

Abstract
Traditionally, large scale genotyping projects have used DNA derived from whole blood or lymphoblast cell lines. But over the past several years, a number of investigators have begun to use DNA prepared from saliva for genotyping studies, particularly for use in behavioral genetic studies. However, the comparability of DNA from these two sources has not been rigorously analyzed by unbiased sources. In this communication, we compare the single nucleotide polymorphism (SNP) genotyping results from DNA derived from whole blood samples obtained from 474 subjects from the Iowa Adoption Studies with that saliva samples prepared from 555 members of the Strong African American Families project. We found that DNA prepared from whole blood performed significantly better than that prepared from saliva. Genotyping success was significantly associated with the concentration of human DNA in the saliva sample as determined by quantitative PCR, but not with the total amount of DNA as determined by UV spectroscopy. We conclude that investigators contemplating the choice of source materials of DNA for genotyping studies will need to balance the ease and economy of saliva based DNA collection methods with the higher yields and rates of genotyping calls associated with DNA prepared from whole blood.
Keywords: saliva, DNA, blood, genotyping
INTRODUCTION
DNA for genotyping studies can be produced from virtually any tissue. However, for the past 30 years, DNA prepared from whole blood or lymphoblast cell lines has dominated the landscape in human genetic studies. This has occurred despite the fact in order to get blood, subjects must undergo phlebotomy which is a potentially painful procedure that must be conducted by a skilled individual (Fetzer 1999).
However, alternative methods of obtaining material from which to prepare DNA have emerged. One of the most popular of these methods prepares DNA from saliva samples (Steinberg, Beck et al. 2002). This painless method of biomaterial collection is made even more attractive by the availability of inexpensive, easy to use, commercially available kits. Unfortunately, unbiased systematic evaluations comparing the suitability of this DNA derived from saliva versus that derived from blood for genetic studies are not readily available.
Since our consortium is planning large-scale genetic studies, we are examining potential trade-offs between methods in order to determine which DNA collection method will work best for our studies. During pilot genotyping studies in which we amplified the serotonin transporter (5HTT) promoter VNTR (variable nucleotide repeat), the monoamine oxidase A (MAOA) VNTR, the dopamine receptor 4 (DRD4) and the HOPA12bp insertional polymorphism, we found that the use of relatively high amounts of saliva derived (SD) DNA was necessary in order to obtain adequate PCR products for our polyacrylamide based genotyping approaches. Since the amount of DNA needed for VNTR genotyping and the amount needed for fluorescent SNP genotyping approaches may be correlated, our pilot findings with VNTRs could indicate the potential for general and systemic difficulties in our use of SD DNA in our genetic studies.
To help determine the suitability of SD DNA for our studies, we compared the DNA quantities and genotyping efficiencies of DNA prepared from whole blood (WB) with that prepared from saliva.
Methods
DNA was derived from two subject bases with all protocols being approved by the human subjects committees at both the University of Iowa and the University of Georgia. The first set of DNA, which was derived from whole blood (WB), was obtained from biomaterial contributed by probands in the Iowa Adoption Studies (IAS), a large case and control study of the role of genetic factors and gene-environment interactions in the etiology of behavioral illness (Yates, Cadoret et al. 1998). After consent was obtained, IAS subjects were phlebotomized by a certified phlebotomist and resulting tubes of blood were shipped via overnight courier or university car to our laboratory in Iowa City. After receipt in the lab, DNA from a 15 ml sodium citrate tube of blood was obtained using the cold protein precipitation method (Lahiri and Nurnberger 1991).
The second set of DNA samples, which was derived from saliva (SD for saliva derived) was obtained from biomaterial contributed by subjects in the Strong African American Family (SAAF) studies. The SAAF cohort is a longitudinal study of 650 African American caretaker- (e.g. parent or guardian) target (i.e., children, current age 18-19) pairs with respect to the role of behavioral and environmental on psychological well being (Brody, Murry et al. 2004). DNA from these caretaker-samples was processed using materials and methods using Oragene™ kits (DNA Genetek, Canada). Subjects rinsed their mouths, then deposited 4 ml of saliva into the Oragene sample container after receiving instructions from a trained research assistant. The vial was sealed, inverted then shipped via courier to a central laboratory in Iowa City where samples were prepped according to the manufacturer’s specifications including the use of an optional chloroform extraction. Both types of DNA samples were then quantified spectrophotometrically using an Eppendorf Biophotometer (Eppendorf, Hamburg) using the default OD260 nm absorbance algorithm, then stored at -20°C until use.
