

Conspectus

In the panoply of modern synthetic methods for forming carbon-carbon and carbon-heteroatom bonds, the transition metal-catalyzed cross coupling of organometallic nucleophiles with organic electrophiles enjoys a preeminent status. The preparative utility of these reactions is, in large measure, a consequence of the wide variety of organometallic donors that have been conscripted into service. The most common of these reagents are organic derivatives of tin, boron, and zinc, which each possess unique advantages and shortcomings. Because of their low cost, low toxicity, and high chemical stability, organosilanes have emerged as viable alternatives to the conventional reagents in recent years. However, unlike the tin- and zinc-based reactions that require no activation or the boron-based reactions that require only heating with mild bases, silicon-based cross-coupling reactions often require heating in the presence of a fluoride source; this has significantly hampered the widespread acceptance of organosilanes.
To address the “fluoride problem”, we have introduced a new paradigm for palladium-catalyzed, silicon-based cross-coupling reactions that employs organosilanols, a previously underutilized class of silicon reagents. The use of organosilanols either in the presence of Brønsted bases or as their silanolate salts represents a simple and mild alternative to the classic fluoride-based activation method. Organosilanols are easily available by many well-established methods for introducing carbon-silicon bonds onto alkenes, alkynes and arenes, and heteroarenes. Moreover, we have developed four different protocols for the generation of alkali metal salts of, vinyl-, alkenyl-, alkynyl-, aryl-, and heteroarylsilanolates: (1) reversible deprotonation with weak Brønsted bases, (2) irreversible deprotonation with strong Brønsted bases, (3) isolation of the salts from irreversible deprotonation, and (4) silanolate exchange with disiloxanes. We have demonstrated the advantages of each of these methods for a number of different coupling classes.
The defining feature of this new process is the formation of a covalently linked palladium silanolate species that facilitates the critical transmetalation step. We have verified the intermediacy of a critical species that contains the key Si-O-Pd linkage by its identification as the resting state in reaction mixtures, by X-ray analysis, and by demonstrating its competence in thermal cross-coupling with no additives. Our conclusions contradict the long-standing dogma that silicon-based cross-coupling reactions require the generation of a pentacoordinate siliconate prior to transmetalation. This revelation has opened a new vista for discovery of new reactions that involve this critical process.
“...Work in this field (silicon-based cross-coupling) has been quite active since the initial disclosure of siletane cross-coupling from these laboratories... Future studies are focused on several fronts, including the extension of scope to incorporate less reactive substrates such as chlorides and triflates, further optimization of alkenyl/alkenyl coupling, and creation of an efficient biaryl (and heteroaryl) synthesis using silanols. In addition, synthetic endeavors are underway that highlight the power of this cross-coupling though key steps in the total synthesis of natural products. It must also be noted that the mechanistic work is still at its infancy, and more detailed analysis of the reaction through kinetics and reactive-intermediate isolation is underway...”
—Acc. Chem. Res. 2002, 35, 835.
Introduction
Transition-metal-catalyzed, cross-coupling reactions are now considered to be among the most general methods for the formation of carbon-carbon and carbon-heteroatom bonds.1 Since the independent reports from Kumada and Corriu,2 on the reaction of organomagnesium reagents with organic halides, numerous other organometallic nucleophiles have been developed with varying levels of utility. The most popular among these are organotin, organoboron, and organozinc reagents because of their stability and good functional group compatibility.1 Nevertheless, an enduring objective in this field is the search for reactions that proceed under mild conditions without toxic byproducts and that employ cheap, readily available starting materials.
In recent years, the cross-coupling reactions of organosilicon compounds have emerged as viable alternatives.3 The development of organosilanes as cross-coupling partners has been slower than that of the other organometallic compounds because of the lower reactivity of organosilanes. However, in 1989, Hiyama reported a major breakthrough by capitalizing on the high affinity of fluoride for organosilanes,4 to enhance the rate of transmetalation through the formation of pentacoordinate siliconates.5 In the pentacoordinate complex, transmetalation is facilitated by the enhanced polarization of the Si-C bond. Since the pioneering work of Hiyama and Hatanaka,6 organosilanes have achieved broader utility through the introduction of heteroatoms7 on the silicon moiety. In particular, chloro-, and fluorosilanes are more potent cross-coupling partners, but they are also hydrolytically sensitive and thus more difficult to handle. Accordingly, heteroatom surrogates (siletanes, 2-pyridyl-, 2-thienyl-, and benzylsilanes,)8 have been introduced, that have a greater stability and can be converted to more reactive heterofunctional silanes in the presence of fluoride sources.7,8 For example, studies from these laboratories showed that siletane (E)-1 undergoes facile cross-coupling with aryl halides under standard conditions in the presence of tetrabutylammonium fluoride (TBAF·3H2O). Subsequent studies revealed that (E)-1 suffers rapid ring opening with TBAF·3H2O to afford a mixture of silanol 2 and disiloxane 3 (Scheme 1). The dimethyl analogs of both the silanol and the disiloxane were independently synthesized and both underwent smooth cross-coupling with similar substrates to provide comparable yields of coupling products.9
Scheme 1.

Kinetic analysis (of reactions of the dimethyl analogs), provided a mechanistic picture that is consistent with a turnover limiting transmetalation from a fluoride-activated disiloxane (E,E)-7 to the organopalladium halide complex (Scheme 2) in line with the Hiyama-Hatanaka paradigm.10
Scheme 2.

Many useful cross-coupling reactions have been developed on the basis of these findings. For example, cross-coupling of siletanes, silanols, silyl hydrides, cyclic silyl ethers, disiloxanes, and oligosiloxanes, all proceed efficiently and stereospecifically under mild reaction conditions, with good functional group compatibility and easily removed byproducts.11 Although the scope and selectivity of these transformations are high, the requirement for superstoichiometric amounts of fluoride is a significant drawback. To achieve the same breadth and utility enjoyed by organostannanes and organoboranes, the requirement to use fluoride as a means of activation must be eliminated so that the reaction would be compatible with substrates bearing silylprotecting groups and be amenable to large-scale operation.
The development of non-fluoride-based cross-coupling reactions of organosilanes was a challenging prospect given the reigning dogma that a pentacoordinate siliconate was required to effect transmetalation to an organopalladium halide. How else can the Si-C bond be activated for transmetalation in the absence of fluoride? Initial hypotheses proposed that the conjugate base of silanol (E)-4 could serve this function in two ways (Figure 1): (1) the in-situ-generated silanolate (i) could form an organopalladium(II) silanolate complex (ii) by the displacement of the halide from the organopalladium(II) complex (ArylPdLnX), and (2) a second equiv of i could serve as the nucleophilic activator to form the desired pentacoordinate siliconate (iii). This species would undergo intramolecular transmetalation with simultaneous formation of polysiloxanes. In this proposal, the role of the second silanolate is analogous to the established role of fluoride in the preceding transformation.
Figure 1.

