the structure–function relationships, combined with mathematical modeling and fundamental studies of viral function and cellular processes, will permit the molecular scale design of synthetic vectors. Ultimately, efficient vectors will expand the traditional applications of gene therapy within the clinic, and would provide an enabling technology for numerous other opportunities.
Acknowledgments
We would like to thank Kevin Rice, Todd Giorgio, and Theresa Reineke for helpful comments and discussions. Partial support was provided by NIH (RO1 GM066830 (LDS), R01 EB003806-01 (LDS)) and a Ford Foundation Fellowship (JCR).
Footnotes
Editor’s Note: Leading Opinions: This paper is one of a newly instituted series of scientific articles that provide evidence-based scientific opinions on topical and important issues in biomaterials science. They have some features of an invited editorial but are based on scientific facts, and some features of a review paper, without attempting to be comprehensive. These papers have been commissioned by the Editor-in-Chief and reviewed for factual, scientific content by referees.
References
- 1.http://www.wiley.co.uk/wileychi/genmed/clinical/index.html. In.
- 2.Ziauddin J, Sabatini DM. Microarrays of cells expressing defined cDNAs. Nature. 2001;411(6833):107–110. doi: 10.1038/35075114. [DOI] [PubMed] [Google Scholar]
- 3.Martin-Herranz A, Ahmad A, Evans HM, Ewert K, Schulze U, Safinya CR. Surface functionalized cationic lipid-DNA complexes for gene delivery: PEGylated lamellar complexes exhibit distinct DNA-DNA interaction regimes. Biophys J. 2004;86(2):1160–1168. doi: 10.1016/S0006-3495(04)74190-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boulanger C, Di Giorgio C, Gaucheron J, Vierling P. Transfection with fluorinated lipoplexes based on new fluorinated cationic lipids and in the presence of a bile salt surfactant. Bioconjug Chem. 2004;15(4):901–908. doi: 10.1021/bc049942+. [DOI] [PubMed] [Google Scholar]
- 5.Segura T, Shea LD. Materials for non-viral gene delivery. Annu Rev Mater Res. 2001;31:25–46. [Google Scholar]
- 6.Anderson DG, Lynn DM, Langer R. Semi-automated synthesis and screening of a large library of degradable cationic polymers for gene delivery. Angew Chem Int Ed Engl. 2003;42(27):3153–3158. doi: 10.1002/anie.200351244. [DOI] [PubMed] [Google Scholar]
- 7.Ewert K, Ahmad A, Evans HM, Safinya CR. Cationic lipid-DNA complexes for non-viral gene therapy: relating supramolecular structures to cellular pathways. Expert Opin Biol Ther. 2005;5(1):33–53. doi: 10.1517/14712598.5.1.33. [DOI] [PubMed] [Google Scholar]
- 8.Tranchant I, Thompson B, Nicolazzi C, Mignet N, Scherman D. Physicochemical optimisation of plasmid delivery by cationic lipids. J Gene Med. 2004;6 Suppl 1:S24–S35. doi: 10.1002/jgm.509. [DOI] [PubMed] [Google Scholar]
- 9.Boussif O, Lezoualc’h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, et al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci USA. 1995;92(16):7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Braun CS, Vetro JA, Tomalia DA, Koe GS, Koe JG, Russell Middaugh C. Structure/function relationships of polyamidoamine/DNA dendrimers as gene delivery vehicles. J Pharm Sci. 2005;94(2):423–436. doi: 10.1002/jps.20251. [DOI] [PubMed] [Google Scholar]
- 11.Wang J, Zhang PC, Mao HQ, Leong KW. Enhanced gene expression in mouse muscle by sustained release of plasmid DNA using PPE-EA as a carrier. Gene Ther. 2002;9(18):1254–1261. doi: 10.1038/sj.gt.3301794. [DOI] [PubMed] [Google Scholar]