Genotyping was performed using our standard methods (Philibert, Gunter et al. 2006). Adoptee DNA was diluted to 10 ng/ul and saliva DNA was diluted to 100 ng/ul in 10 mM Tris, 0.1 mM EDTA, pH 8.0. DNA solutions were then robotically aliquotted to 384 well plates, which were centrifuged, visually inspected to insure the presence of sample in each well, then finally left to dry overnight at room temperature. Each 384 well plate contains 8 “no template” controls to help assess the possibility of systematic contaminations. SD and WB DNA were treated identically with two major exceptions. First, because in our initial genotyping experiments, SD DNA did not perform well and increasing amounts of SD DNA gave better results, we plated up to 100 ng of SD DNA in each well with the actual amount of DNA pipetted being retained for all calculations. Second, as internal controls, each of the SD DNA plates had 5 WB DNA samples at concentration of 10 ng per well.
For one set of experiments, the genotyping of DNA aliquots from a series of serial dilutions 24 SD DNA samples was compared. These serial dilutions were prepared by diluting the original plating concentration of each DNA sample (up to 100 ng/μl; see Table I) 1:5, 1:25 and 1:100 with a buffer solution containing 10 mM Tris and 0.1 mM EDTA, pH 8.0. One μl of each solution, including the original, were then plated robotically and processed as described above.
Table I.
Distribution of Saliva DNA concentrations prior to plating.
| 100 ng/ μl | 283 |
| > 50 ng/μl | 153 |
| > 25 ng/μl | 84 |
| > 10 ng/μl | 32 |
| > 1 to 10 ng/μl | 5 |
DNA concentrations were determined spectrophotometrically as described in the methods.
For a second set of experiments, the total human DNA concentration of 188 samples was assessed using standard quantitative PCR techniques and reagents, including a primer probe set recognizing the RNase P locus and human DNA standards, from Applied Biosystems, used according to the manufacturer’s suggestions (Applied Biosystems, Foster City, CA). DNA concentration for each sample was imputed using the 2-ΔΔCT method (Livak and Schmittgen 2001).
These samples were then PCR amplified in 5 μl volumes using the following ABI primer probe sets (the ABI reference ID is given in parentheses) and Universal Master mix in accordance with the manufacturer’s directions (Applied Biosystems, Foster, CA): rs5905613 (hCV30227161), rs3788862 (hCV11180605), rs909525 (hCV8817688), rs302739 (hCV11180591), rs2283724 (hCV15959449), rs1800659 (hCV9459989), rs979606 (hCV8878819), rs206407 (hCV2491071), rs1799836 (hCV8878790), rs2283729 (hCV15959461). Genotype was then determined for each of the samples using an ABI 7900 HT Real Time system using the SDS version 2.1 software at the default settings.
Success of genotyping at each locus, which was defined by the proportion of samples whose genotypes were called by the SDS genotyping program using the default status, was compared using Chi-square or ANOVA analysis using the JMP suite of programs (SAS Institute, Cary, N.C.) as indicated in the text. The relationship between UV and RNase P DNA concentrations was analyzed using Pearson’s correlation coefficient (Fleiss 1981).
RESULTS
As expected, the amount of DNA collected as measured by UV spectroscopy using each method differed. The average amount of DNA derived from blood collected using a 15 ml citrate venipuncture collection tube was 277 ± 214 μg of DNA (n= 474). The amount of DNA obtained from saliva using the commercially derived kit was lower 92 ± 74 (SD) μg per 4 ml saliva sample (n=555).
The lower amounts of DNA derived from saliva samples presented an initial challenge to systematic handling. In our preliminary “pilot” VNTR genotyping experiments we found that the use of 30 ng of SD DNA per 10 ul reaction was associated with a much lower success rate than the use of 100 ng per 10 ul reaction. Therefore, since the saliva kit manufacturer has suggested that UV spectroscopy may not adequately assess the true concentration of DNA prepared using their methods and material, for these experiments we plated the saliva DNA in amounts up to 100 ng per well. Unfortunately, because some of SD DNA yields were very low and we wished to use sytematic (e.g., robotic fluid handling) approaches to both methods of preparing and aliquoting DNA, we were not able to prepare stock SD DNA solutions with concentrations ≥ 100 ng/μl using standardized methods for some samples. Hence, were we not able to robotically dispense 100 ng of SD DNA to each well of the 384 well sample plates for some of the samples in this set of experiments. Table I gives the distribution of the concentration of the stock SD DNA used in these experiments. In contrast because of the higher yields of DNA obtained from whole blood and the high success rate of VNTR genotyping using 30 ng of DNA per 10 ul PCR reaction, we were able to make 10 ng/μl stocks for all WB samples and dispense 10 ng of DNA to each well for all WB samples.