Early proposal for the mechanism for the fluoride-free cross-coupling of silanols.
In the event, the use of potassium trimethylsilanolate (KOSiMe3) with (E)-4 and (Z)-4 or their independently prepared conjugate bases underwent clean cross-coupling in DME with a range of aryl iodides (Scheme 3).12 This discovery was a watershed event in the development of a new paradigm for silicon-based cross-coupling reactions. These early observations launched campaigns on both preparative and mechanistic fronts to map out the uncharted landscape before us. This Account chronicles the most recent advances in the evolution of alkali organosilanolate cross-coupling reactions, provides mechanistic insight into reactive intermediates, and summarizes the scope and applicability of this new method. Although we thought that a simple solution to the “fluoride problem” had been found, we did not anticipate how intellectually stimulating this investigation would be, and what surprises lay waiting to be discovered.
Scheme 3.

Kinetic Analysis and Mechanistic Implications
The palladium-catalyzed cross-coupling reactions of various organometallic donors involve three elementary steps: (1) oxidative addition the transition metal catalyst to the halide or pseudohalide, (2) transmetalation of the organometallic reagent to provide a diorganopalladium(II) complex and (3) reductive elimination of the diorganopalladium(II) complex to form the product with regeneration of the palladium(0) catalyst.13 Of the three elementary steps, the transmetalation remains the most controversial and likely depends upon the organometallic donor.14
Unlike the fluoride-promoted reaction of organosilanols, the Brønsted base-promoted reaction exhibits significant rate dependence on the reaction conditions and on the steric and electronic properties of the silicon center.15 Accordingly, it was not prudent to assume that the fluoride-free process operates by the same mechanism as the TBAF-promoted cross-coupling.10,16 Thus, a full kinetic analysis was undertaken with the following objectives in mind: (1) determine the intermediacy of a tetracoordinate species (ii, Figure 1) (2) determine if ii undergoes anionic activation by a second equivalent of silanolate, (3) provide insight into the transmetalation event of organosilanols and, (4) isolate or spectroscopically characterize the catalyst resting state.
To test the validity of the proposed mechanism, the reaction order with respect to each component in the cross-coupling reaction of K+(E)-4- with 2-iodothiophene was determined (Figure 2).17 The experimentally derived rate equation is:16
| (eq. 1) |
| (eq. 2) |
| (eq. 3) |
Figure 2.

Revised mechanistic proposal for the fluoride-free cross-coupling.
This equation is consistent with the saturation of a reactive intermediate prior to transmetalation, but it was not clear whether that species is ii or iii. These possibilities were distinguished by kinetic analysis using a superstoichiometric amount of palladium relative to K+(E)-4-. Under these conditions, a first-order dependence on silanolate was observed. These critical experiments revealed an inconsistency with our original proposal (Figure 1).16 If iii were a reactive intermediate prior to transmetalation a second order dependence on K+(E)-4- should be observed with a superstoichiometric loading of palladium and substoichiometric amount of K+(E)-4-. This result suggests that the transmetalation proceeds directly from an arylpalladium(II)silanolate ii complex (Figure 2). These data also imply that the first-order dependence on K+(E)-4- (under catalytic conditions) is consistent with a turnover-limiting bimolecular displacement of halide by K+(E)-4- (i to ii). Therefore, at high concentrations of K+(E)-4- intramolecular transmetalation from an arylpalladium(II) silanolate (ii to iv) becomes turnover limiting.
These results mandated a revision of the original mechanistic proposal to incorporate an intramolecular transmetalation from a neutral species (Figure 2, ii to iv), thus contradicting the dogma that silicon-based cross-coupling reactions require the generation of a pentacoordinate silicate prior to transmetalation (Hiyama-Hatanaka paradigm). Furthermore, these observations illustrate the critical importance of the Si-O-Pd linkage for this new transmetalation pathway. To confirm this new hypothesis, additional evidence was needed to support the intermediacy of ii by its isolation and demonstration of its kinetic competence in the absence of additional silanolate. Unfortunately, the high reactivity of K+(E)-4- precluded the isolation of any intermediates.
The problem was addressed by making recourse to the palladium-catalyzed cross-coupling reactions of aryldimethylsilanolates, which are considerably slower compared to their alkenyl congeners (Figure 3).18
Figure 3.
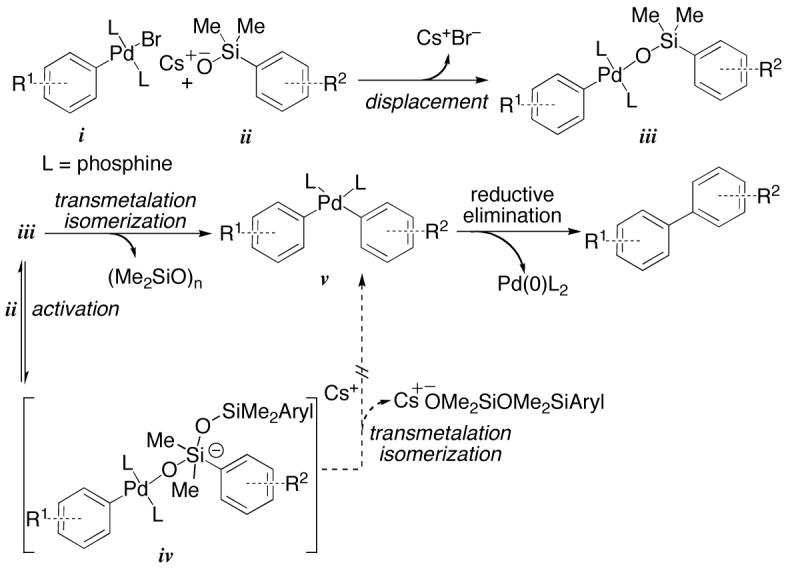
Mechanistic proposal for the coupling of aryldimethylsilanols.
Kinetic analysis of the reaction of cesium silanolate (Cs+10-) with 2-bromotoluene in the presence of allylpalladium chloride dimer ([allylPdCl]2), at 100 °C established that the cross-coupling is in the same mechanistic regime as K+(E)-4- (equation 4).
 |
(eq. 4) |
This rate equation is consistent with a turnover-limiting transmetalation of an arylpalladium(II) silanolate and is similar to the observed rate equation for the cross-coupling of K+(E)-4- (equation 3). These data once again rule out activation by a second equivalent of silanolate to generate a pentacoordinate siliconate prior to transmetalation. Moreover, the key arylpalladium(II) silanolate intermediate (iii) could be detected by 31P NMR spectroscopy in reaction mixtures (L-L = dppp). Finally, complex 12 was independently synthesized and fully characterized including single crystal X-ray analysis (Figure 4).19
Figure 4.
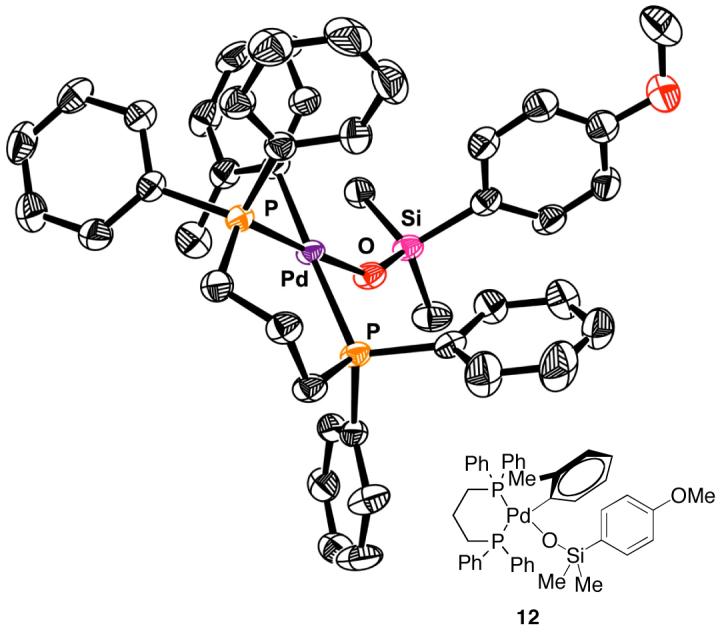
X-ray crystal structure of complex 12 (ORTEP drawing with thermal ellipsoids drawn at 50% probability level).
The isolation of 12 allowed for the demonstration that arylpalladium(II) silanolate complexes can undergo transmetalation in the absence of an activator. Heating 12 to 100 °C provides the biaryl product in quantitative yield. This result unambiguously demonstrates that a neutral arylpalladium(II) silanolate complex can undergo direct transmetalation to give a cross-coupled product.
The mechanistic studies forced a radical rethinking of how the fluoride-free cross-coupling reactions occur. Through a combination of kinetics, spectroscopy and synthesis, a new pathway was elucidated in which transmetalation occurs from a tetracoordinate species containing an Si-O-Pd linkage. This discovery led to the design of new silicon-based cross-coupling reactions, improvements upon existing methods, and application in total synthesis.
Methods Development and Applications
Methods for Fluoride-Free Activation of Organosilanols
The crucial discovery that the fluoride-free cross-coupling of silanols proceeds through an Si-O-Pd linkage, stimulated development of conditions for the formation of metal silanolate precursors. To date a wide variety of Brønsted bases have been implemented for the formation of silanolates including: KOSiMe3, Cs2CO3, NaOt-Bu, NaH, NaHMDS, KH, and Cs(0). Presently, four different modes of activation have been developed: (1) reversible deprotonation, (2) irreversible deprotonation, (3) isolation, and (4) silanolate exchange. The experimental protocol, scope and utility of each mode of activation are detailed below.
1. Reversible Deprotonation
1.1. Alkenyl Metal Silanolates
The first studies to expand the scope of silanolate cross-coupling reactions focused on alkenyldimethylsilanols such as (E)-4 and (Z)-4.12,20 With 2.0 equiv of KOSiMe3 in DME at ambient temperature, the cross-coupling of (E)-4 and (Z)-4 with a variety of aryl iodides proceeds with high generality and stereospecificity (Scheme 4). These conditions are mild enough to engage halides bearing esters, ketones and silyl-protected substrates for the generation of both (E)-13, and (Z)-13.
Scheme 4.