- 12.Akinc A, Lynn DM, Anderson DG, Langer R. Parallel synthesis and biophysical characterization of a degradable polymer library for gene delivery. J Am Chem Soc. 2003;125(18):5316–5323. doi: 10.1021/ja034429c. [DOI] [PubMed] [Google Scholar]
- 13.Neu M, Fischer D, Kissel T. Recent advances in rational gene transfer vector design based on poly(ethylene imine) and its derivatives. J Gene Med. 2005;7(8):992–1009. doi: 10.1002/jgm.773. [DOI] [PubMed] [Google Scholar]
- 14.Schaffer DV, Fidelman NA, Dan N, Lauffenburger DA. Vector unpacking as a potential barrier for receptor-mediated polyplex gene delivery. Biotechnol Bioeng. 2000;67(5):598–606. doi: 10.1002/(sici)1097-0290(20000305)67:5<598::aid-bit10>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 15.Agarwal A, Unfer R, Mallapragada SK. Novel cationic pentablock copolymers as non-viral vectors for gene therapy. J Control Release. 2005;103(1):245–258. doi: 10.1016/j.jconrel.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 16.Reineke TM, Davis ME. Structural effects of carbohydrate-containing polycations on gene delivery. 1. Carbohydrate size and its distance from charge centers. Bioconjug Chem. 2003;14(1):247–254. doi: 10.1021/bc025592k. [DOI] [PubMed] [Google Scholar]
- 17.Popielarski SR, Mishra S, Davis ME. Structural effects of carbohydrate-containing polycations on gene delivery. 3. Cyclodextrin type and functionalization. Bioconjug Chem. 2003;14(3):672–678. doi: 10.1021/bc034010b. [DOI] [PubMed] [Google Scholar]
- 18.Liu Y, Reineke TM. Hydroxyl stereochemistry and amine number within poly(glycoamidoamine)s affect intracellular DNA delivery. J Am Chem Soc. 2005;127(9):3004–3015. doi: 10.1021/ja0436446. [DOI] [PubMed] [Google Scholar]
- 19.Leong KW, Mao HQ, Truong-Le VL, Roy K, Walsh SM, August JT. DNA-polycation nanospheres as non-viral gene delivery vehicles. J Control Release. 1998;53(1–3):183–193. doi: 10.1016/s0168-3659(97)00252-6. [DOI] [PubMed] [Google Scholar]
- 20.Gupta M, Gupta AK. Hydrogel pullulan nanoparticles encapsulating pBUDLacZ plasmid as an efficient gene delivery carrier. J Control Release. 2004;99(1):157–166. doi: 10.1016/j.jconrel.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 21.Anderson DG, Akinc A, Hossain N, Langer R. Structure/property studies of polymeric gene delivery using a library of poly(beta-amino esters) Mol Ther. 2005;11(3):426–434. doi: 10.1016/j.ymthe.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 22.Hwa Kim S, Hoon Jeong J, Joe CO, Gwan Park T. Folate receptor mediated intracellular protein delivery using PLL-PEG-FOL conjugate. J Control Release. 2005;103(3):625–634. doi: 10.1016/j.jconrel.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 23.Ogris M, Brunner S, Schuller S, Kircheis R, Wagner E. PEGylated DNA/transferring-PEI complexes: reduced interaction with blood components, extended circulation in blood and potential for systemic gene delivery. Gene Ther. 1999;6(4):595–605. doi: 10.1038/sj.gt.3300900. [DOI] [PubMed] [Google Scholar]
- 24.Kunath K, Merdan T, Hegener O, Haberlein H, Kissel T. Integrin targeting using RGD-PEI conjugates for in vitro gene transfer. J Gene Med. 2003;5(7):588–599. doi: 10.1002/jgm.382. [DOI] [PubMed] [Google Scholar]