In order to systematically assess the suitability for large scale efforts, we genotyped each of these DNA samples at 10 SNP polymorphisms at or near the Xp11 MAOA locus (Table I). A total of 474 IAS and 555 SAAF DNA samples were genotyped for the 10 SNP loci. Overall, 12 IAS samples and 85 SAAF samples failed at two or more of these SNP loci, and 4 IAS and 40 SAAF samples failed at all loci. The summed success rate at all 10 loci combined was 98.0% for WB DNA and 93.7% for SD DNA. Table II lists the MAOA SNPs that were genotyped and their overall success rate using the default 95% confidence setting of the ABI SDS allele calling software for both IAS and SAAF subjects. In nine of the 10 SNPs examined, WB DNA performed significantly better than SD DNA. The difference was not attributable to problems with adolescent samples. As can be seen in table II, there was no significant difference between adolescents and adults in success rate using SD DNA.
Table II.
Genotyping Failure Rates at 10 SNP loci in and around MAOA
| rs5905613 | rs3788862 | rs909525 | rs979606 | rs206407 | rs2283729 | rs1799836 | rs302739 | rs1800659 | rs2283724 | |
|---|---|---|---|---|---|---|---|---|---|---|
| WB DNA | 1.5% (7/474) | 1.9% (9/474) | 3.0% (14/474) | 3.4% (16/474) | 1.7% (8/474) | 1.3% (6/474) | 1.7% (8/474) | 2.3% (11/474) | 1.3% (6/474) | 1.1% (5/474) |
| SD DNA Total | 6.7% (37/555) | 4% (22/555) | 4.1% (23/555) | 9.9% (55/555) | 3.8% (21/555) | 5.4% (30/555) | 6.8% (38/555) | 7.2% (40/555) | 5.2% (29/555) | 6.7% (37/555) |
| SD ADULT | 6.8% (19/278) | 4.3% (12/278) | 4.7% (13/278) | 10.4% (29/278) | 4% (11/278) | 6.5% (18/278) | 6.8% (19/278) | 7.6% (21/278) | 5% (14/278) | 6.8% (19/278) |
| SD ADOLESCENT | 6.5% (18/277) | 3.6% (10/277) | 3.6% (10/277) | 9.4% (26/277) | 3.6% (10/277) | 4.3% (12/277) | 6.9% (19/277) | 6.9% (19/277) | 5.4% (15/277) | 6.5% (18/277) |
| Test between WB and SD DNA | z=4.10 p=.000 | z=1.93 p=.053 | z=1.02 p=.306 | z=4.14 p=.000 | z=2.03 p=.042 | z=3.60 p=.000 | z=3.99 p=.000 | z=3.60 p=.000 | z=3.49 p=.000 | z=4.53 p=.000 |
| Test between SD for Adult and Adolecent | z=.16 p=.873 | z=.43 p=.699 | z=.63 p=.528 | z=.41 p=.68 | z=.21 p=.83 | z=1.12 p=.264 | z=.11 p=.99 | z=.32 p=.751 | z=.20 p=.84 | z=.16 p=.873 |
Abreviations: WB: Whole Blood; SD: Saliva Derived
The marked contrast in genotyping success rates between the two types of DNA observed above could be secondary to several reasons. However, since the ability of the ABI SDS program to accurately determine genotypes is dependent on the strength and uniformity of fluorescent signal, we first compared and contrasted the clustering diagrams and signal strengths obtained using the SD DNA with the WB DNA for the hemizygous males and homozygous females at each of the SNP loci. For each genotype at each SNP locus, the average signal strength was greater and the distribution of the resulting fluorescent signals tighter for those samples prepared from blood than from saliva (data not shown). Visual inspection of the allelic discrimination plots for each of the loci confirmed this observation (Figure 1). These differences were not secondary to variability in inter-run PCR efficiency since in each case the 5 internal IAS WB DNA samples embedded as controls in the SD DNA plates gave excellent signal. So, it appears that differences in fluorescent signals were the likely source of different call rates for SD DNA and WB DNA.