1.2. Alkynyl Silanolates
Conjugated alkynes play a major role in synthetic, pharmaceutical, and polymer industries.21 Since the introduction of the Sonogashira reaction22 a wide variety of alkynyl organometallic reagents have been developed as cross-coupling partners.23 Alkynylsilicon reagents do participate in the Sonogashira reaction; however, they are usually activated by fluoride ion24a-b and / or silver salts.24c-d Hence, it was of interest to investigate if alkynylsilanols could be activated by KOSiMe3 for this process.
Initial studies revealed that dimethyl(1-heptynyl)silanol 14 is a much more effective coupling partner than 1-heptyne or (1-heptynyl)trimethylsilane under silanolate activation.25 With 2.0 equiv of KOSiMe3, (Ph3P)2PdCl2, and CuI, the cross-coupling reaction of 14 with 4-iodoanisole is complete after 3 h whereas 1-heptyne, (1-heptynyl)trimethyl silane afford <20% conversion. Clearly, KOSiMe3 must activate the silanol without cleavage of the Si-C bond, because in-situ deprotection would give identical rates for alkynes 14 and 1-heptyne. The scope of the electrophile in this process is good; both electron rich and electron deficient aryl iodides bearing a variety of functional groups provide the desired products in high yields (Scheme 5).
Scheme 5.

Aryl Silanolates
In contrast to alkenyl- and alkynylsilanolates, arylsilanolates are less reactive cross-coupling partners, and require more forcing conditions to undergo productive reaction. After thorough optimization we found that Cs2CO3 in toluene at 90 °C effects the cross-coupling of dimethyl(4-methoxyphenyl)silanol 16 with ethyl 4-iodobenzoate.18 Optimization studies revealed that the level of hydration of Cs2CO3 significantly influences the overall conversion. The addition of 3.0 equiv of water per equiv of Cs2CO3 proves to be general for reactions of a range of aryl bromides and aryl iodides in the presence of [allylPdCl]2 with dppb or Ph3As to suppress homocoupling of the halide (Scheme 6). The scope in the aryl halide partner is broad, however a similar range is not found for the dimethyl(aryl)silanol component. For example, cross-coupling of dimethyl(4-trifluorotolyl)silanol with 4-bromoanisole provides the product with poor selectivity and low yield (54%). A solution to these problems will be addressed in the Isolation section below.
Scheme 6.

Five-Membered Heterocyclic Silanolates
Five-membered heterocycles are well represented within the fields of pharmaceutical, materials and natural product chemistry. Moreover, indole-containing compounds are among the most common heterocycles in nature, and the broad range of biological activity has stimulated research into their construction for decades.26
Given the importance of 2-substituted indoles, N-Boc-dimethyl(2-indolyl)silanol (18) and N-methyl-dimethyl(2-indolyl)silanol (19) were prepared to evaluate the utility of silanol-based reagents.27 The sodium silanolates of 18 and 19 are formed in situ under the action of NaOt-Bu and these salts are excellent coupling partners for a range of aryl halides. Optimal conditions for the cross-coupling reaction of 18 and 19 employ NaOt-Bu (2.0 equiv), [Pd2(dba)3]·CHCl3 (5 mol %), and CuI (1.0 equiv). A variety of substrates including electron-rich, electron-poor, and 2-substituted aryl iodides react smoothly under these conditions (Scheme 7).
Scheme 7.