- 25.Blessing T, Kursa M, Holzhauser R, Kircheis R, Wagner E. Different strategies for formation of pegylated EGF-conjugated PEI/DNA complexes for targeted gene delivery. Bioconjug Chem. 2001;12(4):529–537. doi: 10.1021/bc0001488. [DOI] [PubMed] [Google Scholar]
- 26.Kircheis R, Kichler A, Wallner G, Kursa M, Ogris M, Felzmann T, et al. Coupling of cell-binding ligands to polyethylenimine for targeted gene delivery. Gene Ther. 1997;4(5):409–418. doi: 10.1038/sj.gt.3300418. [DOI] [PubMed] [Google Scholar]
- 27.Kichler A. Gene transfer with modified polyethylenimines. J Gene Med. 2004;6 Suppl 1:S3–S10. doi: 10.1002/jgm.507. [DOI] [PubMed] [Google Scholar]
- 28.Pannier AK, Shea LD. Controlled release systems for DNA delivery. Mol Ther. 2004;10(1):19–26. doi: 10.1016/j.ymthe.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 29.Kong HJ, Liu J, Riddle K, Matsumoto T, Leach K, Mooney DJ. Non-viral gene delivery regulated by stiffness of cell adhesion substrates. Nat Mater. 2005 doi: 10.1038/nmat1392. [DOI] [PubMed] [Google Scholar]
- 30.Jang JH, Rives CB, Shea LD. Plasmid delivery in vivo from porous tissue engineering scaffolds: transgene expression and cellular transfection. Mol Ther. 2005;12(3):475–483. doi: 10.1016/j.ymthe.2005.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang YC, Riddle K, Rice KG, Mooney DJ. Long-term in vivo gene expression via delivery of PEI-DNA condensates from porous polymer scaffolds. Hum Gene Ther. 2005;16(5):609–617. doi: 10.1089/hum.2005.16.609. [DOI] [PubMed] [Google Scholar]
- 32.Scherer F, Schillinger U, Putz U, Stemberger A, Plank C. Nonviral vector loaded collagen sponges for sustained gene delivery in vitro and in vivo. J Gene Med. 2002;4(6):634–643. doi: 10.1002/jgm.298. [DOI] [PubMed] [Google Scholar]
- 33.Zhang J, Chua LS, Lynn DM. Multilayered thin films that sustain the release of functional DNA under physiological conditions. Langmuir. 2004;20(19):8015–8021. doi: 10.1021/la048888i. [DOI] [PubMed] [Google Scholar]
- 34.Bajaj B, Lei P, Andreadis ST. High efficiencies of gene transfer with immobilized recombinant retrovirus: kinetics and optimization. Biotechnol Prog. 2001;17(4):587–596. doi: 10.1021/bp010039n. [DOI] [PubMed] [Google Scholar]
- 35.Pannier AK, Anderson BC, Shea LD. Substrate-mediated delivery from self-assembled monolayers: effect of surface ionization, hydrophilicity, and patterning. Acta Biomater. 2005;1(5):511–522. doi: 10.1016/j.actbio.2005.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Segura T, Volk MJ, Shea LD. Substrate-mediated DNA delivery: role of the cationic polymer structure and extent of modification. J Control Release. 2003;93(1):69–84. doi: 10.1016/j.jconrel.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 37.Segura T, Chung PH, Shea LD. DNA delivery from hyaluronic acid-collagen hydrogels via a substrate-mediated approach. Biomaterials. 2005;26(13):1575–1584. doi: 10.1016/j.biomaterials.2004.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Varga CM, Tedford NC, Thomas M, Klibanov AM, Griffith LG, Lauffenburger DA. Quantitative comparison of polyethylenimine formulations and adenoviral vectors in terms of intracellular gene delivery processes. Gene Ther. 2005 doi: 10.1038/sj.gt.3302495. [DOI] [PubMed] [Google Scholar]
- 39.Morris MC, Chaloin L, Heitz F, Divita G. Translocating peptides and proteins and their use for gene delivery. Curr Opin Biotechnol. 2000;11(5):461–466. doi: 10.1016/s0958-1669(00)00128-2. [DOI] [PubMed] [Google Scholar]