Figure 1.
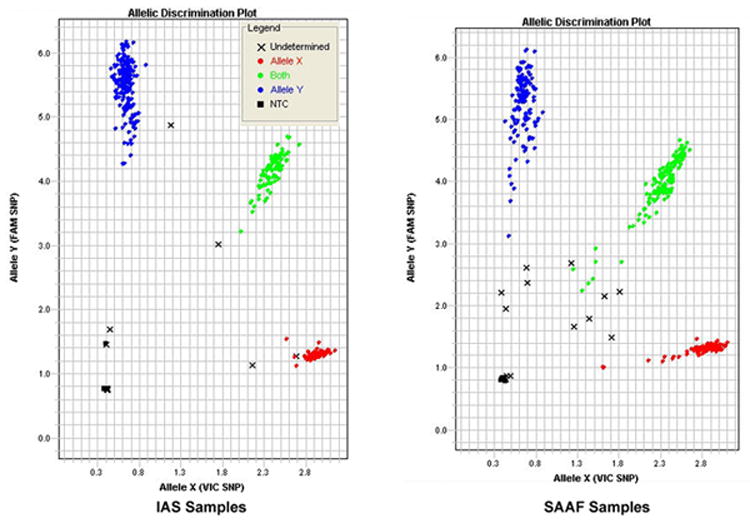
A representative comparison of allelic discrimination plots. These are the results at rs2064040 using single 384 well plates containing 376 IAS whole blood derived (left) and 371 SAAF saliva derived (right) DNA samples. Fluorescent Fam (T) signal is on the Y axis. Fluorescent Vic Signal is on the X axis. Blue and red dots represent DNA samples determined by the ABI SDS software to be T or A homo- or hemizygous DNA samples respectively, while green dots represent DNA from heterozygous females. “X” represent uncalled alleles. Filled boxes represent no template controls.
However, it remained to determined the source of the difference in strength and uniformity of fluorescent signal between the two types of samples. In our experience, the most common reason for such differences and resulting genotyping failures are the use of inadequate amounts or impure forms of DNA. So, we examined the relationship between SD DNA concentration and fluorescent signal in a subset of 24 SD DNA samples for 4 separate individual SNP markers, rs2064070, rs909525, rs979606 and rs5905613. For each of the markers, we found that there was a strong positive, linear relationship between total DNA concentration, as assessed by UV spectroscopy, and fluorescent signal with the highest concentrations of DNA always giving the best signal (p <0.0001 for each marker). As an example of the strength of this relationship, Figure 2 show a bivariate scatterplot demonstrating the relationship between Vic (A allele) fluorescence and DNA concentration at the rs5905613 locus for the 8 saliva samples that are either homozygous or hemizygous for the A(Vic) allele and their respective serial dilutions. As the figure demonstrates, there is a strong relationship between DNA concentration and Vic fluorescence (p<0.0001). So, differences in DNA concentration may be a potential source of genotyping failure.
Figure 2.
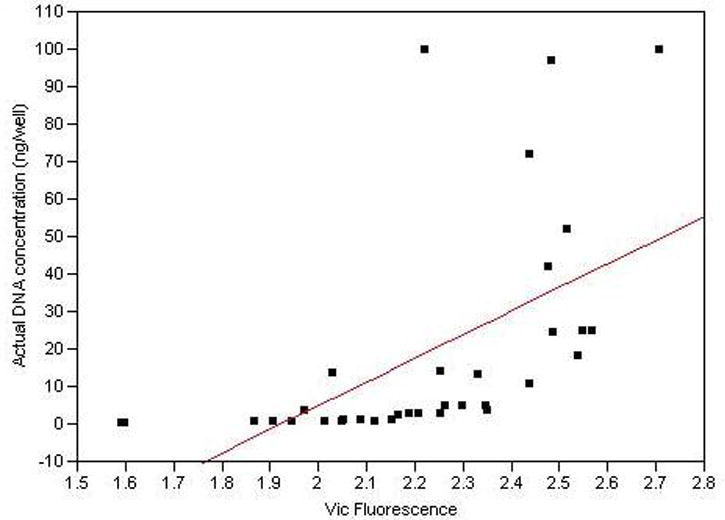
A typical plot demonstrating the relationship between fluorescence and UV determined SD DNA concentration. Twenty four SD DNA samples were serially diluted, PCR amplified and endpoint reads determined as described for four MAOA loci. This plot demonstrates the relationship between Vic fluorescence and DNA concentration for eight SD DNA samples either homo or hemizygous for the A allele at rs5905613 (p<0.0001)
To directly assess this possibility, we examined the relationship between genotyping success and various spectrophotometric measures with respect to the SD DNA. Surprisingly, there was no relationship between genotyping failure and UV determined DNA concentration, 260/280 or 260/230 ratios (data not shown). Most intriguingly, there was only one genotyping failure in the six SD DNA samples with the lowest DNA concentrations.