However, the cross-coupling of 19 with electron-deficient aryl iodides affords the desired product in lower yields because of the formation of the 2,3-bisaryl indole as the major product. Fortunately, the less reactive aryl bromides led to the generation of the desired products in good yields (Scheme 8).
Scheme 8.

4-Isoxazolylsilanols (22) are also viable substrates for the cross-coupling reaction under NaOt-Bu activation. The isoxazole is constructed by dipolar cycloaddition of alkynyldimethylsilyl ethers (23) and in-situ prepared aryl nitrile oxides (Scheme 9). The silicon function controls the regioselectivity of the [3+2] cycloaddition and it enables further functionalization through the cross-coupling reaction. Optimal conditions for the coupling employ NaOt-Bu and [Pd2(dba)3]·CHCl3 in toluene or dioxane at 80 °C to produce the 3,4,5-trisubstituted isoxazoles 25.28
Scheme 9.

1.6. Synthetic Application
The reversible deprotonation protocol was highlighted in the total synthesis of the antifungal agent, (+)-papulacandin D (26).29 One of the key steps in the synthesis is the cross-coupling reaction of glucal silanol 27 with aromatic iodide 28 (Scheme 10).
Scheme 10.

This cross-coupling reaction is very challenging because the aromatic iodide is hindered and the protected glucal silanol is sensitive to strongly basic conditions. Fortunately, it was found that the use of NaOt-Bu (2.0 equiv) and Pd2(dba)3·CHCl3 (5 mol %) with an equal molar ratio of 27 and 28 in toluene at 50 °C provides the desired (1-aryl)hexenopyranose 29 in 82% yield. In this single transformation the entire carbon-skeleton of the spiroketal portion of (+)- papulacandin D was assembled. The mildness of this method is highlighted by the compatibility of the sodium silanolate of 27 with different silicon protecting groups.
Finally, a strategy that combines both fluoride-free and fluoride-promoted reactions has been developed for the sequential cross-coupling reaction of differently functionalized 1,4-bissilylbutadienes.30 One end of the 1,4-bissilylbutadiene bears a silanol that can easily be converted to the potassium silanolate with KOSiMe3. The other end bears a “safety-catch” silanol surrogate that is revealed to the corresponding silanol upon treatment with TBAF.
Under KOSiMe3 activation, silanol 30 exhibits reactivity similar to the alkenylsilanol (E)-4 discussed above. With KOSiMe3 (2.0 equiv) and [Pd(dba)2] (2.5 mol %) in dioxane at ambient temperature a wide range of substituted aryl iodides provide the dienylsilanes 31 in good yields (Scheme 11). The presence of electron-rich or deficient groups on the aryl iodide has little to no effect on the rate of the cross-coupling reaction. Under fluoride activation, the silanol is revealed from the benzylsilane and the cross-coupling proceeds smoothly with a variety of aryl iodides in good yields (Scheme 11). A more dramatic substituent effect was observed in the fluoride-promoted reaction manifold. For example, it was found that the cross-coupling reaction with electron-rich substrates is significantly faster that the corresponding reactions with the electronically neutral dienylsilanes, and electron-poor dienylsilanes (e.g. 31a) undergo a competitive migration of the benzyl group to the diene.
Scheme 11.

This problem was remedied by replacing the benzyl with a 2-thienyl group in 33 (Scheme 12). This group survives the initial Brønsted base-promoted cross-coupling and provides the desired silanol upon treatment with TBAF without the undesired migration.
Scheme 12.

2. Irreversible Deprotonation
2.1. Heterocyclic Silanolates
Although the cross-coupling reaction of 18 is successful with NaOt-Bu alone (no CuI), a minor amount of the protiodesilylated product is observed. This problem can be avoided by the stoichiometric generation of sodium N-Boc-dimethyl (2-indolyl)silanolate (Na+18-) through deprotonation of 18 with NaH.27 The in-situ preparation of Na+18- provides an active reagent for the cross-coupling of aryl iodides and extends the scope of the cross-coupling partners to aryl iodides containing esters and nitriles without the need for CuI (Scheme 13).
Scheme 13.

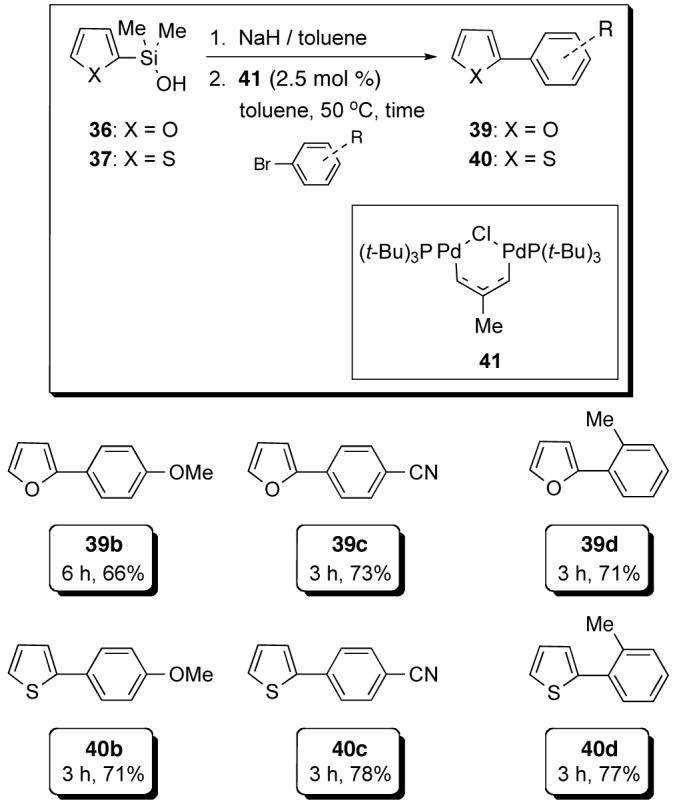
The irreversible deprotonation protocol can be employed in the cross-coupling reactions of other π-rich heterocycles with aryl iodides and bromides.27 The preformed sodium N-Boc-dimethyl(2-pyrrolyl)silanolate Na+35- cross-couples smoothly electron-deficient, electron-rich and 2-substituted aryl iodides (Scheme 14). The cross-coupling reactions of both preformed sodium 2-furylsilanolate Na+36- and 2-thienylsilanolate Na+37- proceed in an analogous fashion to Na+35- with electron-deficient and electron-rich aryl iodides. In addition, the cross-coupling of Na+36- and Na+37- with aryl bromides can be effected with the Pd(I) catalyst 41 (Scheme 15).
Scheme 14.