- 40.Roth CM, Sundaram S. Engineering synthetic vectors for improved DNA delivery: insights from intracellular pathways. Annu Rev Biomed Eng. 2004;6:397–426. doi: 10.1146/annurev.bioeng.6.040803.140203. [DOI] [PubMed] [Google Scholar]
- 41.Kichler A, Mechtler K, Behr JP, Wagner E. Influence of membrane-active peptides on lipospermine/DNA complex mediated gene transfer. Bioconjug Chem. 1997;8(2):213–221. doi: 10.1021/bc970009z. [DOI] [PubMed] [Google Scholar]
- 42.Wyman TB, Nicol F, Zelphati O, Scaria PV, Plank C, Szoka FC., Jr. Design, synthesis, and characterization of a cationic peptide that binds to nucleic acids and permeabilizes bilayers. Biochemistry. 1997;36(10):3008–3017. doi: 10.1021/bi9618474. [DOI] [PubMed] [Google Scholar]
- 43.Baru M, Nahum O, Jaaro H, Sha’anani J, Nur I. Lysosome-disrupting peptide increases the efficiency of in-vivo gene transfer by liposome-encapsulated DNA. J Drug Target. 1998;6(3):191–199. doi: 10.3109/10611869808997893. [DOI] [PubMed] [Google Scholar]
- 44.Dean DA, Strong DD, Zimmer WE. Nuclear entry of nonviral vectors. Gene Ther. 2005;12(11):881–890. doi: 10.1038/sj.gt.3302534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Y, Lenert P, Weeratna R, McCluskie M, Wu T, Davis HL, et al. Identification of methylated CpG motifs as inhibitors of the immune stimulatory CpG motifs. Gene Ther. 2001;8(13):1024–1032. doi: 10.1038/sj.gt.3301482. [DOI] [PubMed] [Google Scholar]
- 46.Li S, Wu SP, Whitmore M, Loeffert EJ, Wang L, Watkins SC, et al. Effect of immune response on gene transfer to the lung via systemic administration of cationic lipidic vectors. Am J Physiol. 1999;276(5 Pt 1):L796–L804. doi: 10.1152/ajplung.1999.276.5.L796. [DOI] [PubMed] [Google Scholar]
- 47.Gruneich JA, Price A, Zhu J, Diamond SL. Cationic corticosteroid for nonviral gene delivery. Gene Ther. 2004;11(8):668–674. doi: 10.1038/sj.gt.3302214. [DOI] [PubMed] [Google Scholar]
- 48.Zhang X, Sawyer GJ, Dong X, Qiu Y, Collins L, Fabre JW. The in vivo use of chloroquine to promote non-viral gene delivery to the liver via the portal vein and bile duct. J Gene Med. 2003;5(3):209–218. doi: 10.1002/jgm.340. [DOI] [PubMed] [Google Scholar]
- 49.Kelkar SA, Pfister KK, Crystal RG, Leopold PL. Cytoplasmic dynein mediates adenovirus binding to microtubules. J Virol. 2004;78(18):10122–10132. doi: 10.1128/JVI.78.18.10122-10132.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Check E. Harmful potential of viral vectors fuels doubts over gene therapy. Nature. 2003;423(6940):573–574. doi: 10.1038/423573a. [DOI] [PubMed] [Google Scholar]
- 51.Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005 doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- 52.Liu Y, Wenning L, Lynch M, Reineke TM. New poly(d-glucaramidoamine) s induce DNA nanoparticle formation and efficient gene delivery into mammalian cells. J Am Chem Soc. 2004;126(24):7422–7423. doi: 10.1021/ja049831l. [DOI] [PubMed] [Google Scholar]
- 53.Ritter W, Plank C, Lausier J, Rudolph C, Zink D, Reinhardt D, et al. A novel transfecting peptide comprising a tetrameric nuclear localization sequence. J Mol Med. 2003;81(11):708–717. doi: 10.1007/s00109-003-0483-2. [DOI] [PubMed] [Google Scholar]
- 54.Bremner KH, Seymour LW, Logan A, Read ML. Factors influencing the ability of nuclear localization sequence peptides to enhance nonviral gene delivery. Bioconjug Chem. 2004;15(1):152–161. doi: 10.1021/bc034140k. [DOI] [PubMed] [Google Scholar]