However, since UV spectroscopy does not discriminate between human and non-human (e.g. bacterial) DNA, we next assessed the amount of human DNA in 188 samples using a quantitative RTPCR approach and a primer probe set that recognizes the RNAse P locus, a well characterized single copy number gene (McNees, White et al. 2005). There was only a modest correlation between the amount of total DNA as determined by UV spectroscopy and the amount of human DNA as determined by RTPCR of the RNase P locus (Pearson r = 0.50; figure 3). More interestingly, in contrast to the lack of relationships between UV determined DNA values and genotyping success, there was a significant relationship between the amount of human DNA, as determined by the RNase P assay, and genotyping success at each of the 10 loci MAOA (p<0.005 for each locus; supplementary data).
Figure 3.
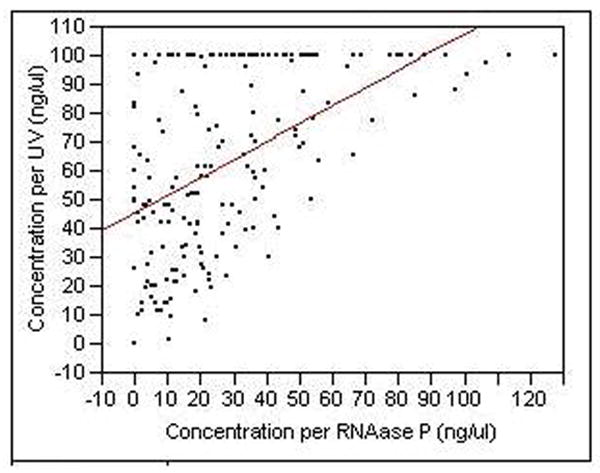
The concentration of DNA as assessed by UV spectroscopy and RTPCR at the RNase P locus for 188 of the DNA samples (ANOVA, p< 0.0001, Pearson correlation coefficient between the two sets of values is 0.50).
Finally, since factors affecting the suitability of SD DNA could be caused by heritable biological factors (e.g., PCR inhibitors in the saliva) segregating in the families of our subjects, we examined whether there was a relationship between genetic relatedness of subjects and the quality of the DNA prepared from their saliva. Relatedness of subjects predicted the success or failure of the DNA analysis, with mother’s and biological offspring showing greater than expected concordance on failure for two or more SNPs (Fisher’s exact test, P = .045, data not shown). However, there was no significant relationship between genotyping success and the individual research assistants who collected the DNA.
DISCUSSION
To our knowledge, this is the first large scale, systematic, non-industry affiliated comparison of genotyping results obtained using SD DNA with that obtained from WB DNA. However, before these results are discussed, a few caveats are in order. First, we used only one method for preparing saliva DNA. Use of other methods or other materials may provide better (or worse) results. Second, we only used one method of SNP genotyping. Although the ABI Taqman™ based approach is robust in our hands and those of others, the use of other methods could significantly improve genotyping yields. Third and finally, the saliva DNA and whole blood DNA were derived from different cohorts rendering direct comparison of DNA from the same subjects impossible and raising concerns that DNA sampling method and ethnicity of the sample are confounded. However, we are not aware of any data that suggests that global genotyping success rate varies with ethnicity for any method of DNA sampling. Nor were there any differences in genotyping success between those of European and African American ethnicity in the IAS cohorts (data not shown), indicating that the success rates reported for whole blood are representative of success rates for both European American and African American subsamples. Hence, we believe that the results obtained from the IAS whole blood samples are very similar to that what we would have obtained if we had genotyped whole blood DNA from the SAAF population.