Scheme 15.
Unfortunately, the preformation protocol could not be extended to N-Boc-dimethyl[(5-methoxy)-2-indolyl]silanol 42. Attempts to prepare Na+42- with 1.0 equiv of NaH led to ∼20% protiodesilylation. The problem of protiodesilylation in the preformation of 42 could be solved by the use of NaHMDS which provided an 82% yield of the desired cross-coupling product 43 after 12 h at 40 °C (equation 5). This form of activation can also be used with other silanolate precursors that are sensitive to protiodesilylation such as 44 and 45 (Scheme 16).27
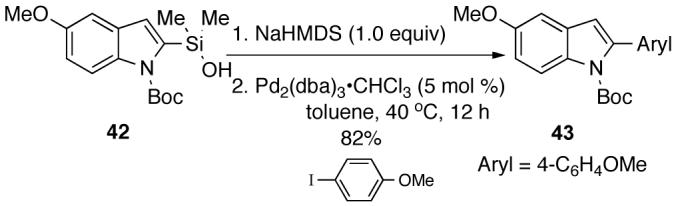 |
(eq. 5) |
Scheme 16.

2.2. Cross-Coupling of Alkenylsilanolates with Aryl Chlorides
Although silanols (E)-4 and (Z)-4 couple easily with a range of aryl iodides, these electrophiles have obvious disadvantages compared to the corresponding aryl chlorides (higher cost, lesser availability, higher molecular weight). In recent years, the development of ligands that facilitate the oxidative additions of the stronger C-Cl bonds31 to palladium(0) catalysts has made the cross-coupling of aryl chlorides possible.32 However, the use of silanolate salts in cross-coupling reactions would still require the displacement of a stronger Pd-Cl bond and as such could change the turnover limiting step.
Optimization experiments began with the combination of K+(E)-4-with the biphenyl-based ligands developed by Buchwald33 in the presence of [allylPdCl]2.34 The use of 1.3 equiv of K+(E)-4-, [allylPdCl]2, and 47 (S-Phos) in THF at 60 °C provides excellent yield of 13 with a variety of aryl chlorides bearing nitrile, ester, nitro and ketone substituents. Furthermore, pyridines react smoothly, as do mono- and diortho-substituted substrates (Scheme 17). In all cases the cross-coupling reaction is highly stereospecific, even for K+(Z)-4-. The scope in silanolate was further expanded to (E)- and (Z)-styrylsilanolates, and tri- and tetrasubstituted alkenylsilanolates with similar results.
Scheme 17.

2.3. Application of Irreversible Deprotonation in Total Synthesis
The synthetic utility of the irreversible deprotonation protocol was illustrated in the total synthesis of the polyene macrolide RK-397.35 Retrosynthetic analysis reveled that the polyene chain could be accessed rapidly through the sequential cross-coupling reaction of 30 to prepare the tetraene portion of the polyene phosphonate 48 (Scheme 18).
Scheme 18.


The first cross-coupling reaction is initiated by the preformation of Na+30- with NaH. The resulting silanolate reacts smoothly with 50 in the presence of Pd2(dba)3·CHCl3, to afford triene 51 in 77% yield. The second cross-coupling reaction is initiated by in-situ unmasking of the benzylsilane, to the corresponding silanol in with TBAF·3H2O. The cross-coupling reaction or 51 with (E)-iodopropenoate 49 provides the key tetraene 52 in 79% yield (Scheme 18).
3. Isolation
To streamline the manipulation of (aryl)- and (heteroaryl)dimethyl-silanolates, their salts can be isolated from the irreversible deprotonation protocol. The salts are stable, storable, solids that can be charged directly into a reaction mixture and are competent nucleophiles for a broad range of organic halides.
3.1. Sodium N-SEM-Dimethyl(2-indolyl)silanolate
The sodium salt of N-SEM-dimethyl(2-indolyl)silanolate (53) is easily prepared following the irreversible deprotonation protocol using NaH. Removal of the solvent affords Na+53- as a colorless, free-flowing solid. This silanolate undergoes smooth cross coupling with aryl chlorides (under conditions developed for K+4-) to provide excellent yields of 2-substituted indoles 54 with a broad substrate scope (Scheme 19).27
Scheme 19.

3.2. Aryldimethylsilanolates
Although the rates and yields of the cross-coupling reactions of 16 promoted by Cs2CO3 were good, other arylsilanols are poor substrates. The potassium salt K+16- was prepared to evaluate the influence of experimental conditions on rate of cross-coupling of a stoichiometrically formed silanolate with aryl bromides. Not surprisingly, a substantial rate increase was observed (compared to the use of KOSiMe3) when the isolated arylsilanolate is used. Moreover, a significant rate effect was observed upon the addition of 1-5 mol % of dppbO2 to a mixture of K+16- with 4-iodoanisole and 2.5 mol % of [allylPdCl]2 in toluene at 90 °C.36 With this additive, complete conversion is observed in 15 min compared to ca. 180 min under the previous conditions (equation 6).
 |
(eq. 6) |
The general utility of phosphine oxides in preparative reactions of aryl bromides was established by a survey in the presence of 5 mol % of Ph3P=O with 2.5 mol % of [allylPdCl]2. Both electron-deficient and electron-rich aryl bromides reacted within 1 h to afford moderate yields of the desired unsymmetrical biaryl products (Scheme 20).36
Scheme 20.

Preliminary results show that a broader range silanolates can participate in aryl-aryl cross-coupling reactions when (t-Bu3P)2Pd is employed as the catalyst.37 Substrates such as potassium (4-trifluorotolyl)dimethylsilanolate, potassium (4-fluorophenyl)dimethylsilanolate, and potassium (1-naphthyl)dimethylsilanolate all cross-couple successfully to give high yields of the desired unsymmetrical biaryls (Scheme 21). Current efforts are focused on expanding the scope in both the aryldimethylsilanolate and aryl halide.
Scheme 21.

4. Silanolate Exchange. Vinylation of Aryl Iodides and Bromides
Logically, dimethylvinylsilanol (55) would be the reagent of choice for the vinylation of aryl halides, but this compound suffers spontaneous dehydrative dimerization to form divinyltetramethyldisiloxane (56, DVDS). Given the facility of siloxane formation from unhindered silanols, we considered the intriguing possibility of employing an in-situ generation of a dimethylvinylsilanolate (or its equivalent) from readily available precursors (Chart 1).
Chart 1.