The present findings strongly suggest that genotyping efforts using SD DNA have higher failure rates than those using WB DNA. However, the reasons for this difference may be multifactorial. Certainly factors in the collection and processing of the saliva could be relevant and could be a target for improvement in the future. But these data strongly suggest that low quantities of usable human DNA template may be the critical factor in many failures. This assertion is supported by two lines of evidence. First, there was a very strong relationship between genotyping success and the amount of human DNA as assessed by the RNase P assay. Second, the serial dilution of SD DNA experiment demonstrated that the relatively high amount of SD DNA actually improved the amount of fluorescent signal suggests that excess of DNA template was not a factor in the genotyping failures.
We also found a modestly significant relationship between genotyping success and subject relatedness (Exact p=0.045). Unfortunately, since the caretakers and targets share both genetic and environmental factors and we have only limited information about critical variables such as oral hygiene of the subjects, we cannot say whether this association arises from factors such as heritable inhibitors of PCR being present in the saliva of some subjects or environmental factors such as shared patterns of diet or oral hygiene.
We found only a modest relationship between the amount of SD DNA as determined by UV (r=0.50; figure 3) as compared to that by quantitative RTPCR. Therefore, the question arises as to what is the UV signal actually measuring? First, some of the signal is usable human template. Second, since two previous studies of saliva derived DNA have shown that only 62% and 49% of the total DNA was of human origin with very little degraded DNA being present, the UV signal is also measuring significant amounts of bacterial DNA (Garcia-Closas, Egan et al. 2001; Rylander-Rudqvist, Hakansson et al. 2006). Third and finally, since the manufacturer (Oragene) also states that co-purifying polysaccharides from the saliva interfere with UV assessment of DNA concentrations, a portion of the UV signal also is probably secondary to these polysaccharides with the exact proportion of each of these components varying from sample to sample.
Could the use of other easily performed benchtop methods more aptly quantify the DNA concentrations immediately after preparation? Some investigators prefer the use of pico-green fluoroscopy rather than UV spectroscopy to measure DNA concentrations immediately after initial DNA preparation and prior to plating for genotyping (or quantitative PCR) . But the use of pico-green is primarily advantageous at low DNA concentrations (less than 5 ng/ μl) and its use does not discriminate between DNA from human vs bacteria origin (Ahn, Costa et al. 1996). Therefore, since the vast majority of samples that failed had DNA concentrations of greater than 50 ng/μl and at those concentrations, values derived using UV and pico-green are highly correlated (Rylander-Rudqvist, Hakansson et al. 2006), it is unlikely that pico-green assessment of DNA concentration prior to genotyping would have substantially improved our success rate using SD DNA
Our success rate with SD DNA (93.7%, n=555) here is slightly lower than that obtained recently by Rylander and colleagues (96%, n=81) using the same kit and same DNA quantification methods (Rylander-Rudqvist, Hakansson et al. 2006). However, differences in subject characteristics and higher rate of calls afforded by their method of genotyping (matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy (Pusch, Wurmbach et al. 2002)) may account for the slight differences in the success rates. Unfortunately, since Rylander and colleagues did not compare results for saliva with results obtained using DNA from other sources, it is unknown whether they would have replicated the relative advantage of blood observed in the current investigation.
Reassuringly, re-sampling of the subjects after these experiments were completed provided usable amounts of DNA in 3 of the 5 samples tested so far. This suggests that the problems encountered in this study may not be static for each subject and that usable DNA can be obtained from the majority of subjects whose initial material was unsatisfactory if one round of follow-up saliva re-sampling is permitted in the clinical protocol. The success rate could also be augmented by the simultaneous use of buccal swabs in 1st round failures. These data provide both reassurance regarding the feasibility of protocols to limit loss of subjects in saliva based DNA collection as well as a strong rationale for including re-sampling in cost estimates for this approach.
How do the current findings impact research practice? If correct, they suggest that both whole blood and saliva based approaches to genotyping are viable and the choice of DNA collection methods largely depends on the type of study being conducted. Each of the DNA collection methods has drawbacks. In our experience, we have found that phlebotomy is more expensive to perform and is more poorly received by study subjects than collecting saliva. Furthermore, in our experience research assistants in behavioral studies are more loathe to draw blood than their counterparts in more “medically” oriented studies. But the current findings suggest that the likelihood of obtaining greater amounts of higher quality DNA blood is also greater. Furthermore, immortalized lymphoblast lines, which have a variety of uses, can be prepared from blood but not saliva. Therefore investigators pondering which method to use need to consider a variety of factors before making their decision and should keep in mind that a hybrid approach using both of these methods of sample collection is often possible.
In summary, we have compared the genotyping results obtained using DNA derived from whole blood with that from saliva and found that the use of DNA prepared from whole blood was associated with greater genotyping success. However, since the use of SD DNA has a number of advantages, we suggest that the choice of method for obtaining DNA largely depends on the balance between future analytic needs that may require use of whole blood, receptivity of the target population to phlebotomy vs. collection of saliva, and the feasibility of re-sampling. In addition, future investigations of the performance of SD DNA may identify ways to reduce potential interference from bacterial DNA or PCR inhibitors in saliva, thereby increasing the efficiency of genotyping from SD DNA and shifting the relative balance of costs and benefits.
Acknowledgments
This work was supported by NIH grant R01DA015789 (RAP), NIDA grants R01DA019230-01 and R01DA021736-01 (GHB), and NIAAA grant 2R01AA012768-06 (GHB).
References
- Ahn SJ, Costa J, et al. PicoGreen quantitation of DNA: effective evaluation of samples pre- or post-PCR. 1996;24:2623–2625. doi: 10.1093/nar/24.13.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody GH, Murry VM, et al. The Strong African American Families Program: translating research into prevention programming. Child Dev. 2004;75(3):900–17. doi: 10.1111/j.1467-8624.2004.00713.x. [DOI] [PubMed] [Google Scholar]
- Fetzer S. Reducing the pain of venipuncture. J Perianesth Nurs. 1999;14(2):95–101. doi: 10.1016/s1089-9472(99)80024-0. [DOI] [PubMed] [Google Scholar]
- Fleiss JL. Statistical Methods for Rates and Proportions. New York, NY: John Wiley & Sons Inc; 1981. [Google Scholar]
- Garcia-Closas M, Egan KM, et al. Collection of genomic DNA from adults in epidemiological studies by buccal cytobrush and mouthwash. Cancer Epidemiol Biomarkers Prev. 2001;10(6):687–96. [PubMed] [Google Scholar]
- Lahiri DK, Nurnberger JI., Jr A rapid non-enzymatic method for the preparation of HMW DNA from blood for RFLP studies. Nucleic Acids Research. 1991;19(19):5444. doi: 10.1093/nar/19.19.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- McNees AL, White ZS, et al. Specific and quantitative detection of human polyomaviruses BKV, JCV, and SV40 by real time PCR. Journal of Clinical Virology. 2005;34(1):52–62. doi: 10.1016/j.jcv.2004.12.018. [DOI] [PubMed] [Google Scholar]
- Philibert R, Gunter T, et al. No association of the C677T methylenetetrahydrofolate reductase polymorphism with schizophrenia. Psychiatr Genet. 2006;16(5):221–3. doi: 10.1097/01.ypg.0000242192.28526.fa. [DOI] [PubMed] [Google Scholar]
- Pusch W, Wurmbach J-H, et al. MALDI-TOF mass spectrometry-based SNP genotyping. 2002;3:537–548. doi: 10.1517/14622416.3.4.537. [DOI] [PubMed] [Google Scholar]
- Rylander-Rudqvist T, Hakansson N, et al. Quality and Quantity of Saliva DNA Obtained from the Self-administrated Oragene Method--A Pilot Study on the Cohort of Swedish Men. 2006;15:1742–1745. doi: 10.1158/1055-9965.EPI-05-0706. [DOI] [PubMed] [Google Scholar]
- Steinberg K, Beck J, et al. DNA banking for epidemiologic studies: a review of current practices. Epidemiology. 2002;13(3):246–54. doi: 10.1097/00001648-200205000-00003. [DOI] [PubMed] [Google Scholar]
- Yates W, Cadoret R, et al. The Iowa Adoption Studies Methods and Results. In: LaBuda M, Grigorenko E, editors. On the way to individuality: Methodological Issues in Behavioral Genetics. Hauppauge NY: Nova Science Publishers; 1998. pp. 95–125. [Google Scholar]