For this strategy to be successful, the active silanolate must be generated in equilibrium by the action of a non-participating activator with a siloxane precursor. A series of orienting experiments revealed that KOSiMe3 facilitates rapid exchange with DVDS in DMF at room temperature to form potassium dimethylvinylsilanolate (K+55-) in-situ (equation 7).38
 |
(eq. 7) |
 |
(eq. 8) |
Optimization studies demonstrated that the combination of DVDS, 4.0 equiv of KOSiMe3, [allylPdCl]2 and a phosphine oxide efficiently converts 4-iodoanisole to the corresponding styrene 61a in 95% yield (equation 8). For preparative reactions, KOSiMe3 (3.5 equiv, per iodide), DVDS (0.75 equiv), 2.5 mol % of Pd(dba)2, and 5 mol % of Ph3P=O at room temperature provide facile vinylations with a variety of aryl iodides (Scheme 22).
Scheme 22.

Only minor modifications of these conditions were required to extend the reaction to include aryl bromides (Scheme 23).38 Still milder reaction conditions were developed that employ 3.0 equiv of KOSiEt3, 2.5 mol % of [allylPdCl]2 with 5 mol % of ligand 62. These conditions proved to be general for a wider range of aryl bromides (Scheme 24).
Scheme 23.

Scheme 24.

Conclusion and Outlook
The initial efforts to develop a fluoride-free method of cross-coupling of organosilanols were based upon a simple hypothesis that ultimately led to the development of a new paradigm for cross-coupling reactions. The defining feature of this process is the competency of a neutral palladium silanolate that refutes the longstanding notion that pentacoordinate silicates are required for successful transmetalation of organosilanes. With this knowledge, four different protocols were developed for the formation of the active silanolate species ranging from alkenyl, alkynyl, aryl, heteroaryl, to bissilylbutadienes. In addition, the unique nature of silicon has enabled for the amalgamation of the fluoride-promoted cross-coupling of organosilanes and this new silanolate-based process for the development of sequential cross-coupling reactions.
The discovery of the fluoride-free process continues to stimulate the development of milder and more diverse conditions and reagents for organosilicon cross-coupling reactions. Current studies are focused on the demonstration of vinyl-vinyl cross-coupling and the more difficult cross-coupling of alkylsilanols. Mechanistic studies are underway to elucidate the molecular detail of the elementary steps in the transmetalation of palladium silanolates as well as the potential similarities to the transmetalation process for organoboron compounds. Finally, more exploratory endeavors involve the use of silanolates as precursors for the generation of organometallic species derived from other elements, such as gold, nickel, copper, and rhodium.
From our current vantage point, it is remarkable that our initial foray into the field of cross-coupling chemistry began modestly with the testing of the strain-release Lewis acidity concept and now has developed into a stimulating and rewarding endeavor that continues to surprise, entice, and challenge us at many different levels.
Acknowledgements
We are pleased to acknowledge the experimental and intellectual contributions of our co-workers in the cross-coupling subgroup, Aaron Bailey, John Baird, Christopher Butler, Timothy Chang, Christophe Eggertswyler, Jack Liu, Jeff Kallemeyn, Tetsuya Kobayashi, Joseck Muhuhi, Michael Ober, Russell Smith, Ramzi Sweis, Steve Tymonko, and Nathan Werner, whose efforts have forged ideas into realities. Funding for this research was provided by the National Institutes of Health (GM63167), Johnson and Johnson, Merck, and Aldrich. We are also grateful to Johnson Mathey for gifts of palladium catalysts.
Biography
Scott E. Denmark was born in Lynbrook, New York on 17 June 1953. He obtained an S.B. degree from MIT in 1975 and his D.Sc.Tech from the ETH Zürich in 1980. That same year he began his career at the University of Illinois and since 1991 he has been the Reynold C. Fuson Professor of Chemistry. He is primarily interested in the invention of new synthetic reactions and the origin of stereocontrol in fundamental bond forming processes. Professor Denmark is currently the Editor-in-Chief of Organic Reactions, he is on the Board of Editors of Organic Syntheses and is a Co-Editor of Topics in Stereochemistry.
Christopher S. Regens was born in Chicago IL, on 18 November 1980. He received a B.A. degree in chemistry from Lake Forest College in 2003. He is currently a graduate student in the Denmark group at the University of Illinois Urbana-Champaign, where he is applying silicon-based cross-coupling reactions in the total synthesis of complex natural products.
References
- (1).For reviews, see:de Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. 2nd ed. Wiley-VCH; Weinheim: 2004. Negishi E, editor. Handbook of Organopalladium Chemistry. Wiley-Interscience; New York: 2002.
- (2).(a) Corriu RJP, Massé JP. Activation of Grignard Reagents by Transition-Metal Complexes. A New and Simple Synthesis of Trans-Stilbenes and Polyphenyls. J. Chem. Soc. Chem. Commun. 1972:144a. [Google Scholar]; (b) Tamao K, Sumitani K, Kumada M. Selective Carbon-Carbon Bond Formation by Cross-Coupling of Grignard Reagents with Organic Halides. Catalysis by Nickel-Phosphine Complexes. J. Am. Chem. Soc. 1972;94:4374–4376. [Google Scholar]
- (3).For reviews on silicon-based cross-coupling, see:Hiyama T. Organosilicon Compounds in Cross-coupling Reactions. In: Diederich F, Stang PJ, editors. Metal-Catalyzed Cross-Coupling Reactions. Wiley-VCH; Weinheim: 1998. Chapter 10.Denmark SE, Sweis RF. Organosilicon Compounds in Cross-Coupling Reactions. In: de Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. Vol. 1. Wiley-VCH; Weinheim Germany: 2004. Chapter 4.Spivey AC, Gripton CJG, Hannah JP. Recent Advances in Group 14 Cross-Coupling: Si and Ge-Based Alternatives to the Stille Reaction. Curr. Org. Synth. 2004;1:211–226.
- (4).Walsh R. Bond Dissociation Energy Values in Silicon-Containing Compounds and Some of Their Implications. Acc. Chem. Res. 1981;14:246–252. [Google Scholar]
- (5).Damrauer R, Danahey SE. Preparation and NMR Studies of Pentacoordinated Silicon Anions. Organometallics. 1986;5:1490–1494. [Google Scholar]
- (6).Hatanaka Y, Hiyama T. Cross-Coupling of Organosilanes with Organic Halides Mediated by Palladium Catalyst and Tris(diethy1amino)sulfonium Difluorotrimethylsilicate. J. Org. Chem. 1988;53:918–920. [Google Scholar]
- (7).For other examples of silicon-based cross-coupling, see:Hirabayashi K, Kondo T, Toriyama F, Nishihara Y, Mori A. Substituent Effect of 3,3,3-Trifluoropropyl Group on Organic Silanols. Palladium-Mediated Mizoroki-Heck Type and Cross-Coupling Reactions. Bull. Chem. Soc. Jpn. 2000;73:749–750.Lee HM, Nolan SP. Efficient Cross-Coupling Reactions of Aryl Chlorides and Bromides with Phenyl- or Vinyltrimethoxysilane Mediated by a Palladium/Imidazolium Chloride System. Org. Lett. 2000;2:2053–2055. doi: 10.1021/ol005956t.Shibata K, Miyazawa K, Goto Y. Preparation of Functionalized Biaryl Compounds via Cross-coupling Reactions of Aryltrialkoxysilanes with Aryl Bromides. Chem. Commun. 1997:1309–1310.Handy CJ, Manoso AS, McElroy WT, Seganish WM, DeShong P. Recent Advances in Siloxane-Based Aryl-Aryl Coupling Reactions: Focus on Heteroaromatic Systems. Tetrahedron. 2005;61:12201–12225.
- (8).(a) Itami K, Nokami T, Yoshida J. Palladium-Catalyzed Cross-Coupling Reaction of Alkenyldimethyl(2-pyridyl)silanes with Organic Halides: Complete Switch from the Carbometalation Pathway to the Transmetalation Pathway. J. Am. Chem. Soc. 2001;123:5600–5601. doi: 10.1021/ja015655u. [DOI] [PubMed] [Google Scholar]; (b) Hosoi K, Nozaki K, Hiyama T. Alkenyldimethyl(2-thienyl)silanes, Excellent Coupling Partner for the Palladium-Catalyzed Cross-Coupling Reaction. Chem. Lett. 2002:138–139. [Google Scholar]; (c) Trost BM, Machacek MR, Ball ZT. Ruthenium-Catalyzed Vinylsilane Synthesis and Cross-Coupling as a Selective Approach to Alkenes: Benzyldimethylsilyl as a Robust Vinylmetal Functionality. Org. Lett. 2003;5:1895–1898. doi: 10.1021/ol034463w. [DOI] [PubMed] [Google Scholar]
- (9).Denmark SE, Wehrli D, Choi JY. Convergence of Mechanistic Pathways in the Palladium(0)-Catalyzed Cross-Coupling of Alkenylsilacyclobutanes and Alkenylsilanols. Org. Lett. 2000;2:2491–2494. doi: 10.1021/ol006170y. [DOI] [PubMed] [Google Scholar]
- (10).Denmark SE, Sweis RF, Wehrli D. Fluoride-Promoted Cross-Coupling Reactions of Alkenylsilanols. Elucidation of the Mechanism through Spectroscopic and Kinetic Analysis. J. Am. Chem. Soc. 2004;126:4865–4875. doi: 10.1021/ja037234d. [DOI] [PubMed] [Google Scholar]
- (11).(a) Denmark SE, Sweis RF. Cross-Coupling Reactions of Organosilicon Compounds: New Concepts and Recent Advances. Chem. Pharm. Bull. 2002;50:1531–1541. doi: 10.1248/cpb.50.1531. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Sweis RF. Design and Implementation of New, Silicon-Based, Cross Coupling Reactions: Importance of Silicon-Oxygen Bonds. Acc. Chem. Res. 2002;35:835–846. doi: 10.1021/ar020001r. [DOI] [PubMed] [Google Scholar]; (c) Denmark SE, Ober MH. Organosilicon Reagents: Synthesis and Application to Palladium-Catalyzed Cross-Coupling Reactions. Aldrichimica Acta. 2003;36:75–85. [Google Scholar]; (d) Denmark SE, Baird JD. Palladium-Catalyzed Cross-Coupling Reactions of Silanolates: A Paradigm Shift in Silicon-Based Cross-Coupling Reactions. Chem. Eur. J. 2006;12:4954–4963. doi: 10.1002/chem.200600034. [DOI] [PubMed] [Google Scholar]
- (12).Denmark SE, Sweis RF. Fluoride-Free Cross-Coupling of Organosilanols. J. Am. Chem. Soc. 2001;123:6439–6440. doi: 10.1021/ja016021q. [DOI] [PubMed] [Google Scholar]
- (13).Echavarren AM, Cardenas DJ. Mechanistic Aspects of Metal-Catalyzed C, C- and C,X-Bond Forming Reations. In: de Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. 2nd Ed. Vol. 1. Wiley-VCH; Weinheim: 2004. Chapter 1. [Google Scholar]
- (14).Espinet P, Echavarren AM. The Mechanism of the Stille Reaction. Angew. Chem., Int. Ed. 2004;43:4704–4734. doi: 10.1002/anie.200300638. [DOI] [PubMed] [Google Scholar]
- (15).Denmark SE, Neuville L, Christy MEL, Tymonko SA. A Qualitative Examination of the Effects of Silicon Substituents on the Efficiency of Cross-Coupling Reactions. J. Org. Chem. 2006;71:8500–8509. doi: 10.1021/jo061481t. [DOI] [PubMed] [Google Scholar]
- (16).Denmark SE, Sweis RF. Cross-Coupling Reactions of Alkenylsilanolates. Investigation of the Mechanism and Identification of Key Intermediates through Kinetic Analysis. J. Am. Chem. Soc. 2004;126:4876–4882. doi: 10.1021/ja0372356. [DOI] [PubMed] [Google Scholar]
- (17).Although the kinetic experiments were performed for 2-iodothiophene (to facilitate the kinetic analysis) we believe that this scheme is general for coupling of all aromatic halides. Moreover, this picture allows a clearer insight into how the turnover-limiting step can change with structure.
- (18).(a) Denmark SE, Ober MH. Cross-Coupling Reactions of Arylsilanols with Substituted Aryl Halide. Org. Lett. 2003;5:1357–1360. doi: 10.1021/ol034328j. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Ober MH. Palladium-Catalyzed Cross-Coupling Reactions of Substituted Aryl(dimethyl)silanols. Adv. Synth. Catal. 2004;346:1703–1714. [Google Scholar]
- (19).Denmark SE, Tymonko SA, Smith RC, Ober MH. Palladium-Catalyzed Cross-Coupling Reactions of Alkenyl- and Aryl(dimethyl)silanolates Proceed via Neutral (8-Si-4) Arylpalladium Silanolate Complexes. Manuscript submitted [Google Scholar]
- (20).Denmark SE, Wang Z. Palladium Catalyzed Cross-Coupling of (Z)-1-Heptenyldimethylsilanol with 4-iodoanisole: (Z)-1-Heptenyl-4-methoxybenzene. Org. Synth. 2004;81:42–53. [Google Scholar]
- (21).Stang PJ, Diederich F, editors. Modern Acetylene Chemistry. Wiley-VCH; Weinheim: 1995. [Google Scholar]
- (22).Sonogashira K. Cross-Coupling Reactions to sp Carbon Atoms. In: Diederich F, Stang PJ, editors. Metal-Catalyzed, Cross-Coupling Reactions. Wiley-VCH; Weinheim: 1995. Chapter 5. [Google Scholar]
- (23).For a recent review of organometallic reagents in the Sonogashira reaction, see:Negishi E, Anastasia L. Palladium-Catalyzed Alkynylation. Chem. Rev. 2003;103:1979–2018. doi: 10.1021/cr020377i.
- (24).(a) Hatanaka Y, Matsui K, Hiyama T. A One-Pot Synthesis of Conjugated Dienynes by Palladium-Mediated Three Component Cross-Coupling Reaction. Tetrahedron Lett. 1989;30:2403–2406. [Google Scholar]; (b) Koseki Y, Omino K, Anzai S, Nagasaka T. Silver Salt-Promoted Direct Cross-Coupling Reactions of Alkynylsilanes with Aryl Iodides: Synthesis of Aryl-Substituted Alkynylamides. Tetrahedron Lett. 2000;41:2377–2380. [Google Scholar]; (c) Bertus P, Halbes U, Pale P. Pd/Ag-Catalyzed Direct Coupling of 1-Trimethylsilyl Alkynes with Vinyl Triflates. Eur. J. Org. Chem. 2001:4391–4393. [Google Scholar]
- (25).Denmark SE, Tymonko SA. Cross-Coupling of Alkynylsilanols with Aryl Halides Promoted by Potassium Trimethylsilanolate. J. Org. Chem. 2003;68:9151–9154. doi: 10.1021/jo0351771. [DOI] [PubMed] [Google Scholar]
- (26).(a) Cacchi S, Fabrizi G. Synthesis and Functionalization of Indoles Through Palladium-Catalyzed Reactions. Chem. Rev. 2005;105:2873–2920. doi: 10.1021/cr040639b. [DOI] [PubMed] [Google Scholar]; (b) Humphrey GR, Kuethe JT. Practical Methodologies for the Synthesis of Indoles. Chem. Rev. 2006;106:2875–2911. doi: 10.1021/cr0505270. [DOI] [PubMed] [Google Scholar]
- (27).Denmark SE, Baird JD, Regens CS. Palladium-Catalyzed Cross-Coupling of Five-Membered Heterocyclic Silanolates. J. Org. Chem. 2008;73:1440–1455. doi: 10.1021/jo7023784.For the preparation of 18 and 19 see references within.
- (28).Denmark SE, Kallemeyn JM. Synthesis of 3,4,5-Trisubstituted Isoxazoles via Sequential [3+2] Cycloaddition/Silicon-Based Cross-Coupling Reactions. J. Org. Chem. 2005;70:2839–2842. doi: 10.1021/jo047755z. [DOI] [PubMed] [Google Scholar]
- (29).Denmark SE, Regens CS, Kobayashi T. Total Synthesis of Papulacandin D. J. Am. Chem. Soc. 2007;129:2774–2776. doi: 10.1021/ja070071z. [DOI] [PubMed] [Google Scholar]
- (30).Denmark SE, Tymonko SA. Sequential Cross-Coupling of 1,4-Bissilylbutadienes: Synthesis of Unsymmetrical 1,4-Disubstituted 1,3-Butadienes. J. Am. Chem. Soc. 2005;127:8004–8005. doi: 10.1021/ja0518373. [DOI] [PubMed] [Google Scholar]
- (31).Littke AF, Fu GC. Palladium-Catalyzed Coupling Reactions of Aryl Chlorides. Angew. Chem. Int. Ed. 2002;41:4176–4211. doi: 10.1002/1521-3773(20021115)41:22<4176::AID-ANIE4176>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- (32).(a) Old DW, Wolfe JP, Buchwald SL. A Highly Active Catalyst for Palladium-Catalyzed Cross-Coupling Reactions: Room-Temperature Suzuki Couplings and Amination of Unactivated Aryl Chlorides. J. Am. Chem. Soc. 1998;120:9722–9723. [Google Scholar]; (b) Littke AF, Fu GC. The First General Method for Stille Cross-Couplings of Aryl Chlorides. Angew. Chem. Int. Ed. 1999;38:2411–2413. doi: 10.1002/(sici)1521-3773(19990816)38:16<2411::aid-anie2411>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]; (c) Navarro O, Kelly RA, III., Nolan SP. A General Method for the Suzuki-Miyaura Cross-Coupling of Sterically Hindered Aryl Chlorides: Synthesis of Di- and Tri-ortho-substituted Biaryls in 2-Propanol at Room Temperature. J. Am. Chem. Soc. 2003;125:16194–16195. doi: 10.1021/ja038631r. [DOI] [PubMed] [Google Scholar]; (d) Bedford RB, Cazin CSJ, Holder D. The Development of Palladium Catalysts for C-C and C-Heteroatom bond Forming Reactions of Aryl Chloride Substrates. Coord. Chem. Rev. 2004;248:2283–2321. [Google Scholar]
- (33).Barder TE, Walker SD, Martinelli JR, Buchwald SL. Catalysts for Suzuki-Miyaura Coupling Processes: Scope and Studies of the Effect of Ligand Structure. J. Am. Chem. Soc. 2005;127:4685–4696. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]
- (34).Denmark SE, Kallemeyn JM. Stereospecific Palladium-Catalyzed Cross-Coupling of (E)- and (Z)-Alkenylsilanolates with Aryl Chlorides. J. Am. Chem. Soc. 2006;128:15958–15959. doi: 10.1021/ja065988x. [DOI] [PubMed] [Google Scholar]
- (35).Denmark SE, Fujimori S. Total Synthesis of RK-397. J. Am. Chem. Soc. 2005;127:8971–8973. doi: 10.1021/ja052226d. [DOI] [PubMed] [Google Scholar]
- (36).Denmark SE, Smith RC, Tymonko SA. Phosphine Oxides as Stabilizing Ligands for the Palladium-Catalyzed Cross-Coupling of Potassium Aryldimethylsilanolates. Tetrahedron. 2007;63:5730–5738. doi: 10.1016/j.tet.2007.02.017.The origin of the beneficial effect of phosphine oxides is still not certain. In our current hypothesis, phosphine oxides stabilize palladium(0) nanoparticles and prevent precipitation of palladium black.
- (37).Denmark SE, Smith RC, Muhuhi JM, Chang T. Cross-Coupling of Substituted Arylsilanolates. manuscript in preparation. [Google Scholar]
- (38).Denmark SE, Butler CR. A Practical Method for the Vinylation of Aromatic Halides using Inexpensive Organosilicon Reagents. J. Am. Chem. Soc. 2008;130:3690–3704. doi: 10.1021/ja7100888. [DOI] [PMC free article] [PubMed] [Google Scholar]